
Phosphorylation-Induced Ubiquitination and Degradation of PXR through CDK2-TRIM21 Axis


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids and siRNA
2.3. Cell Viability Assay
2.4. Western Blot Analysis
2.5. Quantitative Reverse Transcription-Polymerase Chain Reaction
2.6. Immunoprecipitation and Ubiquitination Assays
2.7. Immunofluorescence Analysis
2.8. Primary Hepatocyte Isolation
2.9. Assay of Oxidative Activity of CYP3A4
2.10. Assay of Intracellular Accumulation of Rhodamine 123
2.11. Data and Statistical Analysis
2.12. Materials
3. Results
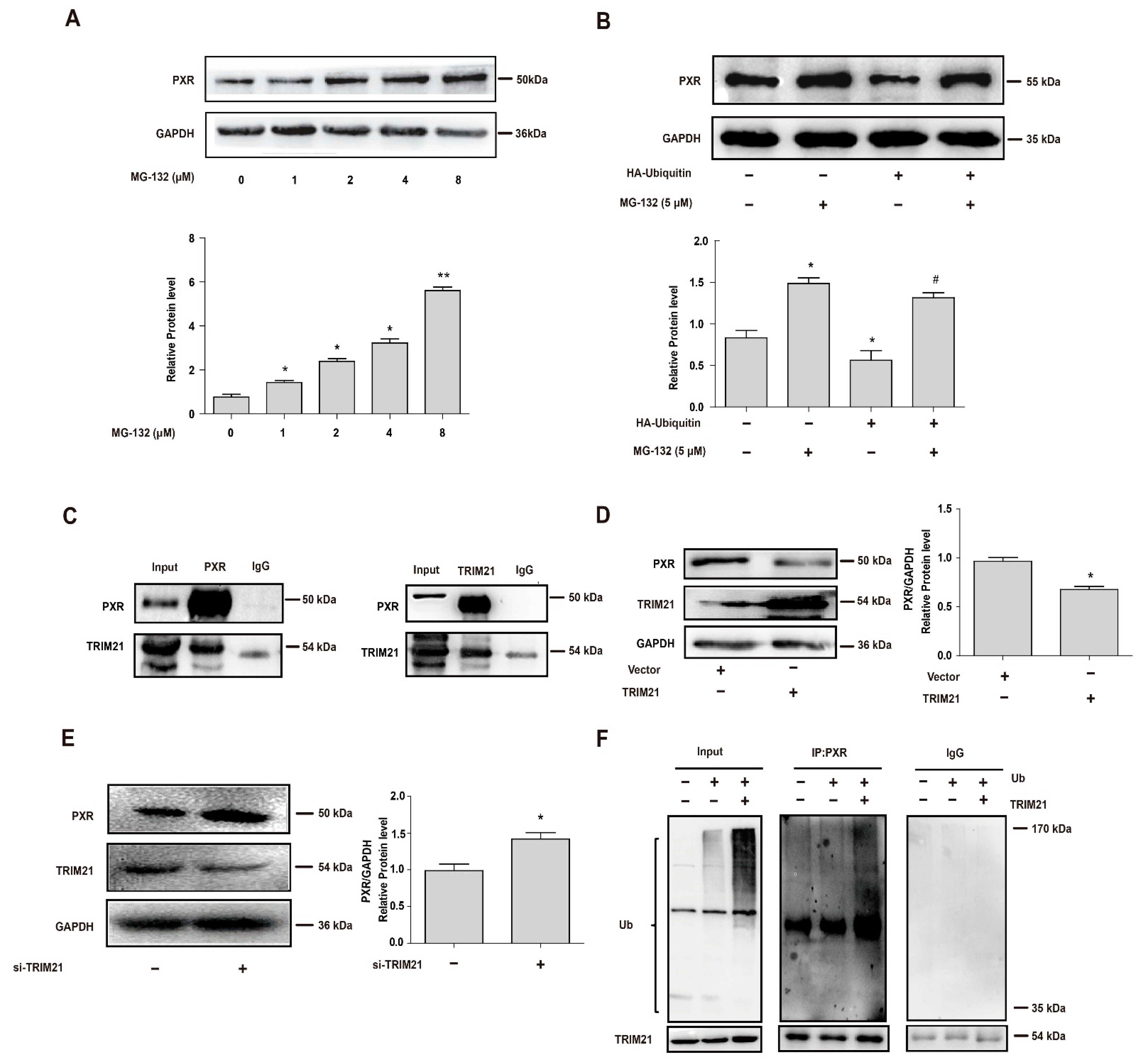
3.1. The E3 Ligase TRIM21 Mediates the Ubiquitination and Degradation of PXR
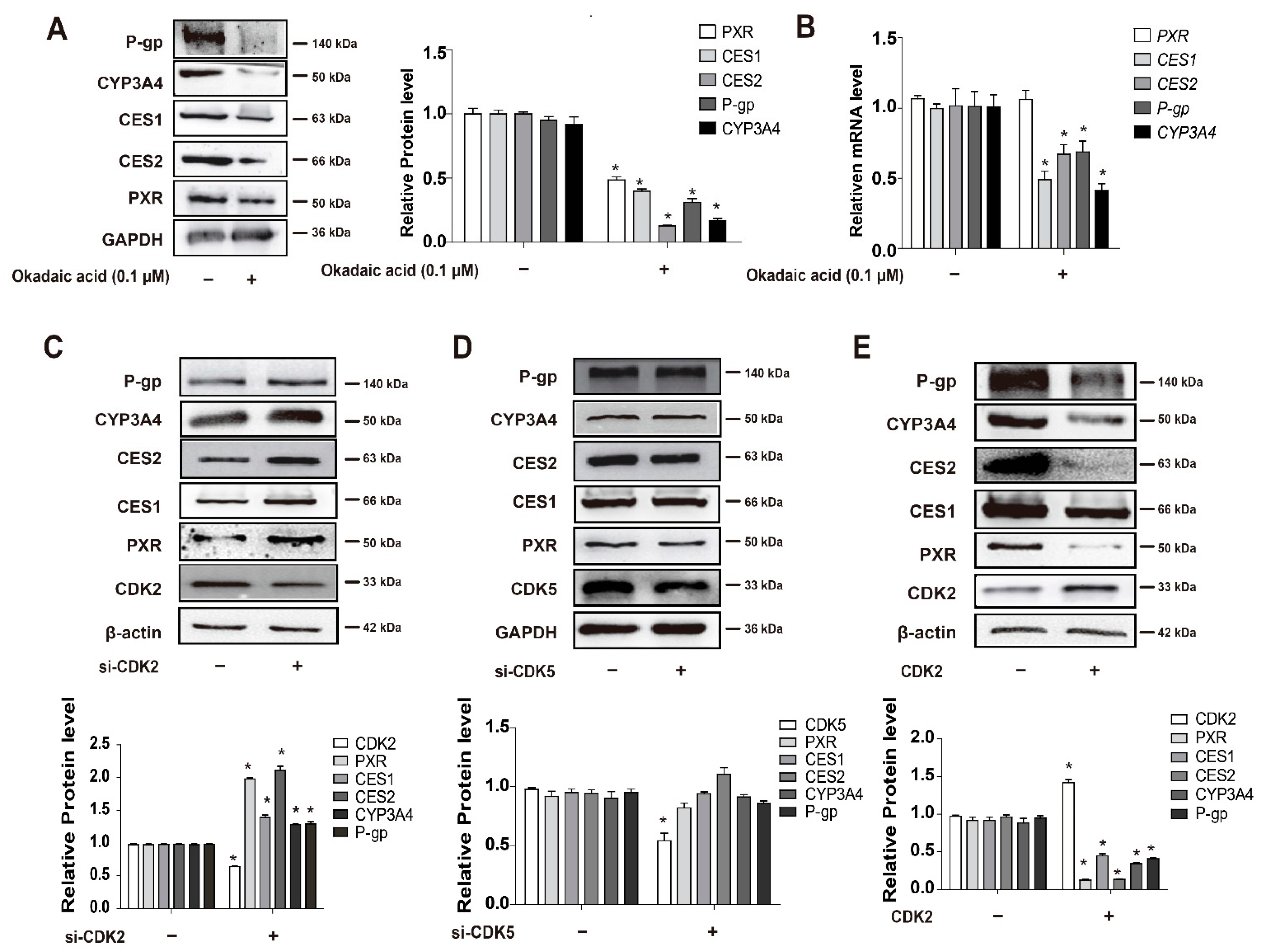
3.2. Inhibition of CDKs Increases the Protein Level of PXR through the Protein Ubiquitination and Proteasomal System
3.3. The Attenuated Interaction of PXR with DNAJC7 and Hsp90 Is Engaged in PXR Degradation by CDKs
3.4. CDKs, and CDK2 Specifically, Decrease PXR Protein Stabilization and Suppress the Expression of PXR-Target Genes
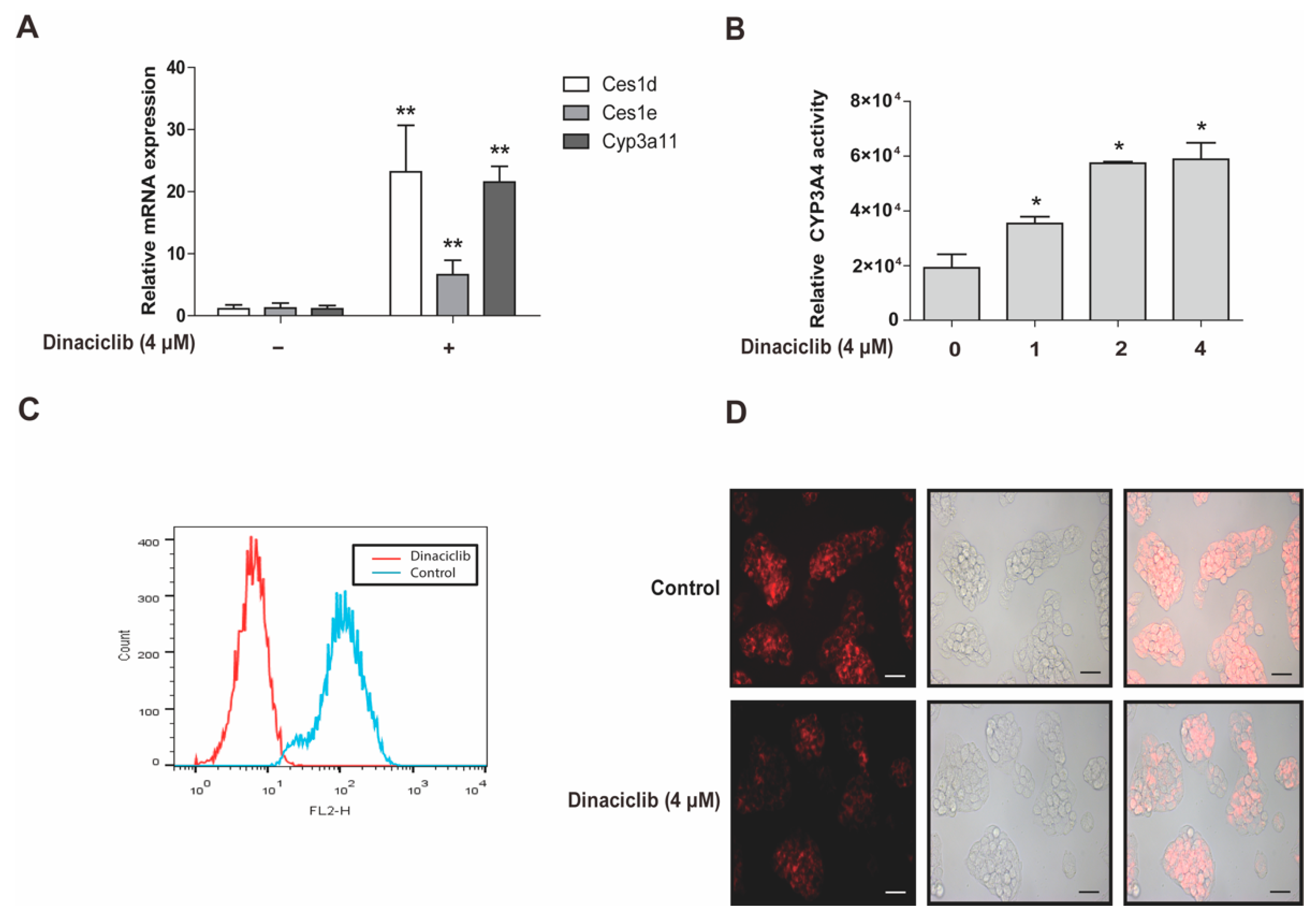
3.5. CDKs Inhibitor Dinaciclib Promotes the Expression and Activity of PXR Target Genes in Primary Hepatocytes and HepG2 Cells
3.6. PXR Phosphorylation at S350 by CDK2 Triggers PXR Degradation via the Ubiquitin-Proteasome Pathway
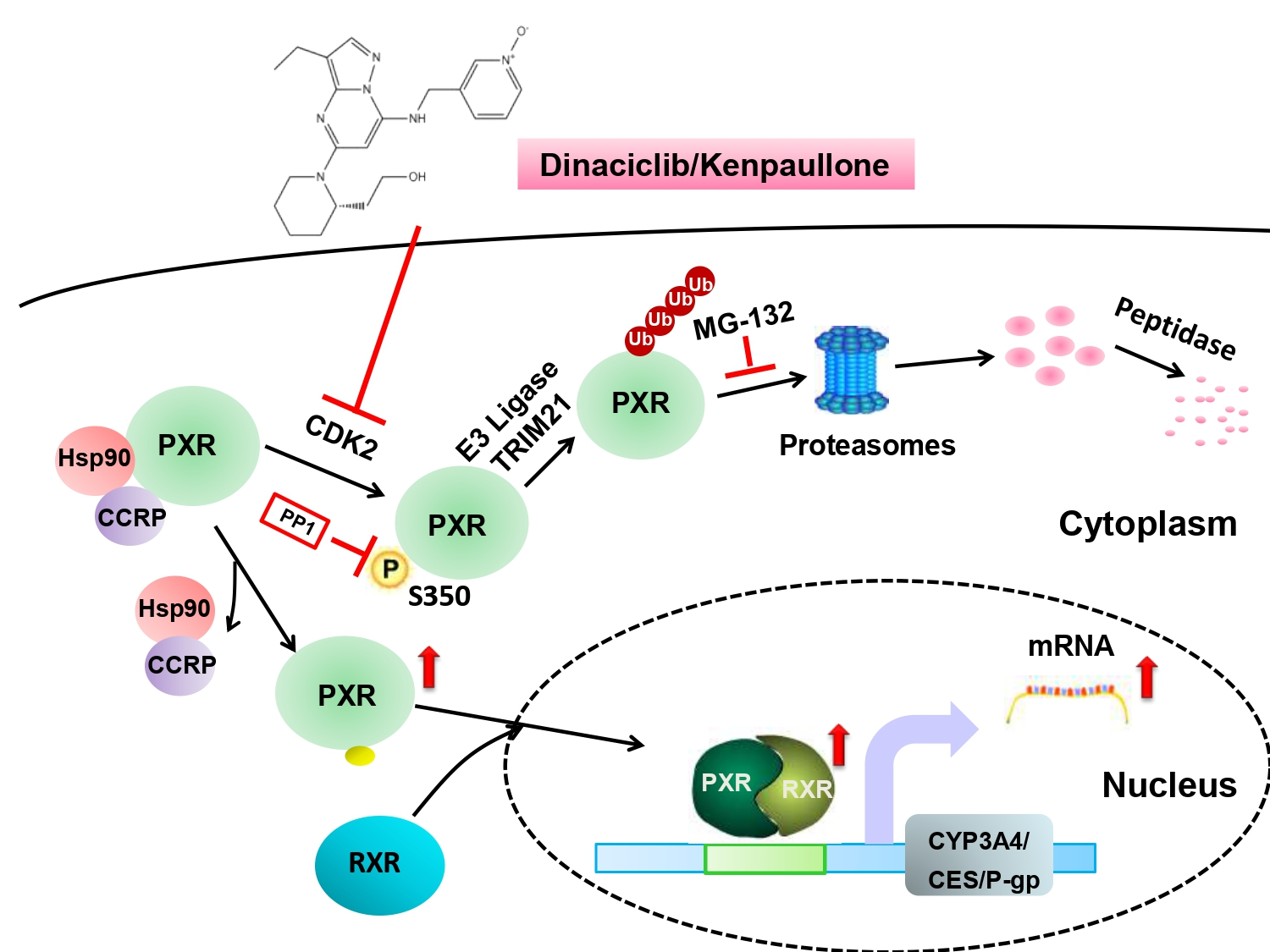
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PXR | pregnane X receptor |
| CYP3A4 | human cytochrome P450 family 3 subfamily A member 4 |
| CES1 | human carboxylesterase 1 |
| CES2 | human carboxylesterase 2 |
| P-gp | P-glycoprotein |
| DMET | drug-metabolizing enzymes and transporters |
| DDIs | drug-drug interactions |
| Cyp3a11 | mouse cytochrome P450 family 3 subfamily A member 11 |
| Ces1d | mouse carboxylesterase 1 |
| Ces1e | mouse carboxylesterase 2 |
References
- Chai, X.; Zeng, S.; Xie, W. Nuclear receptors PXR and CAR: Implications for drug metabolism regulation, pharmacogenomics and beyond. Expert Opin. Drug Metab. Toxicol. 2013, 9, 253–266. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Y.; Guo, C.; Wang, J.; Boral, D.; Nie, D. Nuclear receptors in the multidrug resistance through the regulation of drug-metabolizing enzymes and drug transporters. Biochem. Pharmacol. 2012, 83, 1112–1126. [Google Scholar] [CrossRef] [Green Version]
- Rana, R.; Coulter, S.; Kinyamu, H.; Goldstein, J.A. RBCK1, an E3 ubiquitin ligase, interacts with and ubiquinates the human pregnane X receptor. Drug Metab. Dispos. Biol. Fate Chem. 2013, 41, 398–405. [Google Scholar] [CrossRef]
- Luo, W.; Xin, Y.; Zhao, X.; Zhang, F.; Liu, C.; Fan, H.; Xi, T.; Xiong, J. Suppression of carboxylesterases by imatinib mediated by the down-regulation of pregnane X receptor. Br. J. Pharmacol. 2017, 174, 700–717. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Shen, X.; Agbas, E.; Tompkins, B.; Cameron-Carter, H.; Staudinger, J.L. Phosphorylation Modulates the Coregulatory Protein Exchange of the Nuclear Receptor Pregnane X Receptor. J. Pharmacol. Exp. Ther. 2020, 373, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Pasquel, D.; Doricakova, A.; Li, H.; Kortagere, S.; Krasowski, M.D.; Biswas, A.; Walton, W.G.; Redinbo, M.R.; Dvorak, Z.; Mani, S. Acetylation of lysine 109 modulates pregnane X receptor DNA binding and transcriptional activity. Biochim. Biophys. Acta 2016, 1859, 1155–1169. [Google Scholar] [CrossRef] [Green Version]
- Kotiya, D.; Rana, M.; Subbarao, N.; Puri, N.; Tyagi, R.K. Transcription regulation of nuclear receptor PXR: Role of SUMO-1 modification and NDSM in receptor function. Mol. Cell. Endocrinol. 2016, 420, 194–207. [Google Scholar] [CrossRef]
- Wang, C.; Xu, W.; Zhang, Y.; Huang, D.; Huang, K. Poly(ADP-ribosyl)ated PXR is a critical regulator of acetaminophen-induced hepatotoxicity. Cell Death Dis. 2018, 9, 819. [Google Scholar] [CrossRef] [Green Version]
- Smutny, T.; Mani, S.; Pavek, P. Post-translational and post-transcriptional modifications of pregnane X receptor (PXR) in regulation of the cytochrome P450 superfamily. Curr. Drug Metab. 2013, 14, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- Pondugula, S.R.; Dong, H.; Chen, T. Phosphorylation and protein-protein interactions in PXR-mediated CYP3A repression. Expert Opin. Drug Metab. Toxicol. 2009, 5, 861–873. [Google Scholar] [CrossRef] [Green Version]
- Skandalaki, A.; Sarantis, P.; Theocharis, S. Pregnane X Receptor (PXR) Polymorphisms and Cancer Treatment. Biomolecules 2021, 11, 1142. [Google Scholar] [CrossRef]
- Xing, Y.; Yan, J.; Niu, Y. PXR: A center of transcriptional regulation in cancer. Acta Pharm. Sinica B 2020, 10, 197–206. [Google Scholar] [CrossRef]
- Sivertsson, L.; Edebert, I.; Palmertz, M.P.; Ingelman-Sundberg, M.; Neve, E.P. Induced CYP3A4 expression in confluent Huh7 hepatoma cells as a result of decreased cell proliferation and subsequent pregnane X receptor activation. Mol. Pharmacol. 2013, 83, 659–670. [Google Scholar] [CrossRef]
- Dong, H.; Lin, W.; Wu, J.; Chen, T. Flavonoids activate pregnane x receptor-mediated CYP3A4 gene expression by inhibiting cyclin-dependent kinases in HepG2 liver carcinoma cells. BMC Biochem. 2010, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Pondugula, S.R.; Brimer-Cline, C.; Wu, J.; Schuetz, E.G.; Tyagi, R.K.; Chen, T. A phosphomimetic mutation at threonine-57 abolishes transactivation activity and alters nuclear localization pattern of human pregnane x receptor. Drug Metab. Dispos. Biol. Fate Chem. 2009, 37, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Sugatani, J.; Uchida, T.; Kurosawa, M.; Yamaguchi, M.; Yamazaki, Y.; Ikari, A.; Miwa, M. Regulation of pregnane X receptor (PXR) function and UGT1A1 gene expression by posttranslational modification of PXR protein. Drug Metab. Dispos. Biol. Fate Chem. 2012, 40, 2031–2040. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xie, P.; Lu, L.; Wang, J.; Diao, L.; Liu, Z.; Guo, F.; He, Y.; Liu, Y.; Huang, Q.; et al. An integrated bioinformatics platform for investigating the human E3 ubiquitin ligase-substrate interaction network. Nat. Commun. 2017, 8, 347. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Md, H.; Cho, M.; Kim, N.H.; Choi, H.Y.; Han, J.W.; Park, H.J.; Oh, J.W.; Shin, J.G. Role of 14-3-3 sigma in over-expression of P-gp by rifampin and paclitaxel stimulation through interaction with PXR. Cell. Signal. 2017, 31, 124–134. [Google Scholar] [CrossRef]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Wu, J.; Dong, H.; Bouck, D.; Zeng, F.Y.; Chen, T. Cyclin-dependent kinase 2 negatively regulates human pregnane X receptor-mediated CYP3A4 gene expression in HepG2 liver carcinoma cells. J. Biol. Chem. 2008, 283, 30650–30657. [Google Scholar] [CrossRef] [Green Version]
- Long, F.; He, Y.; Fu, H.; Li, Y.; Bao, X.; Wang, Q.; Wang, Y.; Xie, C.; Lou, L. Preclinical characterization of SHR6390, a novel CDK 4/6 inhibitor, in vitro and in human tumor xenograft models. Cancer Sci. 2019, 110, 1420–1430. [Google Scholar] [CrossRef]
- Sobhani, N.; Corona, S.P.; Zanconati, F.; Generali, D. Cyclin dependent kinase 4 and 6 inhibitors as novel therapeutic agents for targeted treatment of malignant mesothelioma. Genes Cancer 2017, 8, 495–496. [Google Scholar] [CrossRef] [Green Version]
- Blachly, J.S.; Byrd, J.C. Emerging drug profile: Cyclin-dependent kinase inhibitors. Leuk. Lymphoma 2013, 54, 2133–2143. [Google Scholar] [CrossRef]
- Łukasik, P.; Załuski, M.; Gutowska, I. Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development-Review. Int. J. Mol. Sci. 2021, 22, 2935. [Google Scholar] [CrossRef]
- Łukasik, P.; Baranowska-Bosiacka, I.; Kulczycka, K.; Gutowska, I. Inhibitors of Cyclin-Dependent Kinases: Types and Their Mechanism of Action. Int. J. Mol. Sci. 2021, 22, 2806. [Google Scholar] [CrossRef]
- Lin, S.F.; Lin, J.D.; Hsueh, C.; Chou, T.C.; Wong, R.J. A cyclin-dependent kinase inhibitor, dinaciclib in preclinical treatment models of thyroid cancer. PLoS ONE 2017, 12, e0172315. [Google Scholar] [CrossRef] [Green Version]
- Saqub, H.; Proetsch-Gugerbauer, H.; Bezrookove, V.; Nosrati, M.; Vaquero, E.M.; de Semir, D.; Ice, R.J.; McAllister, S.; Soroceanu, L.; Kashani-Sabet, M.; et al. Dinaciclib, a cyclin-dependent kinase inhibitor, suppresses cholangiocarcinoma growth by targeting CDK2/5/9. Sci. Rep. 2020, 10, 18489. [Google Scholar] [CrossRef]
- Roberts, A.G.; Gibbs, M.E. Mechanisms and the clinical relevance of complex drug-drug interactions. Clin. Pharmacol. Adv. Appl. 2018, 10, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Storelli, F.; Samer, C.; Reny, J.L.; Desmeules, J.; Daali, Y. Complex Drug-Drug-Gene-Disease Interactions Involving Cytochromes P450: Systematic Review of Published Case Reports and Clinical Perspectives. Clin. Pharmacokinet. 2018, 57, 1267–1293. [Google Scholar] [CrossRef] [Green Version]
- Ly, J.Q.; Messick, K.; Qin, A.; Takahashi, R.H.; Choo, E.F. Utility of CYP3A4 and PXR-CAR-CYP3A4/3A7 Transgenic Mouse Models To Assess the Magnitude of CYP3A4 Mediated Drug-Drug Interactions. Mol. Pharm. 2017, 14, 1754–1759. [Google Scholar] [CrossRef]
- Ji, L.; Zheng, Z.; Shi, L.; Huang, Y.; Lu, B.; Wang, Z. Andrographolide decreased VEGFD expression in hepatoma cancer cells by inducing ubiquitin/proteasome-mediated cFos protein degradation. Biochim. Biophys. Acta 2015, 1850, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Maeda, A.; Tani, S.; Akagawa, M. Palmitate induces insulin resistance in human HepG2 hepatocytes by enhancing ubiquitination and proteasomal degradation of key insulin signaling molecules. Arch. Biochem. Biophys. 2015, 566, 26–35. [Google Scholar] [CrossRef]
- Chen, V.C.; Kristensen, A.R.; Foster, L.J.; Naus, C.C. Association of connexin43 with E3 ubiquitin ligase TRIM21 reveals a mechanism for gap junction phosphodegron control. J. Proteome Res. 2012, 11, 6134–6146. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Vara, E.; Ruiz-Llorente, L.; Casado-Vela, J.; Ruiz-Rodríguez, M.J.; López-Andrés, N.; Pattnaik, A.K.; Quintanilla, M.; Bernabeu, C. Endoglin Protein Interactome Profiling Identifies TRIM21 and Galectin-3 as New Binding Partners. Cells 2019, 8, 1082. [Google Scholar] [CrossRef] [Green Version]
- Anandapadamanaban, M.; Kyriakidis, N.C.; Csizmók, V.; Wallenhammar, A.; Espinosa, A.C.; Ahlner, A.; Round, A.R.; Trewhella, J.; Moche, M.; Wahren-Herlenius, M.; et al. E3 ubiquitin-protein ligase TRIM21-mediated lysine capture by UBE2E1 reveals substrate-targeting mode of a ubiquitin-conjugating E2. J. Biol. Chem. 2019, 294, 11404–11419. [Google Scholar] [CrossRef] [PubMed]
- Elias, A.; High, A.A.; Mishra, A.; Ong, S.S.; Wu, J.; Peng, J.; Chen, T. Identification and characterization of phosphorylation sites within the pregnane X receptor protein. Biochem. Pharmacol. 2014, 87, 360–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, F.S.; Chen, Y.L.; Hung, M.H.; Chu, P.Y.; Tsai, M.H.; Chen, L.J.; Hsiao, Y.J.; Shih, C.T.; Chang, M.J.; Chao, T.I.; et al. Palbociclib induces activation of AMPK and inhibits hepatocellular carcinoma in a CDK4/6-independent manner. Biochem. Pharmacol. 2017, 11, 1035–1049. [Google Scholar] [CrossRef]
- Timsit, Y.E.; Negishi, M. Coordinated regulation of nuclear receptor CAR by CCRP/DNAJC7, HSP70 and the ubiquitin-proteasome system. PLoS ONE 2014, 9, e96092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.M.; Chai, S.C.; Lin, W.; Chai, X.; Elias, A.; Wu, J.; Ong, S.S.; Pondugula, S.R.; Beard, J.A.; Schuetz, E.G.; et al. Serine 350 of human pregnane X receptor is crucial for its heterodimerization with retinoid X receptor alpha and transactivation of target genes in vitro and in vivo. Biochem. Pharmacol. 2015, 96, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Xiao, D.; Chen, Y.T.; Yang, D.; Yan, B. Age-related inducibility of carboxylesterases by the antiepileptic agent phenobarbital and implications in drug metabolism and lipid accumulation. Biochem. Pharmacol. 2012, 84, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, X.; Liu, Y.; Xi, T.; Xiong, J. Insulin transcriptionally down-regulates carboxylesterases through pregnane X receptor in an Akt-dependent manner. Toxicology 2019, 422, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Markov, G.V.; Laudet, V. Origin and evolution of the ligand-binding ability of nuclear receptors. Mol. Cell. Endocrinol. 2011, 334, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Lichti-Kaiser, K.; Xu, C.; Staudinger, J.L. Cyclic AMP-dependent protein kinase signaling modulates pregnane x receptor activity in a species-specific manner. J. Biol. Chem. 2009, 284, 6639–6649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugatani, J.; Hattori, Y.; Noguchi, Y.; Yamaguchi, M.; Yamazaki, Y.; Ikari, A. Threonine-290 regulates nuclear translocation of the human pregnane X receptor through its phosphorylation/dephosphorylation by Ca2+/calmodulin-dependent protein kinase II and protein phosphatase 1. Drug Metab. Dispos. Biol. Fate Chem. 2014, 42, 1708–1718. [Google Scholar] [CrossRef] [Green Version]
- Wallace, B.D.; Betts, L.; Talmage, G.; Pollet, R.M.; Holman, N.S.; Redinbo, M.R. Structural and functional analysis of the human nuclear xenobiotic receptor PXR in complex with RXRα. J. Mol. Biol. 2013, 425, 2561–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal. Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Hasanuzzaman, M.; Cho, M.; Heo, Y.R.; Ryu, M.J.; Ha, N.Y.; Park, H.J.; Park, H.Y.; Shin, J.G. Casein Kinase 2 (CK2)-mediated Phosphorylation of Hsp90β as a Novel Mechanism of Rifampin-induced MDR1 Expression. J. Biol. Chem. 2015, 290, 17029–17040. [Google Scholar] [CrossRef] [Green Version]
- Pondugula, S.R.; Tong, A.A.; Wu, J.; Cui, J.; Chen, T. Protein phosphatase 2Cbetal regulates human pregnane X receptor-mediated CYP3A4 gene expression in HepG2 liver carcinoma cells. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 1411–1416. [Google Scholar] [CrossRef] [Green Version]
- Yao, N.; Zeng, C.; Zhan, T.; He, F.; Liu, M.; Liu, F.; Zhang, H.; Xiong, Y.; Xia, C. Oleanolic Acid and Ursolic Acid Induce UGT1A1 Expression in HepG2 Cells by Activating PXR Rather Than CAR. Front. Pharmacol. 2019, 10, 1111. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Shizu, R.; Sasaki, T.; Shimizu, Y.; Hosaka, T.; Kodama, S.; Matsuzawa, A.; Yoshinari, K. Functional Interaction between Pregnane X Receptor and Yes-Associated Protein in Xenobiotic-Dependent Liver Hypertrophy and Drug Metabolism. J. Pharmacol. Exp. Ther. 2019, 371, 590–601. [Google Scholar] [CrossRef]
- Konecny, G.E. Cyclin-dependent kinase pathways as targets for women’s cancer treatment. Curr. Opin. Obstet. Gynecol. 2016, 28, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martínez, C.; Lallena, M.J.; Sanfeliciano, S.G.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015-2019). Bioorg. Med. Chem. Lett. 2019, 29, 126637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar]
- Roskoski, R., Jr. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol. Res. 2019, 139, 471–488. [Google Scholar] [CrossRef]
- Tarasewicz, E.; Hamdan, R.; Straehla, J.; Hardy, A.; Nunez, O.; Zelivianski, S.; Dokic, D.; Jeruss, J.S. CDK4 inhibition and doxorubicin mediate breast cancer cell apoptosis through Smad3 and survivin. Cancer Biol. Ther. 2014, 15, 1301–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.X.; Gao, F.; Luo, J.; Northfelt, D.W.; Goetz, M.; Forero, A.; Hoog, J.; Naughton, M.; Ademuyiwa, F.; Suresh, R.; et al. NeoPalAna: Neoadjuvant Palbociclib, a Cyclin-Dependent Kinase 4/6 Inhibitor, and Anastrozole for Clinical Stage 2 or 3 Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2017, 23, 4055–4065. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.S.; Stoehr, J.; Dokic, D.; Wan, L.; Decker, J.T.; Konopka, K.; Thomas, A.L.; Wu, J.; Kaklamani, V.G.; Shea, L.D.; et al. Synergistic effect of eribulin and CDK inhibition for the treatment of triple negative breast cancer. Oncotarget 2017, 8, 83925–83939. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Germano, S.; Clements, C.; Samuel, J.; Shelmani, G.; Jayne, S.; Dyer, M.J.; Macip, S. Pro-survival signal inhibition by CDK inhibitor dinaciclib in Chronic Lymphocytic Leukaemia. Br. J. Haematol. 2016, 175, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Danilov, A.V.; Hu, S.; Orr, B.; Godek, K.; Mustachio, L.M.; Sekula, D.; Liu, X.; Kawakami, M.; Johnson, F.M.; Compton, D.A.; et al. Dinaciclib Induces Anaphase Catastrophe in Lung Cancer Cells via Inhibition of Cyclin-Dependent Kinases 1 and 2. Mol. Cancer Ther. 2016, 15, 2758–2766. [Google Scholar] [CrossRef] [Green Version]
- Rajput, S.; Khera, N.; Guo, Z.; Hoog, J.; Li, S.; Ma, C.X. Inhibition of cyclin dependent kinase 9 by dinaciclib suppresses cyclin B1 expression and tumor growth in triple negative breast cancer. Oncotarget 2016, 7, 56864–56875. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wang, Z.; Pang, J.C.; Yu, Y.; Bieerkehazhi, S.; Lu, J.; Hu, T.; Zhao, Y.; Xu, X.; Zhang, H.; et al. Multiple CDK inhibitor dinaciclib suppresses neuroblastoma growth via inhibiting CDK2 and CDK9 activity. Sci. Rep. 2016, 6, 29090. [Google Scholar] [CrossRef] [Green Version]
- Nemunaitis, J.J.; Small, K.A.; Kirschmeier, P.; Zhang, D.; Zhu, Y.; Jou, Y.M.; Statkevich, P.; Yao, S.L.; Bannerji, R. A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J. Transl. Med. 2013, 11, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mita, M.M.; Joy, A.A.; Mita, A.; Sankhala, K.; Jou, Y.M.; Zhang, D.; Statkevich, P.; Zhu, Y.; Yao, S.L.; Small, K.; et al. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin. Breast Cancer 2014, 14, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Filppula, A.M.; Laitila, J.; Neuvonen, P.J.; Backman, J.T. Potent mechanism-based inhibition of CYP3A4 by imatinib explains its liability to interact with CYP3A4 substrates. Br. J. Pharmacol. 2012, 165, 2787–2798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Mita, M.; Shapiro, G.I.; Poon, J.; Small, K.; Tzontcheva, A.; Kantesaria, B.; Zhu, Y.; Bannerji, R.; Statkevich, P. Effect of aprepitant on the pharmacokinetics of the cyclin-dependent kinase inhibitor dinaciclib in patients with advanced malignancies. Cancer Chemother. Pharmacol. 2012, 70, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Váradi, A.; Ozvegy-Laczka, C.; Sarkadi, B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Discov. Today 2008, 13, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Cihalova, D.; Ceckova, M.; Kucera, R.; Klimes, J.; Staud, F. Dinaciclib, a cyclin-dependent kinase inhibitor, is a substrate of human ABCB1 and ABCG2 and an inhibitor of human ABCC1 in vitro. Biochem. Pharmacol. 2015, 98, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Seo, H.; Yang, J.Y.; Joo, J.; Im, S.H.; Kim, S.S.; Kim, S.K.; Bae, M.A. Quantitative Evaluation of Cytochrome P450 3A4 Inhibition and Hepatotoxicity in HepaRG 3-D Spheroids. Int. J. Toxicol. 2018, 37, 393–403. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Gene | Sequence of Forward and Reverse Primers |
|---|---|---|
| Homo sapiens | GAPDH | Forward 5′-AAGGTCGGAGTCACCGGATT-3′ |
| Reverse 5′-CTGGAAGATGGTGAGGGATT-3′ | ||
| Homo sapiens | CES1 | Forward 5′-CCAGAGAGAGTCAACCCCTTCT-3′ |
| Reverse 5′-TCCTGCTTGTTAATTCCGACC-3′ | ||
| Homo sapiens | CES2 | Forward 5′-ACCGCAGTGGAGTCAGAGTTTC-3′ |
| Reverse 5′-ATGCTGAGGTACAGGCAGTCCT-3′; | ||
| Homo sapiens | PXR | Forward 5′-GGCAATCCCAGGTTCTCTTT-3′ |
| Reverse 5′-ATGCTTTATGGCAGGTGAGG-3′ | ||
| Homo sapiens | CYP3A4 | Forward 5′-TTCAGCAAGAAGAACAAGGACAA-3′ |
| Reverse 5′-GGTTGAAGAAGTCCTCCTAAGC-3′ | ||
| Homo sapiens | MDR1 | Forward 5′-GAGGCCAACATACATGCCTTC-3′ |
| Reverse 5′-GTCTAACAAGGGCACGAGCTAT-3′ | ||
| Mus musculus | Ces1d | Forward 5′-GAGACCCAAGGCAGTAATAGGA-3′ |
| Reverse 5′-GAGTTGAGGCACCAATCTTCA-3′ | ||
| Mus musculus | Ces1e | Forward 5′-CCAGTGACAGGGCAAATAGTC-3′ |
| Reverse 5′-TCATGCGTAGACAGGACCAGT-3′ | ||
| Mus musculus | Cyp3a11 | Forward 5′-ACAGCACTGGTCAGAGCCTGAA-3′ |
| Reverse 5′-GAGAGCAAACCTCATGCCAAGG-3′ | ||
| Mus musculus | Rplp0 | Forward 5′-GAAACTGCTGCCTCACATCCG-3′ |
| Forward 5′-GCTGGCACAGTGACCTCACACG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, M.; Xin, Y.; Bian, Y.; Yang, X.; Xi, T.; Xiong, J. Phosphorylation-Induced Ubiquitination and Degradation of PXR through CDK2-TRIM21 Axis. Cells 2022, 11, 264. https://doi.org/10.3390/cells11020264
Qin M, Xin Y, Bian Y, Yang X, Xi T, Xiong J. Phosphorylation-Induced Ubiquitination and Degradation of PXR through CDK2-TRIM21 Axis. Cells. 2022; 11(2):264. https://doi.org/10.3390/cells11020264
Chicago/Turabian StyleQin, Mengyao, Yu Xin, Yong Bian, Xuan Yang, Tao Xi, and Jing Xiong. 2022. "Phosphorylation-Induced Ubiquitination and Degradation of PXR through CDK2-TRIM21 Axis" Cells 11, no. 2: 264. https://doi.org/10.3390/cells11020264