
Endolysosomal Cation Channels and Lung Disease

 ,
, {kind=link}
{kind=link}
Abstract
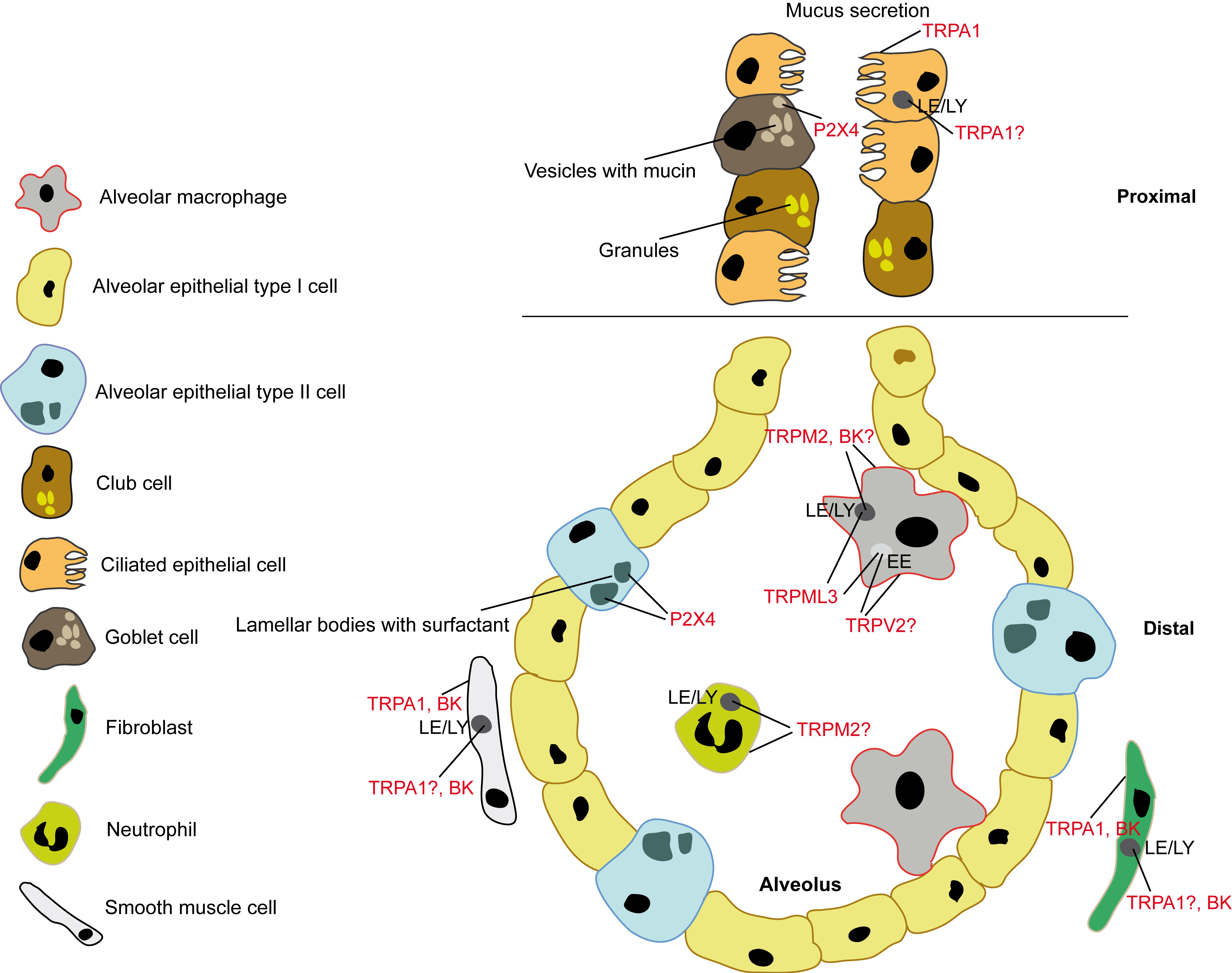
:1. Introduction
2. P2X4—Regulator of Surfactant Release: Lamellar Bodies of ATII Cells
3. TRPML3—Regulator of MMP-12 Levels in Bronchoalveolar Fluid: Early Endosomes of Alveolar Macrophages
4. TRPV2—Critical Player in Alveolar Macrophage Phagocytosis: Cell Membrane or Early Endosome?
5. TRPM2—Protective Role in Lung Inflammation: Cell Membrane or Lysosome?
6. TRPA1—A Target for Cough, Asthma, COPD? Ca2+ from Lysosomes Involved?
7. BK Channel in the Lysosome—Refilling or Releasing Ca2+?
8. Discussion and Conclusions
Funding
Conflicts of Interest
References
- Spix, B.; Butz, E.S.; Chen, C.-C.; Scotto Rosato, A.; Tang, R.; Jeridi, A.; Kudrina, V.; Plesch, E.; Wartenberg, P.; Arlt, E.; et al. Lung emphysema and impaired macrophage elastase clearance in mucolipin 3 deficient mice. Nat. Commun. 2022, 13, 318. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Matalon, S.; Collawn, J.F. Ion channels of the lung and their role in disease pathogenesis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2017, 313, L859–L872. [Google Scholar] [CrossRef]
- Ziȩtkiewicz, E.; Rutkiewicz, E.; Pogorzelski, A.; Klimek, B.; Voelkel, K.; Witt, M. CFTR mutations spectrum and the efficiency of molecular diagnostics in Polish cystic fibrosis patients. PLoS ONE 2014, 9, e89094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotet, V.; L’Hostis, C.; Férec, C. The changing epidemiology of cystic fibrosis: Incidence, survival and impact of the CFTRGene discovery. Genes 2020, 11, 589. [Google Scholar] [CrossRef]
- Huang, F.; Zhang, H.; Wu, M.; Yang, H.; Kudo, M.; Peters, C.J.; Woodruff, P.G.; Solberg, O.D.; Donne, M.L.; Huang, X.; et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc. Natl. Acad. Sci. USA 2012, 109, 16354–16359. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, A. Modulators of transient receptor potential (TRP) channels as therapeutic options in lung disease. Pharmaceuticals 2019, 12, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, J.; Rajan, S.; Schremmer, C.; Chao, Y.K.; Krasteva-Christ, G.; Kannler, M.; Yildirim, A.Ö.; Brosien, M.; Schredelseker, J.; Weissmann, N.; et al. TRPV4 channels are essential for alveolar epithelial barrier function as protection from lung edema. JCI Insight 2020, 5, e134464. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, N.; Sydykov, A.; Kalwa, H.; Storch, U.; Fuchs, B.; Mederos, Y.; Schnitzler, M.; Brandes, R.P.; Grimminger, F.; Meissner, M.; et al. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat. Commun. 2012, 3, 649. [Google Scholar] [CrossRef]
- Chubanov, V.; Ferioli, S.; Wisnowsky, A.; Simmons, D.G.; Leitzinger, C.; Einer, C.; Jonas, W.; Shymkiv, Y.; Bartsch, H.; Braun, A.; et al. Epithelial magnesium transport by TRPM6 is essential for prenatal development and adult survival. eLife 2016, 5, e20914. [Google Scholar] [CrossRef]
- Zech, A.; Wiesler, B.; Ayata, C.K.; Schlaich, T.; Dürk, T.; Hoßfeld, M.; Ehrat, N.; Cicko, S.; Idzko, M. P2rx4 deficiency in mice alleviates allergen-induced airway inflammation. Oncotarget 2016, 7, 80288–80297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miklavc, P.; Mair, N.; Wittekindt, O.H.; Haller, T.; Dietl, P.; Felder, E.; Timmler, M.; Frick, M. Fusion-activated Ca2+ entry via vesicular P2X4 receptors promotes fusion pore opening and exocytotic content release in pneumocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 14503–14508. [Google Scholar] [CrossRef] [Green Version]
- Miklavc, P.; Thompson, K.E.; Frick, M. A new role for P2X4 receptors as modulators of lung surfactant secretion. Front. Cell. Neurosci. 2013, 7, 171. [Google Scholar] [CrossRef] [Green Version]
- Fois, G.; Winkelmann, V.E.; Bareis, L.; Staudenmaier, L.; Hecht, E.; Ziller, C.; Ehinger, K.; Schymeinsky, J.; Kranz, C.; Frick, M. ATP is stored in lamellar bodies to activate vesicular P2X4 in an autocrine fashion upon exocytosis. J. Gen. Physiol. 2018, 150, 277–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkelmann, V.E.; Thompson, K.E.; Neuland, K.; Jaramillo, A.M.; Fois, G.; Schmidt, H.; Wittekindt, O.H.; Han, W.; Tuvim, M.J.; Dickey, B.F.; et al. Inflammation-induced upregulation of P2X4 expression augments mucin secretion in airway epithelia. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2019, 316, L58–L70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Murrell-Lagnado, R.; Zhu, M.X.; Dong, X.P. Calcium release through P2X4 activates calmodulin to promote endolysosomal membrane fusion. J. Cell Biol. 2015, 209, 879–894. [Google Scholar] [CrossRef] [Green Version]
- Suurväli, J.; Boudinot, P.; Kanellopoulos, J.; Rüütel Boudinot, S. P2X4: A fast and sensitive purinergic receptor. Biomed. J. 2017, 40, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.H.; Mathie, A.; Peters, J.A.; Veale, E.L.; Striessnig, J.; Kelly, E.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; et al. The concise guide to pharmacology 2021/22: Ion channels. Br. J. Pharmacol. 2021, 178, S157–S245. [Google Scholar] [CrossRef]
- Nagata, K.; Zheng, L.; Madathany, T.; Castiglioni, A.J.; Bartles, J.R.; García-Añoveros, J. The varitint-waddler (Va) deafness mutation in TRPML3 generates constitutive, inward rectifying currents and causes cell degeneration. Proc. Natl. Acad. Sci. USA 2008, 105, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Grimm, C.; Cuajungco, M.P.; Van Aken, A.F.J.; Schnee, M.; Jörs, S.; Kros, C.J.; Ricci, A.J.; Heller, S. A helix-breaking mutation in TRPML3 leads to constitutive activity underlying deafness in the varitint-waddler mouse. Proc. Natl. Acad. Sci. USA 2007, 104, 19583–19588. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Delling, M.; Li, L.; Dong, X.; Clapham, D.E. Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint-waddler mice. Proc. Natl. Acad. Sci. USA 2007, 104, 18321–18326. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Li, Q.; Tjon-Kon-Sang, S.; So, I.; Kiselyov, K.; Muallem, S. Gain-of-function mutation in TRPML3 causes the mouse varitint-waddler phenotype. J. Biol. Chem. 2007, 282, 36138–36142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jörs, S.; Grimm, C.; Becker, L.; Heller, S. Genetic inactivation of Trpml3 does not lead to hearing and vestibular impairment in mice. PLoS ONE 2010, 5, e14317. [Google Scholar] [CrossRef] [Green Version]
- Remis, N.N.; Wiwatpanit, T.; Castiglioni, A.J.; Flores, E.N.; Cantú, J.A.; García-Añoveros, J. Mucolipin Co-deficiency Causes Accelerated Endolysosomal Vacuolation of Enterocytes and Failure-to-Thrive from Birth to Weaning. PLoS Genet. 2014, 10, e1004833. [Google Scholar] [CrossRef] [Green Version]
- Masubuchi, H.; Ueno, M.; Maeno, T.; Yamaguchi, K.; Hara, K.; Sunaga, H.; Matsui, H.; Nagasawa, M.; Kojima, I.; Iwata, Y.; et al. Reduced transient receptor potential vanilloid 2 expression in alveolar macrophages causes COPD in mice through impaired phagocytic activity. BMC Pulm. Med. 2019, 19, 70. [Google Scholar] [CrossRef] [PubMed]
- Link, T.M.; Park, U.; Vonakis, B.M.; Raben, D.M.; Soloski, M.J.; Caterina, M.J. TRPV2 plays a pivotal role in macrophage particle binding and phagocytosis. Nat. Immunol. 2010, 11, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Wainszelbaum, M.J.; Proctor, B.M.; Pontow, S.E.; Stahl, P.D.; Barbieri, M.A. IL4/PGE2 induction of an enlarged early endosomal compartment in mouse macrophages is Rab5-dependent. Exp. Cell Res. 2006, 312, 2238–2251. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Hanson, P.I.; Schlesinger, P. Luminal chloride-dependent activation of endosome calcium channels: Patch clamp study of enlarged endosomes. J. Biol. Chem. 2007, 282, 27327–27333. [Google Scholar] [CrossRef] [Green Version]
- Sumoza-Toledo, A.; Fleig, A.; Penner, R. TRPM2 channels are not required for acute airway inflammation in OVA-induced severe allergic asthma in mice. J. Inflamm. 2013, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Di, A.; Gao, X.-P.; Qian, F.; Kawamura, T.; Han, J.; Hecquet, C.; Ye, R.D.; Vogel, S.M.; Malik, A.B. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat. Immunol. 2012, 13, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Sumoza-Toledo, A.; Lange, I.; Cortado, H.; Bhagat, H.; Mori, Y.; Fleig, A.; Penner, R.; Partida-Sánchez, S. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011, 25, 3529–3542. [Google Scholar] [CrossRef] [Green Version]
- Lange, I.; Yamamoto, S.; Partida-Sanchez, S.; Mori, Y.; Fleig, A.; Penner, R. TRPM2 Functions as a Lysosomal Ca2+-Release Channel in β Cells. Sci. Signal. 2010, 2, ra23. [Google Scholar] [CrossRef] [Green Version]
- Sumoza-Toledo, A.; Penner, R. TRPM2: A multifunctional ion channel for calcium signalling. J. Physiol. 2011, 589, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Talavera, K.; Startek, J.B.; Alvarez-Collazo, J.; Boonen, B.; Alpizar, Y.A.; Sanchez, A.; Naert, R.; Nilius, B. Mammalian transient receptor potential TRPA1 channels: From structure to disease. Physiol. Rev. 2020, 100, 725–803. [Google Scholar] [CrossRef]
- Shang, S.; Zhu, F.; Liu, B.; Chai, Z.; Wu, Q.; Hu, M.; Wang, Y.; Huang, R.; Zhang, X.; Wu, X.; et al. Intracellular TRPA1 mediates Ca2+ release from lysosomes in dorsal root ganglion neurons. J. Cell Biol. 2016, 215, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, V.; Dijk, F.N.; Holloway, J.W.; Ring, S.M.; Koppelman, G.H.; Postma, D.S.; Strachan, D.P.; Granell, R.; de Jongste, J.C.; Jaddoe, V.W.V.; et al. TRPA1 gene polymorphisms and childhood asthma. Pediatr. Allergy Immunol. 2017, 28, 191–198. [Google Scholar] [CrossRef]
- Geppetti, P.; Patacchini, R.; Nassini, R.; Materazzi, S. Cough: The emerging role of the TRPA1 channel. Lung 2010, 188, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Belvisi, M.G.; Dubuis, E.; Birrell, M.A. Transient receptor potential A1 channels: Insights into cough and airway inflammatory disease. Chest 2011, 140, 1040–1047. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.H.; Taggart, M.; Borthwick, L.; Fisher, A.; Brodlie, M.; Sassano, M.F.; Tarran, R.; Gray, M.A. Acute cigarette smoke or extract exposure rapidly activates TRPA1-mediated calcium influx in primary human airway smooth muscle cells. Sci. Rep. 2021, 11, 9643. [Google Scholar] [CrossRef]
- Rasmussen, J.E.; Sheridan, J.T.; Polk, W.; Davies, C.M.; Tarran, R. Cigarette smoke-induced Ca2+ release leads to cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction. J. Biol. Chem. 2014, 289, 7671–7681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.X. A well-known potassium channel plays a critical role in lysosomes. J. Cell Biol. 2017, 216, 1513–1515. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhang, X.; Gao, Q.; Lawas, M.; Yu, L.; Cheng, X.; Gu, M.; Sahoo, N.; Li, X.; Li, P.; et al. A voltage-dependent K+ channel in the lysosome is required for refilling lysosomal Ca2+ stores. J. Cell Biol. 2017, 216, 1715–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Zhang, Z.; Toro, L.; Dong, X.P. BK Channels Alleviate Lysosomal Storage Diseases by Providing Positive Feedback Regulation of Lysosomal Ca2+ Release. Dev. Cell 2015, 33, 427–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, L.; Hoa, N.T.; Wilson, Z.; Arismendi-Morillo, G.; Kong, X.T.; Tajhya, R.B.; Beeton, C.; Jadus, M.R. Big Potassium (BK) ion channels in biology, disease and possible targets for cancer immunotherapy. Int. Immunopharmacol. 2014, 22, 427–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Xu, M.; Cao, Q.; Huang, P.; Zhu, X.; Dong, X.P. A lysosomal K+ channel regulates large particle phagocytosis by facilitating lysosome Ca2+ release. Sci. Rep. 2020, 10, 1038. [Google Scholar] [CrossRef] [PubMed]
- Goldklang, M.P.; Perez-Zoghbi, J.F.; Trischler, J.; Nkyimbeng, T.; Zakharov, S.I.; Shiomi, T.; Zelonina, T.; Marks, A.R.; D’Armiento, J.M.; Marx, S.O. Treatment of experimental asthma using a single small molecule with anti-inflammatory and BK channelactivating properties. FASEB J. 2013, 27, 4975–4986. [Google Scholar] [CrossRef] [Green Version]
- Scruggs, A.M.; Grabauskas, G.; Huang, S.K. The role of KCNMB1 and BK channels in myofibroblast differentiation and pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 191–203. [Google Scholar] [CrossRef]
- Chen, C.C.; Cang, C.; Fenske, S.; Butz, E.; Chao, Y.K.; Biel, M.; Ren, D.; Wahl-Schott, C.; Grimm, C. Patch-clamp technique to characterize ion channels in enlarged individual endolysosomes. Nat. Protoc. 2017, 12, 1639–1658. [Google Scholar] [CrossRef]
- Rühl, P.; Rosato, A.S.; Urban, N.; Gerndt, S.; Tang, R.; Abrahamian, C.; Leser, C.; Sheng, J.; Jha, A.; Vollmer, G.; et al. Estradiol analogs attenuate autophagy, cell migration and invasion by direct and selective inhibition of TRPML1, independent of estrogen receptors. Sci. Rep. 2021, 11, 8313. [Google Scholar] [CrossRef]
- Huang, P.; Xu, M.; Wu, Y.; Rizvi Syeda, A.K.; Dong, X.P. Multiple facets of TRPML1 in autophagy. Cell Calcium 2020, 88, 10–12. [Google Scholar] [CrossRef]
- Scotto Rosato, A.; Montefusco, S.; Soldati, C.; Di Paola, S.; Capuozzo, A.; Monfregola, J.; Polishchuk, E.; Amabile, A.; Grimm, C.; Lombardo, A.; et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKβ/VPS34 pathway. Nat. Commun. 2019, 10, 5630. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signaling regulates autophagy via calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plesch, E.; Chen, C.C.; Butz, E.; Rosato, A.S.; Krogsaeter, E.K.; Yinan, H.; Bartel, K.; Keller, M.; Robaa, D.; Teupser, D.; et al. Selective agonist of TRPML2 reveals direct role in chemokine release from innate immune cells. eLife 2018, 7, e39720. [Google Scholar] [CrossRef] [PubMed]
- Galione, A.; Davis, L. Revealing the secrets of secretion. eLife 2018, 7, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Hua, Y.; Vergarajauregui, S.; Diab, H.I.; Puertollano, R. Novel Role of TRPML2 in the Regulation of the Innate Immune Response. J. Immunol. 2015, 195, 4922–4932. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, K.; Wong, C.-O.; Zhu, M.X. The Role of TRPMLs in Endolysosomal Trafficking and Function. Cell Calcium 2016, 58, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosato, A.S.; Tang, R.; Grimm, C. Two-pore and TRPML cation channels: Regulators of phagocytosis, autophagy and lysosomal exocytosis. Pharmacol. Ther. 2021, 220, 107713. [Google Scholar] [CrossRef]
- Davis, L.C.; Morgan, A.J.; Galione, A. NAADP-regulated two-pore channels drive phagocytosis through endo-lysosomal Ca2+ nanodomains, calcineurin and dynamin. EMBO J. 2020, 39, e104058. [Google Scholar] [CrossRef]
- Gerndt, S.; Chen, C.C.; Chao, Y.K.; Yuan, Y.; Burgstaller, S.; Rosato, A.S.; Krogsaeter, E.; Urban, N.; Jacob, K.; Nguyen, O.N.P.; et al. Agonist-mediated switching of ion selectivity in TPC2 differentially promotes lysosomal function. eLife 2020, 9, e54712. [Google Scholar] [CrossRef]
- Di Paola, S.; Medina, D.L. TRPML1-/TFEB-Dependent Regulation of Lysosomal Exocytosis. Methods Mol. Biol. 2019, 1925, 143–144. [Google Scholar] [CrossRef]
- Samie, M.; Wang, X.; Zhang, X.; Goschka, A.; Li, X.; Cheng, X.; Gregg, E.; Azar, M.; Zhuo, Y.; Garrity, A.; et al. A TRP Channel in the Lysosome Regulates Large Particle Phagocytosis via Focal Exocytosis. Dev. Cell 2017, 32, 736–740. [Google Scholar] [CrossRef] [Green Version]
- Mizumura, K.; Cloonan, S.; Choi, M.E.; Hashimoto, S.; Nakahira, K.; Ryter, S.W.; Choi, A.M.K. Autophagy: Friend or foe in lung disease? Ann. Am. Thorac. Soc. 2016, 13, S40–S47. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Maruoka, S.; Shimizu, T.; Gon, Y. Autophagy, selective autophagy, and necroptosis in COPD. Int. J. COPD 2018, 13, 3165–3172. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W.; Chen, Z.H.; Hong, P.K.; Choi, A.M.K. Autophagy in chronic obstructive pulmonary disease: Homeostatic or pathogenic mechanism? Autophagy 2009, 5, 235–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryter, S.W.; Choi, A.M.K. Autophagy in the lung. Proc. Am. Thorac. Soc. 2010, 7, 13–21. [Google Scholar] [CrossRef]
- Monick, M.M.; Powers, L.S.; Walters, K.; Lovan, N.; Zhang, M.; Gerke, A.; Hansdottir, S.; Hunninghake, G.W. Identification of an Autophagy Defect in Smokers’ Alveolar Macrophages. J. Immunol. 2010, 185, 5425–5435. [Google Scholar] [CrossRef] [Green Version]
- Borie, R.; Crestani, B.; Guyard, A.; Lidove, O. Interstitial lung disease in lysosomal storage disorders. Eur. Respir. Rev. 2021, 30, 1–16. [Google Scholar] [CrossRef]
- Faverio, P.; Stainer, A.; De Giacomi, F.; Gasperini, S.; Motta, S.; Canonico, F.; Pieruzzi, F.; Monzani, A.; Pesci, A.; Biondi, A. Molecular pathways and respiratory involvement in lysosomal storage diseases. Int. J. Mol. Sci. 2019, 20, 327. [Google Scholar] [CrossRef] [Green Version]
- Staretz-Chacham, O.; Aviram, M.; Morag, I.; Goldbart, A.; Hershkovitz, E. Pulmonary involvement in Niemann-Pick C type 1. Eur. J. Pediatr. 2018, 177, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.-K.; Chang, S.-Y.; Grimm, C. Endo-Lysosomal Cation Channels and Infectious Diseases. Rev. Physiol. Biochem. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Rinkenberger, N.; Schoggins, W. Mucolipin-2 Cation Channel Increases Trafficking Efficiency of Endocytosed Viruses. MBio 2018, 9, e02314-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-schott, C.; Biel, M.; Davey, R.A. Two pore channels control Ebolavirus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castonguay, J.; Orth, J.H.C.; Müller, T.; Sleman, F.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Mallmann, R.T.; Bildl, W.; Schulte, U.; et al. The two-pore channel TPC1 is required for efficient protein processing through early and recycling endosomes. Sci. Rep. 2017, 7, 10038. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spix, B.; Jeridi, A.; Ansari, M.; Yildirim, A.Ö.; Schiller, H.B.; Grimm, C. Endolysosomal Cation Channels and Lung Disease. Cells 2022, 11, 304. https://doi.org/10.3390/cells11020304
Spix B, Jeridi A, Ansari M, Yildirim AÖ, Schiller HB, Grimm C. Endolysosomal Cation Channels and Lung Disease. Cells. 2022; 11(2):304. https://doi.org/10.3390/cells11020304
Chicago/Turabian StyleSpix, Barbara, Aicha Jeridi, Meshal Ansari, Ali Önder Yildirim, Herbert B. Schiller, and Christian Grimm. 2022. "Endolysosomal Cation Channels and Lung Disease" Cells 11, no. 2: 304. https://doi.org/10.3390/cells11020304