

Heparanase: A Novel Therapeutic Target for the Treatment of Atherosclerosis




Abstract
1. Introduction
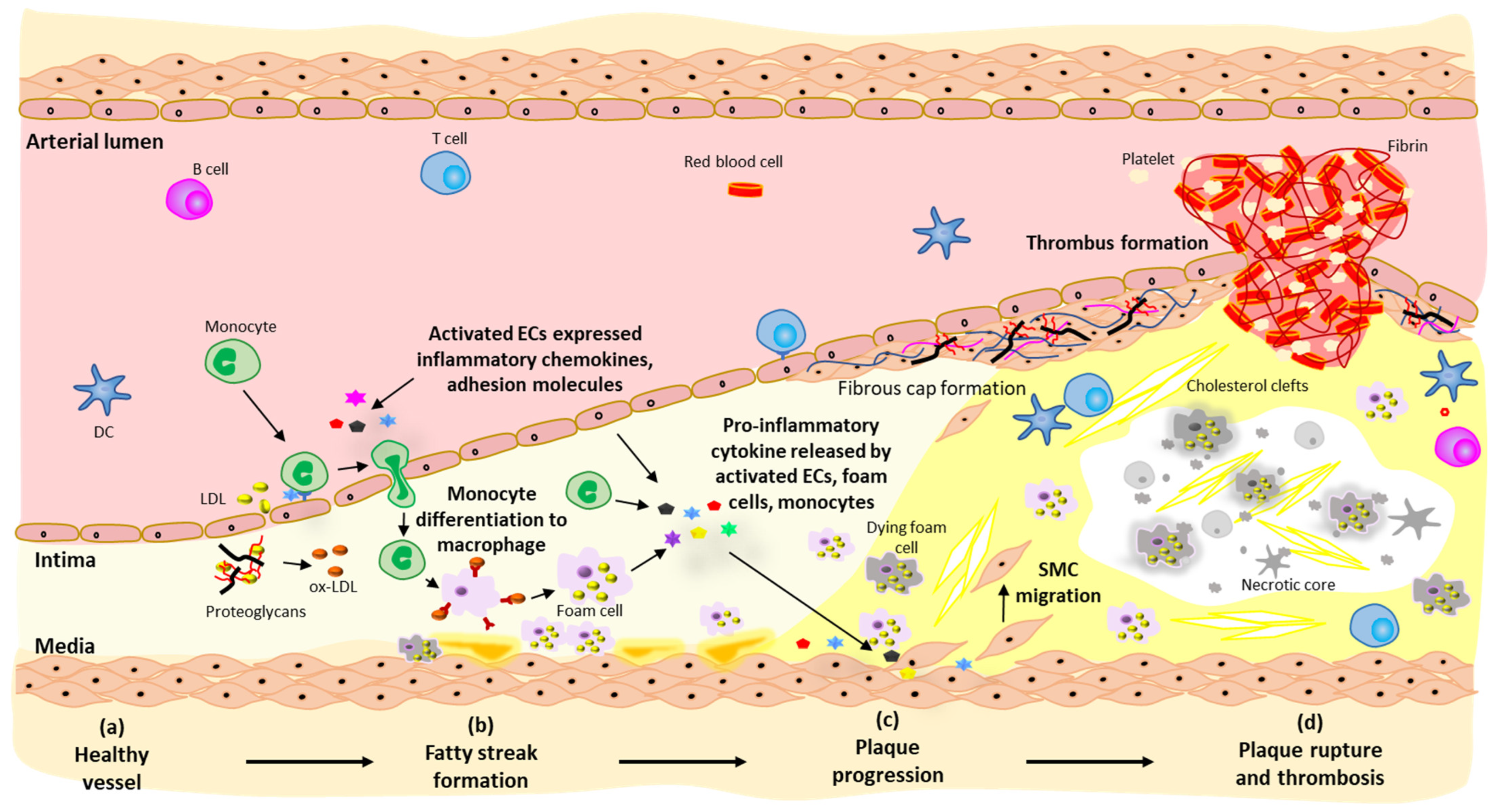
2. Pathogenesis of Atherosclerosis
2.1. Fatty Streak Formation
2.2. Plaque Progression
2.3. Plaque Rupture and Thrombosis
3. Current Therapies for Atherosclerosis
4. HPSE and Atherosclerosis
4.1. Heparan Sulfate Proteoglycans and HPSE
4.2. Physiological and Pathological Functions of HPSE
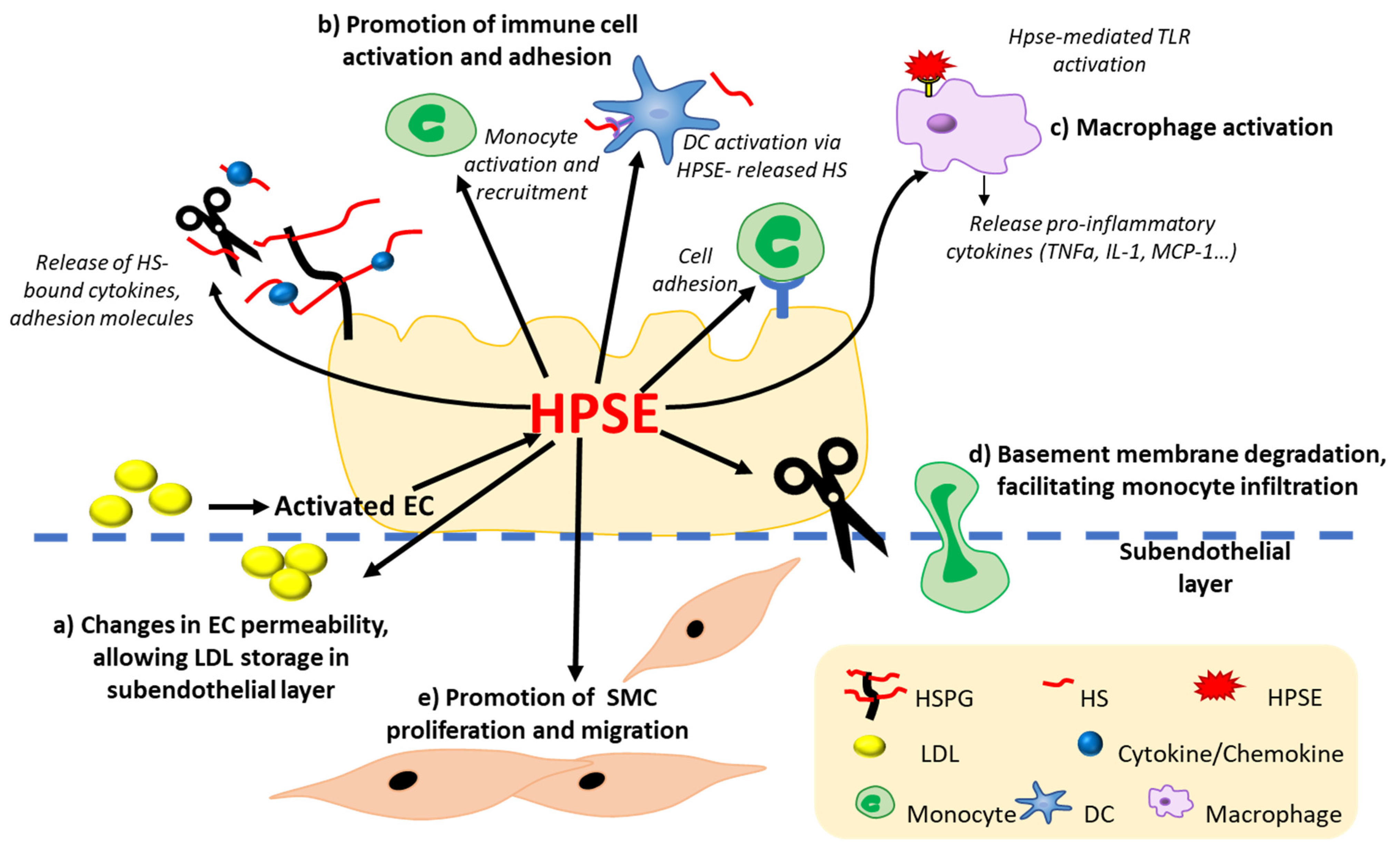
4.3. HPSE Expression and Function in Atherosclerosis
4.3.1. HPSE Modulates Vascular Inflammation by Modifying Immune Cell Activation, Migration, and Adhesion
4.3.2. HPSE Alters the Homeostasis of ECs
4.3.3. HPSE Promotes Monocyte Binding- and Macrophage-Mediated Inflammation
4.3.4. HPSE Promotes SMC Proliferation and Plaque Rupture
5. HPSE in Models of Atherosclerosis and Therapeutics Targeting HPSE
6. Concluding Remarks and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ilhan, F.; Kalkanli, S.T. Atherosclerosis and the role of immune cells. World J. Clin. Cases 2015, 3, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Mayfosh, A.J.; Nguyen, T.K.; Hulett, M.D. The Heparanase Regulatory Network in Health and Disease. Int. J. Mol. Sci. 2021, 22, 11096. [Google Scholar] [CrossRef] [PubMed]
- Zcharia, E.; Zilka, R.; Yaar, A.; Yacoby-Zeevi, O.; Zetser, A.; Metzger, S.; Sarid, R.; Naggi, A.; Casu, B.; Ilan, N.; et al. Heparanase accelerates wound angiogenesis and wound healing in mouse and rat models. FASEB J. 2005, 19, 211–221. [Google Scholar] [CrossRef]
- Zcharia, E.; Philp, D.; Edovitsky, E.; Aingorn, H.; Metzger, S.; Kleinman, H.K.; Vlodavsky, I.; Elkin, M. Heparanase regulates murine hair growth. Am. J. Pathol. 2005, 166, 999–1008. [Google Scholar] [CrossRef]
- Elkin, M.; Ilan, N.; Ishai-Michaeli, R.; Friedmann, Y.; Papo, O.; Pecker, I.; Vlodavsky, I. Heparanase as mediator of angiogenesis: Mode of action. FASEB J. 2001, 15, 1661–1663. [Google Scholar] [CrossRef] [PubMed]
- Parish, C.R.; Freeman, C.; Hulett, M.D. Heparanase: A key enzyme involved in cell invasion. Biochim. Biophys. Acta 2001, 1471, M99–M108. [Google Scholar] [CrossRef]
- Tang, W.; Nakamura, Y.; Tsujimoto, M.; Sato, M.; Wang, X.; Kurozumi, K.; Nakahara, M.; Nakao, K.; Nakamura, M.; Mori, I.; et al. Heparanase: A key enzyme in invasion and metastasis of gastric carcinoma. Mod. Pathol. 2002, 15, 593–598. [Google Scholar] [CrossRef]
- Gohji, K.; Okamoto, M.; Kitazawa, S.; Toyoshima, M.; Dong, J.; Katsuoka, Y.; Nakajima, M. Heparanase protein and gene expression in bladder cancer. J. Urol. 2001, 166, 1286–1290. [Google Scholar] [CrossRef]
- Yang, Y.; Macleod, V.; Bendre, M.; Huang, Y.; Theus, A.M.; Miao, H.Q.; Kussie, P.; Yaccoby, S.; Epstein, J.; Suva, L.J.; et al. Heparanase promotes the spontaneous metastasis of myeloma cells to bone. Blood 2005, 105, 1303–1309. [Google Scholar] [CrossRef]
- Cohen, I.; Pappo, O.; Elkin, M.; San, T.; Bar-Shavit, R.; Hazan, R.; Peretz, T.; Vlodavsky, I.; Abramovitch, R. Heparanase promotes growth, angiogenesis and survival of primary breast tumors. Int. J. Cancer 2006, 118, 1609–1617. [Google Scholar] [CrossRef]
- Khanna, M.; Parish, C.R. Heparanase: Historical Aspects and Future Perspectives. In Heparanase: From Basic Research to Clinical Applications; Vlodavsky, I., Sanderson, R.D., Ilan, N., Eds.; Springer: Cham, Switzerland, 2020; pp. 71–96. [Google Scholar]
- Waterman, M.; Ben-Izhak, O.; Eliakim, R.; Groisman, G.; Vlodavsky, I.; Ilan, N. Heparanase upregulation by colonic epithelium in inflammatory bowel disease. Mod. Pathol. 2007, 20, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; He, Y.; Hu, Z.; Lu, S.; Yin, X.; Ma, X.; Lv, C.; Jin, G. Heparanase Mediates Intestinal Inflammation and Injury in a Mouse Model of Sepsis. J. Histochem. Cytochem. 2017, 65, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Simeonovic, C.; Ziolkowski, A.; Wu, Z.; Choong, F.J.; Freeman, C.; Parish, C. Heparanase and Autoimmune Diabetes. Front. Immunol. 2013, 4, 471. [Google Scholar] [CrossRef] [PubMed]
- Shafat, I.; Ilan, N.; Zoabi, S.; Vlodavsky, I.; Nakhoul, F. Heparanase Levels Are Elevated in the Urine and Plasma of Type 2 Diabetes Patients and Associate with Blood Glucose Levels. PLoS ONE 2011, 6, e17312. [Google Scholar] [CrossRef]
- Gil, N.; Goldberg, R.; Neuman, T.; Garsen, M.; Zcharia, E.; Rubinstein, A.M.; van Kuppevelt, T.; Meirovitz, A.; Pisano, C.; Li, J.-P.; et al. Heparanase is essential for the development of diabetic nephropathy in mice. Diabetes 2012, 61, 208–216. [Google Scholar] [CrossRef]
- Muhammad, R.S.; Abu-Saleh, N.; Kinaneh, S.; Agbaria, M.; Sabo, E.; Grajeda-Iglesias, C.; Volkova, N.; Hamoud, S. Heparanase inhibition attenuates atherosclerosis progression and liver steatosis in E0 mice. Atherosclerosis 2018, 276, 155–162. [Google Scholar] [CrossRef]
- Gutter-Kapon, L.; Alishekevitz, D.; Shaked, Y.; Li, J.-P.; Aronheim, A.; Ilan, N.; Vlodavsky, I. Heparanase is required for activation and function of macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, E7808. [Google Scholar] [CrossRef]
- Osterholm, C.; Folkersen, L.; Lengquist, M.; Ponten, F.; Renne, T.; Li, J.; Hedin, U. Increased expression of heparanase in symptomatic carotid atherosclerosis. Atherosclerosis 2013, 226, 67–73. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Blich, M.; Li, J.-P.; Sanderson, R.D.; Ilan, N. Involvement of heparanase in atherosclerosis and other vessel wall pathologies. Matrix Biol. 2013, 32, 241–251. [Google Scholar] [CrossRef]
- Geovanini, G.R.; Libby, P. Atherosclerosis and inflammation: Overview and updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef]
- Hermida, N.; Balligand, J.L. Low-density lipoprotein-cholesterol-induced endothelial dysfunction and oxidative stress: The role of statins. Antioxid. Redox Signal. 2014, 20, 1216–1237. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.; Channon, K.M. The pathogenesis of atherosclerosis. Medicine 2010, 38, 397–402. [Google Scholar] [CrossRef]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Hori, T.; Ishibashi, T.; Nishio, M.; Aizawa, Y. Effects of chronic cigarette smoking on endothelial function in young men. J. Cardiol. 2010, 56, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Mandal, A.K.; Hiebert, L.M. Endothelial cell injury by high glucose and heparanase is prevented by insulin, heparin and basic fibroblast growth factor. Cardiovasc. Diabetol. 2005, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.; Ding, H.G.; Huang, W.; Le, D.; Maxhimer, J.B.; Oosterhof, A.; van Kuppevelt, T.; Lum, H.; Lewis, E.J.; Reddy, V.; et al. Reactive oxygen species mediate high glucose-induced heparanase-1 production and heparan sulphate proteoglycan degradation in human and rat endothelial cells: A potential role in the pathogenesis of atherosclerosis. Diabetologia 2011, 54, 1527–1538. [Google Scholar] [CrossRef][Green Version]
- Funk, S.D.; Yurdagul, A.; Orr, A.W. Hyperglycemia and Endothelial Dysfunction in Atherosclerosis: Lessons from Type 1 Diabetes. Int. J. Vasc. Med. 2012, 2012, 569654. [Google Scholar] [CrossRef]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Simionescu, M. Implications of early structural-functional changes in the endothelium for vascular disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 266–274. [Google Scholar] [CrossRef]
- Tabas, I.; Williams, K.J.; Boren, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Endemann, D.H.; Schiffrin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Terpstra, V.; Kondratenko, N.; Steinberg, D. Macrophages lacking scavenger receptor A show a decrease in binding and uptake of acetylated low-density lipoprotein and of apoptotic thymocytes, but not of oxidatively damaged red blood cells. Proc. Natl. Acad. Sci. USA 1997, 94, 8127–8131. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell. Mol. Med. 2016, 20, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.-L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.S.; Smyth, D.; et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Stemme, S.; Faber, B.; Holm, J.; Wiklund, O.; Witztum, J.L.; Hansson, G.K. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1995, 92, 3893. [Google Scholar] [CrossRef]
- Koltsova, E.K.; Garcia, Z.; Chodaczek, G.; Landau, M.; McArdle, S.; Scott, S.R.; von Vietinghoff, S.; Galkina, E.; Miller, Y.I.; Acton, S.T.; et al. Dynamic T cell–APC interactions sustain chronic inflammation in atherosclerosis. J. Clin. Investig. 2012, 122, 3114–3126. [Google Scholar] [CrossRef]
- Klingenberg, R.; Gerdes, N.; Badeau, R.M.; Gisterå, A.; Strodthoff, D.; Ketelhuth, D.F.J.; Lundberg, A.M.; Rudling, M.; Nilsson, S.K.; Olivecrona, G.; et al. Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J. Clin. Investig. 2013, 123, 1323–1334. [Google Scholar] [CrossRef]
- Packard, R.R.S.; Maganto-García, E.; Gotsman, I.; Tabas, I.; Libby, P.; Lichtman, A.H. CD11c+ Dendritic Cells Maintain Antigen Processing, Presentation Capabilities, and CD4+ T-Cell Priming Efficacy Under Hypercholesterolemic Conditions Associated with Atherosclerosis. Circ. Res. 2008, 103, 965–973. [Google Scholar] [CrossRef]
- MacRitchie, N.; Grassia, G.; Sabir, S.R.; Maddaluno, M.; Welsh, P.; Sattar, N.; Ialenti, A.; Kurowska-Stolarska, M.; McInnes, I.B.; Brewer, J.M.; et al. Plasmacytoid Dendritic Cells Play a Key Role in Promoting Atherosclerosis in Apolipoprotein E–Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2569–2579. [Google Scholar] [CrossRef] [PubMed]
- Daissormont, I.T.M.N.; Christ, A.; Temmerman, L.; Millares, S.S.; Seijkens, T.; Manca, M.; Rousch, M.; Poggi, M.; Boon, L.; Loos, C.v.d.; et al. Plasmacytoid Dendritic Cells Protect Against Atherosclerosis by Tuning T-Cell Proliferation and Activity. Circ. Res. 2011, 109, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Manthey, H.D.; Drechsler, M.; Lievens, D.; Megens, R.T.A.; Soehnlein, O.; Busch, M.; Manca, M.; Koenen, R.R.; Pelisek, J.; et al. Auto-Antigenic Protein-DNA Complexes Stimulate Plasmacytoid Dendritic Cells to Promote Atherosclerosis. Circulation 2012, 125, 1673–1683. [Google Scholar] [CrossRef]
- Yun, T.J.; Lee, J.S.; Machmach, K.; Shim, D.; Choi, J.; Wi, Y.J.; Jang, H.S.; Jung, I.H.; Kim, K.; Yoon, W.K.; et al. Indoleamine 2,3-Dioxygenase-Expressing Aortic Plasmacytoid Dendritic Cells Protect against Atherosclerosis by Induction of Regulatory T Cells. Cell. Metab. 2016, 23, 852–866. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Bornfeldt, K.E.; Tall, A.R. Atherosclerosis. Circ. Res. 2016, 118, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Investig. 2007, 117, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Randolph, G.J. Proliferating macrophages prevail in atherosclerosis. Nat. Med. 2013, 19, 1094–1095. [Google Scholar] [CrossRef]
- Thorp, E.; Vaisar, T.; Subramanian, M.; Mautner, L.; Blobel, C.; Tabas, I. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cδ, and p38 mitogen-activated protein kinase (MAPK). J. Biol. Chem. 2011, 286, 33335–33344. [Google Scholar] [CrossRef]
- Adiguzel, E.; Ahmad, P.J.; Franco, C.; Bendeck, M.P. Collagens in the progression and complications of atherosclerosis. Vasc. Med. 2009, 14, 73–89. [Google Scholar] [CrossRef]
- Mani, A.J.; Edep, M.E.; Brown, D.L. Chapter 8—Pathophysiology of Acute Coronary Syndromes: Plaque Rupture and Atherothrombosis. In Cardiac Intensive Care, 2nd ed.; Jeremias, A., Brown, D.L., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2010; pp. 73–86. [Google Scholar]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of Vascular Smooth Muscle Cells to Macrophage-Like Cells During Atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Falk, E.; Shah, P.K.; Fuster, V. Coronary plaque disruption. Circulation 1995, 92, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Sukhova, G.K.; Lark, M.W.; Libby, P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Investig. 1994, 94, 2493–2503. [Google Scholar] [CrossRef]
- Newby, A.C. Role of Metalloproteinases in Plaque Rupture. Int. J. Gerontol. 2007, 1, 103–111. [Google Scholar] [CrossRef][Green Version]
- Baker, A.B.; Chatzizisis, Y.S.; Beigel, R.; Jonas, M.; Stone, B.V.; Coskun, A.U.; Maynard, C.; Rogers, C.; Koskinas, K.C.; Feldman, C.L.; et al. Regulation of heparanase expression in coronary artery disease in diabetic, hyperlipidemic swine. Atherosclerosis 2010, 213, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Haslinger-Loffler, B. Multiple effects of HMG-CoA reductase inhibitors (statins) besides their lipid-lowering function. Kidney Int. 2008, 74, 553–555. [Google Scholar] [CrossRef]
- Kim, Y.H.; Her, A.-Y.; Jeong, M.H.; Kim, B.-K.; Hong, S.-J.; Kim, S.; Ahn, C.-M.; Kim, J.-S.; Ko, Y.-G.; Choi, D.; et al. Effect of statin treatment in patients with acute myocardial infarction with prediabetes and type 2 diabetes mellitus: A retrospective observational registry study. Medicine 2021, 100, e24733. [Google Scholar] [CrossRef]
- Ibanez, B.; James, S.; Agewall, S.; Antunes, M.J.; Bucciarelli-Ducci, C.; Bueno, H.; Caforio, A.L.P.; Crea, F.; Goudevenos, J.A.; Halvorsen, S.; et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2017, 39, 119–177. [Google Scholar] [CrossRef]
- Collet, J.P.; Thiele, H.; Barbato, E.; Barthélémy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur. Heart J. 2021, 42, 1289–1367. [Google Scholar] [CrossRef]
- O’Gara, P.T.; Kushner, F.G.; Ascheim, D.D.; Casey, D.E.; Chung, M.K.; Lemos, J.A.d.; Ettinger, S.M.; Fang, J.C.; Fesmire, F.M.; Franklin, B.A.; et al. 2013 ACCF/AHA Guideline for the Management of ST-Elevation Myocardial Infarction. Circulation 2013, 127, e362–e425. [Google Scholar] [CrossRef]
- Liao, J.K.; Laufs, U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 89–118. [Google Scholar] [CrossRef] [PubMed]
- Dongoran, R.A.; Wang, K.H.; Lin, T.J.; Yuan, T.C.; Liu, C.H. Anti-Proliferative Effect of Statins Is Mediated by DNMT1 Inhibition and p21 Expression in OSCC Cells. Cancers 2020, 12, 2084. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.D.; Panza, G.; Zaleski, A.; Taylor, B. Statin-Associated Side Effects. J. Am. Coll. Cardiol. 2016, 67, 2395–2410. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, T.A.; Cheeley, M.K.; Jones, P.H.; La Forge, R.; Maki, K.C.; López, J.A.G.; Xiang, P.; Bushnell, D.M.; Martin, M.L.; Cohen, J.D. The STatin Adverse Treatment Experience Survey: Experience of patients reporting side effects of statin therapy. J. Clin. Lipidol. 2019, 13, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.K.; Ali, S.; Sanghera, R.S. Pharmacological Options in Atherosclerosis: A Review of the Existing Evidence. Cardiol. Ther. 2019, 8, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Bergheanu, S.C.; Bodde, M.C.; Jukema, J.W. Pathophysiology and treatment of atherosclerosis: Current view and future perspective on lipoprotein modification treatment. Neth. Heart J. 2017, 25, 231–242. [Google Scholar] [CrossRef]
- Kong, P.; Cui, Z.-Y.; Huang, X.-F.; Zhang, D.-D.; Guo, R.-J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal. Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Lagace, T.A.; Garuti, R.; Zhao, Z.; McDonald, M.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 2007, 282, 18602–18612. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Blom, D.J.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; Wasserman, S.M.; et al. Efficacy and Safety of Evolocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1500–1509. [Google Scholar] [CrossRef]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and Safety of Alirocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef]
- Tousoulis, D.; Oikonomou, E.; Economou, E.K.; Crea, F.; Kaski, J.C. Inflammatory cytokines in atherosclerosis: Current therapeutic approaches. Eur. Heart J. 2016, 37, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Spronk, H.M.H.; Stouffer, G.A.; Cate, H.t. Dual Anticoagulant and Antiplatelet Therapy for Coronary Artery Disease and Peripheral Artery Disease Patients. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.; Gebhardt, J.; Ramsperger-Gleixner, M.; Jacobi, J.; Weyand, M.; Ensminger, S.M. Clopidogrel significantly lowers the development of atherosclerosis in ApoE-deficient mice in vivo. Heart Vessel. 2016, 31, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Baker, A.B.; Groothuis, A.; Jonas, M.; Ettenson, D.S.; Shazly, T.; Zcharia, E.; Vlodavsky, I.; Seifert, P.; Edelman, E.R. Heparanase alters arterial structure, mechanics, and repair following endovascular stenting in mice. Circ. Res. 2009, 104, 380–387. [Google Scholar] [CrossRef]
- Planer, D.; Metzger, S.; Zcharia, E.; Wexler, I.D.; Vlodavsky, I.; Chajek-Shaul, T. Role of heparanase on hepatic uptake of intestinal derived lipoprotein and fatty streak formation in mice. PLoS ONE 2011, 6, e18370. [Google Scholar] [CrossRef]
- Baker, A.B.; Gibson, W.J.; Kolachalama, V.B.; Golomb, M.; Indolfi, L.; Spruell, C.; Zcharia, E.; Vlodavsky, I.; Edelman, E.R. Heparanase regulates thrombosis in vascular injury and stent-induced flow disturbance. J. Am. Coll. Cardiol. 2012, 59, 1551–1560. [Google Scholar] [CrossRef]
- Blich, M.; Golan, A.; Arvatz, G.; Sebbag, A.; Shafat, I.; Sabo, E.; Cohen-Kaplan, V.; Petcherski, S.; Avniel-Polak, S.; Eitan, A.; et al. Macrophage activation by heparanase is mediated by TLR-2 and TLR-4 and associates with plaque progression. Arterioscler. Thromb. Vasc. Biol. 2013, 33, e56–e65. [Google Scholar] [CrossRef]
- Goldshmidt, O.; Zcharia, E.; Cohen, M.; Aingorn, H.; Cohen, I.; Nadav, L.; Katz, B.Z.; Geiger, B.; Vlodavsky, I. Heparanase mediates cell adhesion independent of its enzymatic activity. FASEB J. 2003, 17, 1015–1025. [Google Scholar] [CrossRef]
- Levy-Adam, F.; Feld, S.; Suss-Toby, E.; Vlodavsky, I.; Ilan, N. Heparanase facilitates cell adhesion and spreading by clustering of cell surface heparan sulfate proteoglycans. PLoS ONE 2008, 3, e2319. [Google Scholar] [CrossRef] [PubMed]
- Gingis-Velitski, S.; Zetser, A.; Flugelman, M.Y.; Vlodavsky, I.; Ilan, N. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J. Biol. Chem. 2004, 279, 23536–23541. [Google Scholar] [CrossRef] [PubMed]
- Lever, R.; Rose, M.J.; McKenzie, E.A.; Page, C.P. Heparanase induces inflammatory cell recruitment in vivo by promoting adhesion to vascular endothelium. Am. J. Physiol. Cell. Physiol. 2014, 306, C1184–C1190. [Google Scholar] [CrossRef] [PubMed]
- Lin, X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development 2004, 131, 6009–6021. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell. Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Bode, L.; Salvestrini, C.; Park, P.W.; Li, J.P.; Esko, J.D.; Yamaguchi, Y.; Murch, S.; Freeze, H.H. Heparan sulfate and syndecan-1 are essential in maintaining murine and human intestinal epithelial barrier function. J. Clin. Investig. 2008, 118, 229–238. [Google Scholar] [CrossRef]
- Coombe, D.R. Biological implications of glycosaminoglycan interactions with haemopoietic cytokines. Immunol. Cell. Biol. 2008, 86, 598–607. [Google Scholar] [CrossRef]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Sanderson, R.D. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J. Cell. Mol. Med. 2011, 15, 1013–1031. [Google Scholar] [CrossRef]
- Lindahl, U.; Li, J.P. Interactions between heparan sulfate and proteins-design and functional implications. Int. Rev. Cell. Mol. Biol. 2009, 276, 105–159. [Google Scholar] [CrossRef] [PubMed]
- Sadir, R.; Imberty, A.; Baleux, F.; Lortat-Jacob, H. Heparan sulfate/heparin oligosaccharides protect stromal cell-derived factor-1 (SDF-1)/CXCL12 against proteolysis induced by CD26/dipeptidyl peptidase IV. J. Biol. Chem. 2004, 279, 43854–43860. [Google Scholar] [CrossRef] [PubMed]
- Dowd, C.J.; Cooney, C.L.; Nugent, M.A. Heparan sulfate mediates bFGF transport through basement membrane by diffusion with rapid reversible binding. J. Biol. Chem. 1999, 274, 5236–5244. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.; Mitsi, M.; Nugent, M.A.; Symes, K. PDGF-A interactions with fibronectin reveal a critical role for heparan sulfate in directed cell migration during Xenopus gastrulation. Proc. Natl. Acad. Sci. USA 2009, 106, 21683–21688. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Moseman, E.A.; Saito, H.; Petryniak, B.; Thiriot, A.; Hatakeyama, S.; Ito, Y.; Kawashima, H.; Yamaguchi, Y.; Lowe, J.B.; et al. Endothelial heparan sulfate controls chemokine presentation in recruitment of lymphocytes and dendritic cells to lymph nodes. Immunity 2010, 33, 817–829. [Google Scholar] [CrossRef]
- Weber, M.; Hauschild, R.; Schwarz, J.; Moussion, C.; de Vries, I.; Legler, D.F.; Luther, S.A.; Bollenbach, T.; Sixt, M. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science 2013, 339, 328–332. [Google Scholar] [CrossRef]
- Wang, L.; Fuster, M.; Sriramarao, P.; Esko, J.D. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat. Immunol. 2005, 6, 902–910. [Google Scholar] [CrossRef]
- Connell, B.J.; Sadir, R.; Baleux, F.; Laguri, C.; Kleman, J.P.; Luo, L.; Arenzana-Seisdedos, F.; Lortat-Jacob, H. Heparan sulfate differentially controls CXCL12alpha- and CXCL12gamma-mediated cell migration through differential presentation to their receptor CXCR4. Sci. Signal. 2016, 9, ra107. [Google Scholar] [CrossRef]
- Freeman, C.; Parish, C.R. Human platelet heparanase: Purification, characterization and catalytic activity. Biochem. J. 1998, 330, 1341–1350. [Google Scholar] [CrossRef]
- Vreys, V.; David, G. Mammalian heparanase: What is the message? J. Cell. Mol. Med. 2007, 11, 427–452. [Google Scholar] [CrossRef]
- Hulett, M.D.; Freeman, C.; Hamdorf, B.J.; Baker, R.T.; Harris, M.J.; Parish, C.R. Cloning of mammalian heparanase, an important enzyme in tumor invasion and metastasis. Nat. Med. 1999, 5, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Kukula, A.K.; Toyoshima, M.; Nakajima, M. Genomic organization and chromosome localization of the newly identified human heparanase gene. Gene 2000, 253, 171–178. [Google Scholar] [CrossRef]
- Fairbanks, M.B.; Mildner, A.M.; Leone, J.W.; Cavey, G.S.; Mathews, W.R.; Drong, R.F.; Slightom, J.L.; Bienkowski, M.J.; Smith, C.W.; Bannow, C.A.; et al. Processing of the human heparanase precursor and evidence that the active enzyme is a heterodimer. J. Biol. Chem. 1999, 274, 29587–29590. [Google Scholar] [CrossRef] [PubMed]
- Vreys, V.; Delande, N.; Zhang, Z.; Coomans, C.; Roebroek, A.; Durr, J.; David, G. Cellular uptake of mammalian heparanase precursor involves low density lipoprotein receptor-related proteins, mannose 6-phosphate receptors, and heparan sulfate proteoglycans. J. Biol. Chem. 2005, 280, 33141–33148. [Google Scholar] [CrossRef]
- Goldshmidt, O.; Nadav, L.; Aingorn, H.; Irit, C.; Feinstein, N.; Ilan, N.; Zamir, E.; Geiger, B.; Vlodavsky, I.; Katz, B.Z. Human heparanase is localized within lysosomes in a stable form. Exp. Cell. Res. 2002, 281, 50–62. [Google Scholar] [CrossRef][Green Version]
- Zetser, A.; Levy-Adam, F.; Kaplan, V.; Gingis-Velitski, S.; Bashenko, Y.; Schubert, S.; Flugelman, M.Y.; Vlodavsky, I.; Ilan, N. Processing and activation of latent heparanase occurs in lysosomes. J. Cell. Sci. 2004, 117, 2249–2258. [Google Scholar] [CrossRef]
- Abboud-Jarrous, G.; Atzmon, R.; Peretz, T.; Palermo, C.; Gadea, B.B.; Joyce, J.A.; Vlodavsky, I. Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J. Biol. Chem. 2008, 283, 18167–18176. [Google Scholar] [CrossRef]
- Hulett, M.D.; Hornby, J.R.; Ohms, S.J.; Zuegg, J.; Freeman, C.; Gready, J.E.; Parish, C.R. Identification of active-site residues of the pro-metastatic endoglycosidase heparanase. Biochemistry 2000, 39, 15659–15667. [Google Scholar] [CrossRef]
- Wu, L.; Viola, C.M.; Brzozowski, A.M.; Davies, G.J. Structural characterization of human heparanase reveals insights into substrate recognition. Nat. Struct. Mol. Biol. 2015, 22, 1016–1022. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Beckhove, P.; Lerner, I.; Pisano, C.; Meirovitz, A.; Ilan, N.; Elkin, M. Significance of heparanase in cancer and inflammation. Cancer Microenviron. 2012, 5, 115–132. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Friedmann, Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J. Clin. Investig. 2001, 108, 341–347. [Google Scholar] [CrossRef]
- Ilan, N.; Elkin, M.; Vlodavsky, I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int. J. Biochem. Cell. Biol. 2006, 38, 2018–2039. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, G.; Nian, J.; Yu, M.; Chen, S.; Zhang, Y.; Yang, G.; Yang, L.; Cheng, P.; Yan, C.; et al. Elevated heparanase expression is associated with poor prognosis in breast cancer: A study based on systematic review and TCGA data. Oncotarget 2017, 8, 43521–43535. [Google Scholar] [CrossRef]
- Vornicova, O.; Naroditsky, I.; Boyango, I.; Shachar, S.S.; Mashiach, T.; Ilan, N.; Vlodavsky, I.; Bar-Sela, G. Prognostic significance of heparanase expression in primary and metastatic breast carcinoma. Oncotarget 2017, 9, 6238–6244. [Google Scholar] [CrossRef][Green Version]
- Boyango, I.; Barash, U.; Fux, L.; Naroditsky, I.; Ilan, N.; Vlodavsky, I. Targeting heparanase to the mammary epithelium enhances mammary gland development and promotes tumor growth and metastasis. Matrix Biol. 2018, 65, 91–103. [Google Scholar] [CrossRef]
- Putz, E.M.; Mayfosh, A.J.; Kos, K.; Barkauskas, D.S.; Nakamura, K.; Town, L.; Goodall, K.J.; Yee, D.Y.; Poon, I.K.; Baschuk, N.; et al. NK cell heparanase controls tumor invasion and immune surveillance. J. Clin. Investig. 2017, 127, 2777–2788. [Google Scholar] [CrossRef]
- Li, R.W.; Freeman, C.; Yu, D.; Hindmarsh, E.J.; Tymms, K.E.; Parish, C.R.; Smith, P.N. Dramatic regulation of heparanase activity and angiogenesis gene expression in synovium from patients with rheumatoid arthritis. Arthritis Rheum. 2008, 58, 1590–1600. [Google Scholar] [CrossRef]
- Schmidt, E.P.; Yang, Y.; Janssen, W.J.; Gandjeva, A.; Perez, M.J.; Barthel, L.; Zemans, R.L.; Bowman, J.C.; Koyanagi, D.E.; Yunt, Z.X.; et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat. Med. 2012, 18, 1217–1223. [Google Scholar] [CrossRef]
- Lerner, I.; Hermano, E.; Zcharia, E.; Rodkin, D.; Bulvik, R.; Doviner, V.; Rubinstein, A.M.; Ishai-Michaeli, R.; Atzmon, R.; Sherman, Y.; et al. Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. J. Clin. Investig. 2011, 121, 1709–1721. [Google Scholar] [CrossRef]
- Roucourt, B.; Meeussen, S.; Bao, J.; Zimmermann, P.; David, G. Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Res. 2015, 25, 412–428. [Google Scholar] [CrossRef]
- Thompson, C.A.; Purushothaman, A.; Ramani, V.C.; Vlodavsky, I.; Sanderson, R.D. Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. J. Biol. Chem. 2013, 288, 10093–10099. [Google Scholar] [CrossRef]
- Shteingauz, A.; Boyango, I.; Naroditsky, I.; Hammond, E.; Gruber, M.; Doweck, I.; Ilan, N.; Vlodavsky, I. Heparanase Enhances Tumor Growth and Chemoresistance by Promoting Autophagy. Cancer Res. 2015, 75, 3946–3957. [Google Scholar] [CrossRef]
- Goldberg, R.; Meirovitz, A.; Hirshoren, N.; Bulvik, R.; Binder, A.; Rubinstein, A.M.; Elkin, M. Versatile role of heparanase in inflammation. Matrix Biol. 2013, 32, 234–240. [Google Scholar] [CrossRef]
- Zetser, A.; Bashenko, Y.; Edovitsky, E.; Levy-Adam, F.; Vlodavsky, I.; Ilan, N. Heparanase induces vascular endothelial growth factor expression: Correlation with p38 phosphorylation levels and Src activation. Cancer Res. 2006, 66, 1455–1463. [Google Scholar] [CrossRef]
- Kodaira, Y.; Nair, S.K.; Wrenshall, L.E.; Gilboa, E.; Platt, J.L. Phenotypic and functional maturation of dendritic cells mediated by heparan sulfate. J. Immunol. 2000, 165, 1599–1604. [Google Scholar] [CrossRef]
- Goodall, K.J.; Poon, I.K.; Phipps, S.; Hulett, M.D. Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PLoS ONE 2014, 9, e109596. [Google Scholar] [CrossRef]
- Sivaram, P.; Obunike, J.C.; Goldberg, I.J. Lysolecithin-induced alteration of subendothelial heparan sulfate proteoglycans increases monocyte binding to matrix. J. Biol. Chem. 1995, 270, 29760–29765. [Google Scholar] [CrossRef]
- Hershkoviz, R.; Marikovsky, M.; Gilat, D.; Lider, O. Keratinocytes-associated chemokines and enzymatically quiescent heparanase induce the binding of resting CD4+ T cells. J. Invest. Dermatol. 1996, 106, 243–248. [Google Scholar] [CrossRef][Green Version]
- Sotnikov, I.; Hershkoviz, R.; Grabovsky, V.; Ilan, N.; Cahalon, L.; Vlodavsky, I.; Alon, R.; Lider, O. Enzymatically quiescent heparanase augments T cell interactions with VCAM-1 and extracellular matrix components under versatile dynamic contexts. J. Immunol. 2004, 172, 5185–5193. [Google Scholar] [CrossRef]
- Spiegel, A.; Zcharia, E.; Vagima, Y.; Itkin, T.; Kalinkovich, A.; Dar, A.; Kollet, O.; Netzer, N.; Golan, K.; Shafat, I.; et al. Heparanase regulates retention and proliferation of primitive Sca-1+/c-Kit+/Lin- cells via modulation of the bone marrow microenvironment. Blood 2008, 111, 4934–4943. [Google Scholar] [CrossRef]
- Chen, X.-P.; Luo, J.-S.; Tian, Y.; Nie, C.-L.; Cui, W.; Zhang, W.-D. Downregulation of Heparanase Expression Results in Suppression of Invasion, Migration, and Adhesion Abilities of Hepatocellular Carcinoma Cells. Biomed. Res. Int. 2015, 2015, 241983. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Tan, Y.X.; Osterholm, C.; Zhang, X.; Hedin, U.; Vlodavsky, I.; Li, J.P. Heparanase expression upregulates platelet adhesion activity and thrombogenicity. Oncotarget 2016, 7, 39486–39496. [Google Scholar] [CrossRef] [PubMed]
- Huegel, J.; Enomoto-Iwamoto, M.; Sgariglia, F.; Koyama, E.; Pacifici, M. Heparanase stimulates chondrogenesis and is up-regulated in human ectopic cartilage: A mechanism possibly involved in hereditary multiple exostoses. Am. J. Pathol. 2015, 185, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Massena, S.; Christoffersson, G.; Hjertstrom, E.; Zcharia, E.; Vlodavsky, I.; Ausmees, N.; Rolny, C.; Li, J.P.; Phillipson, M. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood 2010, 116, 1924–1931. [Google Scholar] [CrossRef]
- Morris, A.; Wang, B.; Waern, I.; Venkatasamy, R.; Page, C.; Schmidt, E.P.; Wernersson, S.; Li, J.-P.; Spina, D. The role of heparanase in pulmonary cell recruitment in response to an allergic but not non-allergic stimulus. PLoS ONE 2015, 10, e0127032. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, A.; Babitz, S.K.; Sanderson, R.D. Heparanase enhances the insulin receptor signaling pathway to activate extracellular signal-regulated kinase in multiple myeloma. J. Biol. Chem. 2012, 287, 41288–41296. [Google Scholar] [CrossRef]
- Riaz, A.; Ilan, N.; Vlodavsky, I.; Li, J.P.; Johansson, S. Characterization of heparanase-induced phosphatidylinositol 3-kinase-AKT activation and its integrin dependence. J. Biol. Chem. 2013, 288, 12366–12375. [Google Scholar] [CrossRef]
- Nadir, Y.; Brenner, B.; Gingis-Velitski, S.; Levy-Adam, F.; Ilan, N.; Zcharia, E.; Nadir, E.; Vlodavsky, I. Heparanase induces tissue factor pathway inhibitor expression and extracellular accumulation in endothelial and tumor cells. Thromb. Haemost. 2008, 99, 133–141. [Google Scholar] [CrossRef]
- Nadir, Y.; Brenner, B.; Fux, L.; Shafat, I.; Attias, J.; Vlodavsky, I. Heparanase enhances the generation of activated factor X in the presence of tissue factor and activated factor VII. Haematologica 2010, 95, 1927–1934. [Google Scholar] [CrossRef]
- Bohdan, N.; Espin, S.; Aguila, S.; Teruel-Montoya, R.; Vicente, V.; Corral, J.; Martinez-Martinez, I. Heparanase Activates Antithrombin through the Binding to Its Heparin Binding Site. PLoS ONE 2016, 11, e0157834. [Google Scholar] [CrossRef]
- Nadir, Y.; Brenner, B.; Zetser, A.; Ilan, N.; Shafat, I.; Zcharia, E.; Goldshmidt, O.; Vlodavsky, I. Heparanase induces tissue factor expression in vascular endothelial and cancer cells. J. Thromb. Haemost. 2006, 4, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Coulson-Thomas, V.J.; Gesteira, T.F.; Esko, J.; Kao, W. Heparan sulfate regulates hair follicle and sebaceous gland morphogenesis and homeostasis. J. Biol. Chem. 2014, 289, 25211–25226. [Google Scholar] [CrossRef] [PubMed]
- Malgouries, S.; Thibaut, S.; Bernard, B.A. Proteoglycan expression patterns in human hair follicle. Br. J. Dermatol. 2008, 158, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kaplan, V.; Naroditsky, I.; Zetser, A.; Ilan, N.; Vlodavsky, I.; Doweck, I. Heparanase induces VEGF C and facilitates tumor lymphangiogenesis. Int. J. Cancer 2008, 123, 2566–2573. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Chen, L.; Yang, Z.; Sun, S. The close correlation between heparanase and COX-2 expression in lymphangiogenesis of cervical cancer. Med. Oncol. 2014, 31, 314. [Google Scholar] [CrossRef]
- Takahashi, H.; Matsumoto, H.; Smirkin, A.; Itai, T.; Nishimura, Y.; Tanaka, J. Involvement of heparanase in migration of microglial cells. Biochim. Biophys. Acta 2008, 1780, 709–715. [Google Scholar] [CrossRef]
- Wood, R.J.; Hulett, M.D. Cell surface-expressed cation-independent mannose 6-phosphate receptor (CD222) binds enzymatically active heparanase independently of mannose 6-phosphate to promote extracellular matrix degradation. J. Biol. Chem. 2008, 283, 4165–4176. [Google Scholar] [CrossRef]
- Poon, I.K.; Goodall, K.J.; Phipps, S.; Chow, J.D.; Pagler, E.B.; Andrews, D.M.; Conlan, C.L.; Ryan, G.F.; White, J.A.; Wong, M.K.; et al. Mice deficient in heparanase exhibit impaired dendritic cell migration and reduced airway inflammation. Eur. J. Immunol. 2014, 44, 1016–1030. [Google Scholar] [CrossRef]
- Sasaki, N.; Higashi, N.; Taka, T.; Nakajima, M.; Irimura, T. Cell surface localization of heparanase on macrophages regulates degradation of extracellular matrix heparan sulfate. J. Immunol. 2004, 172, 3830–3835. [Google Scholar] [CrossRef]
- Gilat, D.; Hershkoviz, R.; Goldkorn, I.; Cahalon, L.; Korner, G.; Vlodavsky, I.; Lider, O. Molecular behavior adapts to context: Heparanase functions as an extracellular matrix-degrading enzyme or as a T cell adhesion molecule, depending on the local pH. J. Exp. Med. 1995, 181, 1929–1934. [Google Scholar] [CrossRef]
- Haimov-Kochman, R.; Friedmann, Y.; Prus, D.; Goldman-Wohl, D.S.; Greenfield, C.; Anteby, E.Y.; Aviv, A.; Vlodavsky, I.; Yagel, S. Localization of heparanase in normal and pathological human placenta. Mol. Hum. Reprod. 2002, 8, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Nadir, Y.; Henig, I.; Naroditzky, I.; Paz, B.; Vlodavsky, I.; Brenner, B. Involvement of Heparanase in early pregnancy losses. Thromb. Res. 2010, 125, e251–e257. [Google Scholar] [CrossRef] [PubMed]
- Hambruch, N.; Kumstel, S.; Haeger, J.D.; Pfarrer, C. Bovine placentomal heparanase and syndecan expression is related to placental maturation. Placenta 2017, 57, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Bitan, M.; Weiss, L.; Zeira, M.; Zcharia, E.; Slavin, S.; Nagler, A.; Vlodavsky, I. Heparanase promotes engraftment and prevents graft versus host disease in stem cell transplantation. PLoS ONE 2010, 5, e10135. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Sterkand, R.T.; Ozbun, M.A. Interaction of human papillomavirus type 16 particles with heparan sulfate and syndecan-1 molecules in the keratinocyte extracellular matrix plays an active role in infection. J. Gen. Virol. 2015, 96, 2232–2241. [Google Scholar] [CrossRef]
- Hopkins, J.; Yadavalli, T.; Agelidis, A.M.; Shukla, D. Host Enzymes Heparanase and Cathepsin L Promote Herpes Simplex Virus 2 Release from Cells. J. Virol. 2018, 92, 23. [Google Scholar] [CrossRef]
- Hadigal, S.R.; Agelidis, A.M.; Karasneh, G.A.; Antoine, T.E.; Yakoub, A.M.; Ramani, V.C.; Djalilian, A.R.; Sanderson, R.D.; Shukla, D. Heparanase is a host enzyme required for herpes simplex virus-1 release from cells. Nat. Commun. 2015, 6, 6985. [Google Scholar] [CrossRef]
- Guo, C.; Zhu, Z.; Guo, Y.; Wang, X.; Yu, P.; Xiao, S.; Chen, Y.; Cao, Y.; Liu, X. Heparanase Upregulation Contributes to Porcine Reproductive and Respiratory Syndrome Virus Release. J. Virol. 2017, 91, 15. [Google Scholar] [CrossRef]
- Crispel, Y.; Ghanem, S.; Attias, J.; Kogan, I.; Brenner, B.; Nadir, Y. Involvement of the heparanase procoagulant domain in bleeding and wound healing. J. Thromb. Haemost. 2017, 15, 1463–1472. [Google Scholar] [CrossRef]
- Pillarisetti, S.; Paka, L.; Obunike, J.C.; Berglund, L.; Goldberg, I.J. Subendothelial retention of lipoprotein (a). Evidence that reduced heparan sulfate promotes lipoprotein binding to subendothelial matrix. J. Clin. Investig. 1997, 100, 867–874. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Hayward, I.P.; Thomas, A.C.; Campbell, G.R.; Campbell, J.H. Matrix metalloproteinase can facilitate the heparanase-induced promotion of phenotype change in vascular smooth muscle cells. Atherosclerosis 1999, 145, 97–106. [Google Scholar] [CrossRef]
- Wijnhoven, T.J.; van den Hoven, M.J.; Ding, H.; van Kuppevelt, T.H.; van der Vlag, J.; Berden, J.H.; Prinz, R.A.; Lewis, E.J.; Schwartz, M.; Xu, X. Heparanase induces a differential loss of heparan sulphate domains in overt diabetic nephropathy. Diabetologia 2008, 51, 372–382. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, F.; Kim, M.S.; Puthanveetil, P.; Kewalramani, G.; Deppe, S.; Ghosh, S.; Abrahani, A.; Rodrigues, B. Endothelial heparanase secretion after acute hypoinsulinemia is regulated by glucose and fatty acid. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1108–H1116. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowski, A.F.; Popp, S.K.; Freeman, C.; Parish, C.R.; Simeonovic, C.J. Heparan sulfate and heparanase play key roles in mouse β cell survival and autoimmune diabetes. J. Clin. Investig. 2012, 122, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, R.; Rubinstein, A.M.; Gil, N.; Hermano, E.; Li, J.P.; van der Vlag, J.; Atzmon, R.; Meirovitz, A.; Elkin, M. Role of heparanase-driven inflammatory cascade in pathogenesis of diabetic nephropathy. Diabetes 2014, 63, 4302–4313. [Google Scholar] [CrossRef]
- Chiu, A.P.; Wan, A.; Lal, N.; Zhang, D.; Wang, F.; Vlodavsky, I.; Hussein, B.; Rodrigues, B. Cardiomyocyte VEGF Regulates Endothelial Cell GPIHBP1 to Relocate Lipoprotein Lipase to the Coronary Lumen During Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 145–155. [Google Scholar] [CrossRef]
- Lider, O.; Baharav, E.; Mekori, Y.A.; Miller, T.; Naparstek, Y.; Vlodavsky, I.; Cohen, I.R. Suppression of experimental autoimmune diseases and prolongation of allograft survival by treatment of animals with low doses of heparins. J. Clin. Investig. 1989, 83, 752–756. [Google Scholar] [CrossRef]
- De Mestre, A.M.; Staykova, M.A.; Hornby, J.R.; Willenborg, D.O.; Hulett, M.D. Expression of the heparan sulfate-degrading enzyme heparanase is induced in infiltrating CD4+ T cells in experimental autoimmune encephalomyelitis and regulated at the level of transcription by early growth response gene 1. J. Leukoc. Biol. 2007, 82, 1289–1300. [Google Scholar] [CrossRef]
- Bitan, M.; Weiss, L.; Reibstein, I.; Zeira, M.; Fellig, Y.; Slavin, S.; Zcharia, E.; Nagler, A.; Vlodavsky, I. Heparanase upregulates Th2 cytokines, ameliorating experimental autoimmune encephalitis. Mol. Immunol. 2010, 47, 1890–1898. [Google Scholar] [CrossRef]
- Van den Hoven, M.J.; Rops, A.L.; Bakker, M.A.; Aten, J.; Rutjes, N.; Roestenberg, P.; Goldschmeding, R.; Zcharia, E.; Vlodavsky, I.; van der Vlag, J.; et al. Increased expression of heparanase in overt diabetic nephropathy. Kidney Int. 2006, 70, 2100–2108. [Google Scholar] [CrossRef]
- Garsen, M.; Benner, M.; Dijkman, H.B.; van Kuppevelt, T.H.; Li, J.P.; Rabelink, T.J.; Vlodavsky, I.; Berden, J.H.; Rops, A.L.; Elkin, M.; et al. Heparanase Is Essential for the Development of Acute Experimental Glomerulonephritis. Am. J. Pathol. 2016, 186, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Garsen, M.; Lenoir, O.; Rops, A.L.; Dijkman, H.B.; Willemsen, B.; van Kuppevelt, T.H.; Rabelink, T.J.; Berden, J.H.; Tharaux, P.L.; van der Vlag, J. Endothelin-1 Induces Proteinuria by Heparanase-Mediated Disruption of the Glomerular Glycocalyx. J. Am. Soc. Nephrol. 2016, 27, 3545–3551. [Google Scholar] [CrossRef] [PubMed]
- Abassi, Z.; Hamoud, S.; Hassan, A.; Khamaysi, I.; Nativ, O.; Heyman, S.N.; Muhammad, R.S.; Ilan, N.; Singh, P.; Hammond, E.; et al. Involvement of heparanase in the pathogenesis of acute kidney injury: Nephroprotective effect of PG545. Oncotarget 2017, 8, 34191–34204. [Google Scholar] [CrossRef] [PubMed]
- Masola, V.; Gambaro, G.; Tibaldi, E.; Onisto, M.; Abaterusso, C.; Lupo, A. Regulation of heparanase by albumin and advanced glycation end products in proximal tubular cells. Biochim. Biophys. Acta 2011, 1813, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Masola, V.; Gambaro, G.; Tibaldi, E.; Brunati, A.M.; Gastaldello, A.; D’Angelo, A.; Onisto, M.; Lupo, A. Heparanase and syndecan-1 interplay orchestrates fibroblast growth factor-2-induced epithelial-mesenchymal transition in renal tubular cells. J. Biol. Chem. 2012, 287, 1478–1488. [Google Scholar] [CrossRef]
- Li, J.; Li, J.P.; Zhang, X.; Lu, Z.; Yu, S.P.; Wei, L. Expression of heparanase in vascular cells and astrocytes of the mouse brain after focal cerebral ischemia. Brain Res. 2012, 1433, 137–144. [Google Scholar] [CrossRef]
- Takahashi, H.; Matsumoto, H.; Kumon, Y.; Ohnishi, T.; Freeman, C.; Imai, Y.; Tanaka, J. Expression of heparanase in nestin-positive reactive astrocytes in ischemic lesions of rat brain after transient middle cerebral artery occlusion. Neurosci. Lett. 2007, 417, 250–254. [Google Scholar] [CrossRef]
- Masola, V.; Zaza, G.; Bellin, G.; Dall’Olmo, L.; Granata, S.; Vischini, G.; Secchi, M.F.; Lupo, A.; Gambaro, G.; Onisto, M. Heparanase regulates the M1 polarization of renal macrophages and their crosstalk with renal epithelial tubular cells after ischemia/reperfusion injury. FASEB J. 2018, 32, 742–756. [Google Scholar] [CrossRef]
- Francis, D.J.; Parish, C.R.; McGarry, M.; Santiago, F.S.; Lowe, H.C.; Brown, K.J.; Bingley, J.A.; Hayward, I.P.; Cowden, W.B.; Campbell, J.H.; et al. Blockade of vascular smooth muscle cell proliferation and intimal thickening after balloon injury by the sulfated oligosaccharide PI-88: Phosphomannopentaose sulfate directly binds FGF-2, blocks cellular signaling, and inhibits proliferation. Circ. Res. 2003, 92, e70–e77. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, B.; O’Callaghan, P.; Hjertstrom, E.; Jia, J.; Gong, F.; Zcharia, E.; Nilsson, L.N.; Lannfelt, L.; Vlodavsky, I.; et al. Heparanase overexpression impairs inflammatory response and macrophage-mediated clearance of amyloid-beta in murine brain. Acta Neuropathol. 2012, 124, 465–478. [Google Scholar] [CrossRef]
- Kovalchuk Ben-Zaken, O.; Nissan, I.; Tzaban, S.; Taraboulos, A.; Zcharia, E.; Matzger, S.; Shafat, I.; Vlodavsky, I.; Tal, Y. Transgenic over-expression of mammalian heparanase delays prion disease onset and progression. Biochem. Biophys. Res. Commun. 2015, 464, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Changyaleket, B.; Chong, Z.Z.; Dull, R.O.; Nanegrungsunk, D.; Xu, H. Heparanase promotes neuroinflammatory response during subarachnoid hemorrhage in rats. J. Neuroinflamm. 2017, 14, 137. [Google Scholar] [CrossRef] [PubMed]
- Lerner, I.; Zcharia, E.; Neuman, T.; Hermano, E.; Rubinstein, A.M.; Vlodavsky, I.; Elkin, M. Heparanase is preferentially expressed in human psoriatic lesions and induces development of psoriasiform skin inflammation in mice. Cell Mol. Life Sci. 2014, 71, 2347–2357. [Google Scholar] [CrossRef]
- Martin, L.; De Santis, R.; Koczera, P.; Simons, N.; Haase, H.; Heinbockel, L.; Brandenburg, K.; Marx, G.; Schuerholz, T. The Synthetic Antimicrobial Peptide 19-2.5 Interacts with Heparanase and Heparan Sulfate in Murine and Human Sepsis. PLoS ONE 2015, 10, e0143583. [Google Scholar] [CrossRef]
- Matan, M.; King, D.; Peled, E.; Ackerman, S.; Bar-Lavi, Y.; Brenner, B.; Nadir, Y. Heparanase level and procoagulant activity are reduced in severe sepsis. Eur. J. Haematol. 2018, 100, 182–188. [Google Scholar] [CrossRef]
- Kim, T.H.; Lee, H.M.; Lee, S.H.; Choi, W.; Kim, H.K.; Lee, J.H.; Oh, K.H.; Lee, S.H. Up-regulation of heparanase in the ethmoid sinus mucosa of patients with chronic sinusitis. Am. J. Rhinol. Allergy 2009, 23, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Digre, A.; Singh, K.; Åbrink, M.; Reijmers, R.M.; Sandler, S.; Vlodavsky, I.; Li, J.-P. Overexpression of heparanase enhances T lymphocyte activities and intensifies the inflammatory response in a model of murine rheumatoid arthritis. Sci. Rep. 2017, 7, 46229. [Google Scholar] [CrossRef]
- Chen, G.; Wang, D.; Vikramadithyan, R.; Yagyu, H.; Saxena, U.; Pillarisetti, S.; Goldberg, I.J. Inflammatory cytokines and fatty acids regulate endothelial cell heparanase expression. Biochemistry 2004, 43, 4971–4977. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428. [Google Scholar] [CrossRef]
- Young, E. The anti-inflammatory effects of heparin and related compounds. Thromb. Res. 2008, 122, 743–752. [Google Scholar] [CrossRef]
- Abdi, K.; Singh, N.J.; Matzinger, P. Lipopolysaccharide-activated dendritic cells: “Exhausted” or alert and waiting? J. Immunol. 2012, 188, 5981–5989. [Google Scholar] [CrossRef] [PubMed]
- Trevejo, J.M.; Marino, M.W.; Philpott, N.; Josien, R.; Richards, E.C.; Elkon, K.B.; Falck-Pedersen, E. TNF-alpha -dependent maturation of local dendritic cells is critical for activating the adaptive immune response to virus infection. Proc. Natl. Acad. Sci. USA 2001, 98, 12162–12167. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sasson, S.Z.; Hu-Li, J.; Quiel, J.; Cauchetaux, S.; Ratner, M.; Shapira, I.; Dinarello, C.A.; Paul, W.E. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 7119–7124. [Google Scholar] [CrossRef] [PubMed]
- Gotte, M. Syndecans in inflammation. FASEB J. 2003, 17, 575–591. [Google Scholar] [CrossRef]
- Axelsson, J.; Xu, D.; Kang, B.N.; Nussbacher, J.K.; Handel, T.M.; Ley, K.; Sriramarao, P.; Esko, J.D. Inactivation of heparan sulfate 2-O-sulfotransferase accentuates neutrophil infiltration during acute inflammation in mice. Blood 2012, 120, 1742–1751. [Google Scholar] [CrossRef]
- Qin, Q.; Niu, J.; Wang, Z.; Xu, W.; Qiao, Z.; Gu, Y. Heparanase induced by advanced glycation end products (AGEs) promotes macrophage migration involving RAGE and PI3K/AKT pathway. Cardiovasc. Diabetol. 2013, 12, 37. [Google Scholar] [CrossRef]
- Smailbegovic, A.; Lever, R.; Page, C.P. The effects of heparin on the adhesion of human peripheral blood mononuclear cells to human stimulated umbilical vein endothelial cells. Br. J. Pharmacol. 2001, 134, 827–836. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Cir. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Thomas, S.R.; Witting, P.K.; Drummond, G.R. Redox control of endothelial function and dysfunction: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2008, 10, 1713–1765. [Google Scholar] [CrossRef]
- Quinn, M.T.; Parthasarathy, S.; Fong, L.G.; Steinberg, D. Oxidatively modified low density lipoproteins: A potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proc. Natl. Acad. Sci. USA 1987, 84, 2995–2998. [Google Scholar] [CrossRef]
- Cunningham, K.S.; Gotlieb, A.I. The role of shear stress in the pathogenesis of atherosclerosis. Lab. Invest. 2005, 85, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Ramella, R.; Boero, O.; Alloatti, G.; Angelone, T.; Levi, R.; Gallo, M.P. Vasostatin 1 activates eNOS in endothelial cells through a proteoglycan-dependent mechanism. J. Cell Biochem. 2010, 110, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Tempel, D.; van Haperen, R.; van der Baan, A.; Grosveld, F.; Daemen, M.J.; Krams, R.; de Crom, R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation 2006, 113, 2744–2753. [Google Scholar] [CrossRef]
- Kim, M.; Kim, S.; Lim, J.H.; Lee, C.; Choi, H.C.; Woo, C.H. Laminar flow activation of ERK5 protein in vascular endothelium leads to atheroprotective effect via NF-E2-related factor 2 (Nrf2) activation. J. Biol. Chem. 2012, 287, 40722–40731. [Google Scholar] [CrossRef]
- Koo, A.; Dewey, C.F., Jr.; Garcia-Cardena, G. Hemodynamic shear stress characteristic of atherosclerosis-resistant regions promotes glycocalyx formation in cultured endothelial cells. Am. J. Physiol. Cell Physiol. 2013, 304, C137–C146. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, N.; Mulligan-Kehoe, M.J.; Corti, F.; Simon, D.D.; Ross, T.D.; Rhodes, J.M.; Wang, T.Z.; Mejean, C.O.; Simons, M.; Humphrey, J.; et al. Syndecan 4 is required for endothelial alignment in flow and atheroprotective signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 17308. [Google Scholar] [CrossRef]
- Florian, J.A.; Kosky, J.R.; Ainslie, K.; Pang, Z.; Dull, R.O.; Tarbell, J.M. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ. Res. 2003, 93, e136–e142. [Google Scholar] [CrossRef]
- Woollard, K.J.; Geissmann, F. Monocytes in atherosclerosis: Subsets and functions. Nat. Rev. Cardiol. 2010, 7, 77–86. [Google Scholar] [CrossRef]
- Čejková, S.; Králová-Lesná, I.; Poledne, R. Monocyte adhesion to the endothelium is an initial stage of atherosclerosis development. Cor. Vasa 2016, 58, e419–e425. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Leitinger, N.; Schulman, I.G. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Perisic, L.; Hedin, E.; Razuvaev, A.; Lengquist, M.; Osterholm, C.; Folkersen, L.; Gillgren, P.; Paulsson-Berne, G.; Ponten, F.; Odeberg, J.; et al. Profiling of atherosclerotic lesions by gene and tissue microarrays reveals PCSK6 as a novel protease in unstable carotid atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2432–2443. [Google Scholar] [CrossRef] [PubMed]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Getz, G.S.; Reardon, C.A. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J. Lipid Res. 2009, 50, S156–S161. [Google Scholar] [CrossRef] [PubMed]
- Lo Sasso, G.; Schlage, W.K.; Boué, S.; Veljkovic, E.; Peitsch, M.C.; Hoeng, J. The Apoe−/− mouse model: A suitable model to study cardiovascular and respiratory diseases in the context of cigarette smoke exposure and harm reduction. J. Transl. Med. 2016, 14, 146. [Google Scholar] [CrossRef]
- Zhang, S.H.; Reddick, R.L.; Piedrahita, J.A.; Maeda, N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992, 258, 468–471. [Google Scholar] [CrossRef]
- Planer, D.; Metzger, S.; Zcharia, E.; Vlodavsky, I.; George, J.; Chajek-Shaul, T. Th-W48:2 Heparanase and atherosclerosis: Lessons from transgenic mice over-expressing the human heparanase gene. Atheroscler. Suppl. 2006, 7, 463. [Google Scholar] [CrossRef]
- Cohen-Mazor, M.; Sela, S.; Mazor, R.; Ilan, N.; Vlodavsky, I.; Rops, A.L.; van der Vlag, J.; Cohen, H.I.; Kristal, B. Are primed polymorphonuclear leukocytes contributors to the high heparanase levels in hemodialysis patients? Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H651–H658. [Google Scholar] [CrossRef][Green Version]
- Dredge, K.; Brennan, T.V.; Hammond, E.; Lickliter, J.D.; Lin, L.; Bampton, D.; Handley, P.; Lankesheer, F.; Morrish, G.; Yang, Y.; et al. A Phase I study of the novel immunomodulatory agent PG545 (pixatimod) in subjects with advanced solid tumours. Br. J. Cancer 2018, 118, 1035–1041. [Google Scholar] [CrossRef]
- Hamoud, S.; Shekh Muhammad, R.; Abu-Saleh, N.; Hassan, A.; Zohar, Y.; Hayek, T. Heparanase Inhibition Reduces Glucose Levels, Blood Pressure, and Oxidative Stress in Apolipoprotein E Knockout Mice. Biomed. Res. Int. 2017, 2017, 7357495. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Physiological Process | HPSE Function | References |
|---|---|---|
| Angiogenesis | Liberates HS-bound angiogenic growth factors VEGF and basic FGF | [5,126] |
| Autophagy | Confers resistance to chemotherapy through regulation of autophagy | [124] |
| Cell activation | Releases soluble HS, HS-bound growth factors, activating DCs and macrophages; signals through toll-like receptors, activating macrophages | [18,81,127,128] |
| Cell adhesion | Facilitates platelet, neutrophil, monocyte, T cell, and hematopoietic progenitor cell adhesion independent of enzymatic activity | [82,83,120,129,130,131,132,133,134] |
| Cell proliferation | Stimulates chondrogenesis in bone growth plates | [135] |
| Cell recruitment | Stimulates eosinophil recruitment to the lungs; influences chemokine gradient established by endothelial HS to alter neutrophil crawling | [85,136,137] |
| Cell signalling | Contributes to cell signalling via Akt, PIK3, ERK, JNK, P38, Src | [18,84,126,138,139] |
| Coagulation | Stimulates tissue factor expression by endothelial and cancer cells facilitating coagulation; dissociates tissue factor pathway inhibitor, increasing cell surface coagulation activity; increases generation of activated factor X; activates anti thrombin at physiologic pH | [140,141,142,143] |
| Exosome biogenesis | Activates syndecan–syntenin–exosome biogenesis pathway and regulates exosome production, composition, and secretion | [122,123] |
| Hair growth | Increases vascularisation in the hair follicle, migration of follicular stem cell progeny, and release of HS-bound growth factors regulating the hair growth cycle; contributes to hair follicle cycling through HS regulation | [3,144,145] |
| Lymphangiogenesis | Upregulates VEGF and cyclooxygenase-2 | [146,147] |
| Migration | Mediates migration of DCs, neutrophils, T cells, monocytes, microglia, and ECs | [84,131,148,149,150,151] |
| Reproduction | Endometrial remodelling essential for embryo implantation; regulates tissue factor and tissue factor pathway inhibitor in trophoblasts during early miscarriages; trophoblast invasion; placental maturation | [152,153,154,155] |
| Transplant rejection | Inhibits T cell activation and modulates cytokine release to facilitate engraftment and suppress graft-versus-host disease | [156] |
| Viral release | Cleaves HS-bound viral progenies to facilitate release of HSV-1, HSV-2, HPV, and PRRSV, preventing viral infection | [157,158,159,160] |
| Wound healing | Increases vascularity to accelerate wound closure | [3,5,161] |
| Disease | HPSE Function | References |
|---|---|---|
| Airway inflammation | Facilitates DC and eosinophil recruitment and allergic inflammatory response; contributes to sepsis-associated acute lung injury by promoting pulmonary glycocalyx loss and neutrophil adhesion | [120,137,150] |
| Atherosclerosis | Contributes to LDL retention in the intima; facilitates monocyte binding to the endothelium; mediates SMC proliferation and migration; promotes release of inflammatory mediators from macrophages | [81,129,162,163] |
| Crohn’s/colitis | Stimulates macrophage activation | [121] |
| Diabetes | Mediates pancreatic β-cell death and macrophage activation; regulates fatty acid use by cardiomyocytes; mediates proteinuria through degradation of the nephron BM | [164,165,166,167,168] |
| Experimental autoimmune encephalomyelitis | Promotes T cell activation; induces upregulation of Th2 cytokines, inhibits inflammation; facilitates CD4+ T cell infiltration into the CNS, promoting inflammation | [169,170,171] |
| Glomerular disease | Promotes HS degradation, increasing glomerular BM permeability, enhancing leukocyte and macrophage influx; upregulates inflammatory mediators | [172,173,174,175] |
| Ischemia reperfusion injury | Regulates FGF-2 and transforming growth factor β-induced epithelial to mesenchymal transition, inducing chronic renal damage; contributes to revascularisation by promoting angiogenesis; promotes astrocyte migration towards ischemic core, resulting in astrogliosis; regulates macrophage polarisation to inflammatory M1 phenotype | [176,177,178,179,180] |
| Neointimal hyperplasia | Promotes SMC proliferation to establish restenosis; stimulates macrophage recruitment and vascular remodelling | [78,181] |
| Neuroinflammation | Prevents immune cell recruitment, activation, and clearance of amyloid-β; delays prion disease onset and progression; increases leukocyte trafficking associated with neurological deficiency after subarachnoid haemorrhage | [182,183,184] |
| Psoriasis | Facilitates activation of skin-infiltrating macrophages and induction of signal transducer and activator of transcription 3, enhanced NF-κB signalling, and increased TNFα expression | [185] |
| Sepsis | Facilitates destruction of mucosal epithelial glycocalyx via HS degradation, promoting neutrophil infiltration and inflammatory cytokine production, acts as a predictor of sepsis severity | [13,186,187] |
| Sinusitis | Contributes to tissue remodelling in nasal polyps | [188] |
| Rheumatoid arthritis | Regulates angiogenesis and stimulates immune cell migration and proliferation | [119,189] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.K.; Paone, S.; Chan, E.; Poon, I.K.H.; Baxter, A.A.; Thomas, S.R.; Hulett, M.D. Heparanase: A Novel Therapeutic Target for the Treatment of Atherosclerosis. Cells 2022, 11, 3198. https://doi.org/10.3390/cells11203198
Nguyen TK, Paone S, Chan E, Poon IKH, Baxter AA, Thomas SR, Hulett MD. Heparanase: A Novel Therapeutic Target for the Treatment of Atherosclerosis. Cells. 2022; 11(20):3198. https://doi.org/10.3390/cells11203198
Chicago/Turabian StyleNguyen, Tien K., Stephanie Paone, Enoch Chan, Ivan K. H. Poon, Amy A. Baxter, Shane R. Thomas, and Mark D. Hulett. 2022. "Heparanase: A Novel Therapeutic Target for the Treatment of Atherosclerosis" Cells 11, no. 20: 3198. https://doi.org/10.3390/cells11203198
APA StyleNguyen, T. K., Paone, S., Chan, E., Poon, I. K. H., Baxter, A. A., Thomas, S. R., & Hulett, M. D. (2022). Heparanase: A Novel Therapeutic Target for the Treatment of Atherosclerosis. Cells, 11(20), 3198. https://doi.org/10.3390/cells11203198