
To Fix or Not to Fix: Maintenance of Chromosome Ends Versus Repair of DNA Double-Strand Breaks
 , , , ,
, , , ,
Abstract
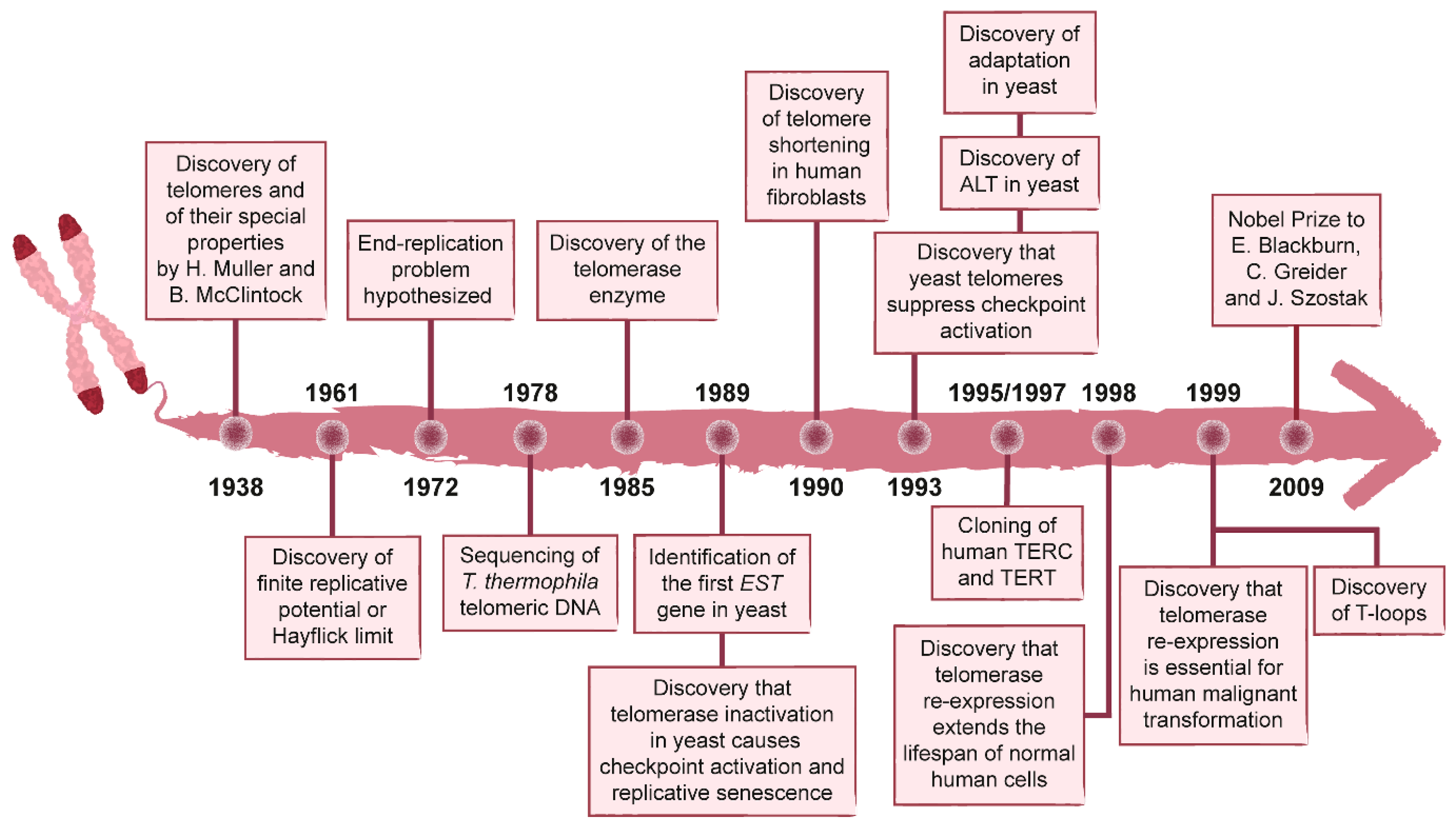
:1. History of the Discovery of Telomeres and Telomerase
2. The DNA Damage Response
3. Telomere Capping and the Consequence of Its Loss
3.1. The CST Complex
3.2. The Ku Complex
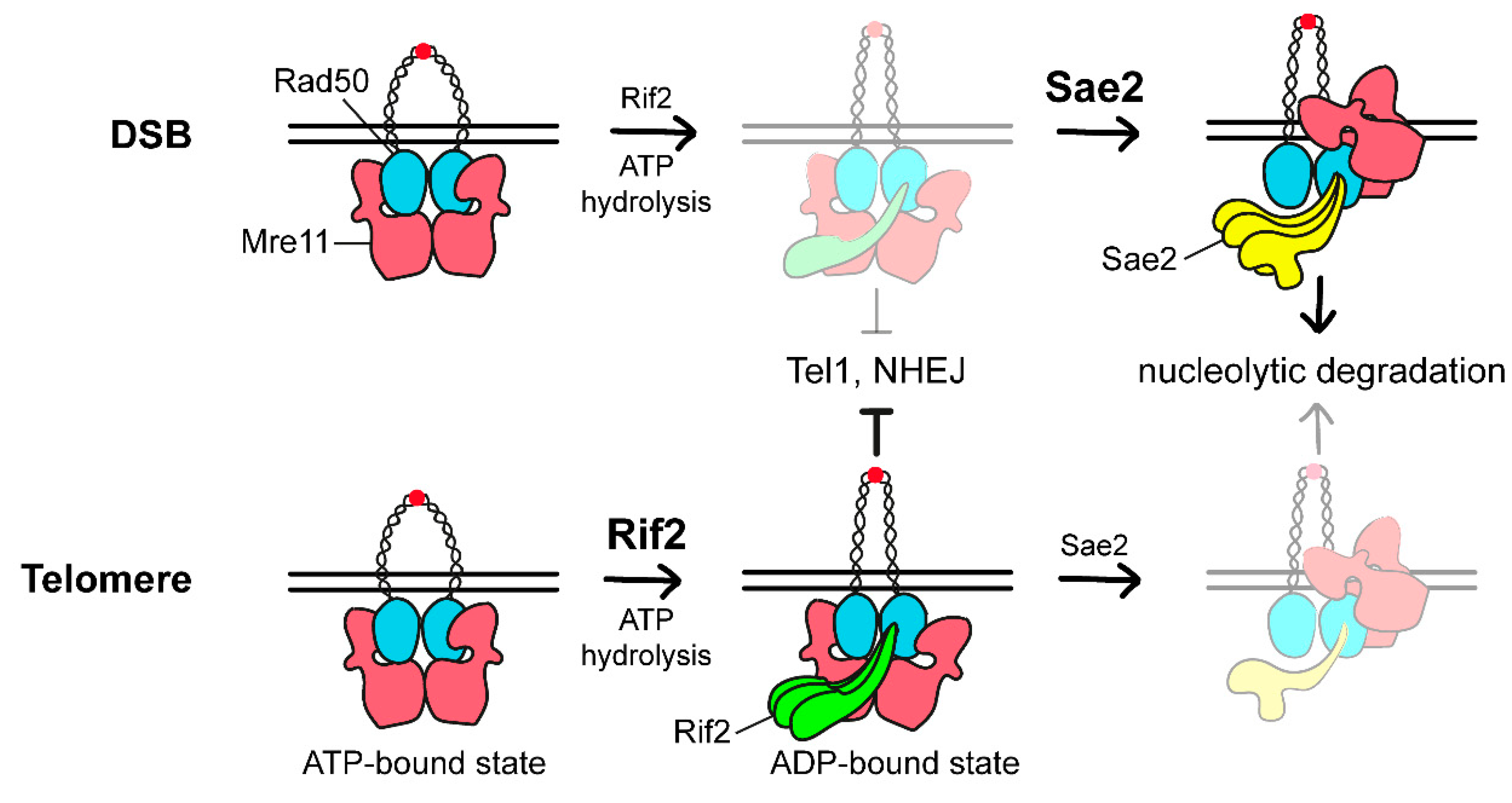
3.3. The Rap1-Rif1-Rif2 Complex
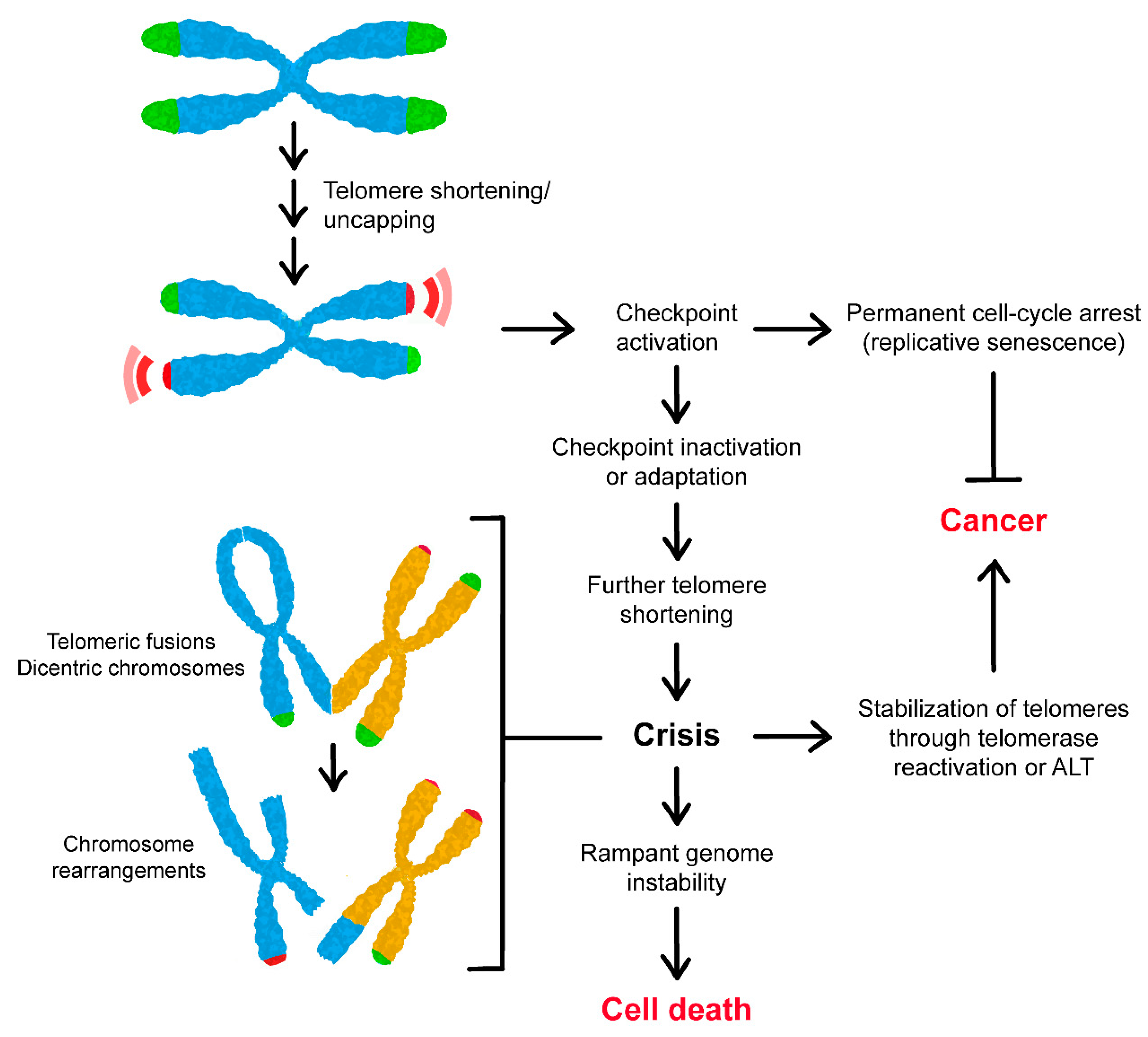
4. Consequences of Telomere Shortening
5. Escape from Telomere-Induced Replicative Senescence
6. Telomerase-Independent Re-Stabilization of Telomere Length
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muller, H.J. The remaking of chromosomes. Collect. Net 1938, 13, 181–198. [Google Scholar]
- McClintock, B. The production of homozygous deficient tissues with mutant characteristics by means of the aberrant mitotic behavior of ring-shaped chromosomes. Genetics 1938, 23, 315–376. [Google Scholar] [CrossRef] [PubMed]
- Sandell, L.L.; Zakian, V.A. Loss of a yeast telomere: Arrest, recovery, and chromosome loss. Cell 1993, 75, 729–739. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Gall, J.G. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J. Mol. Biol. 1978, 120, 33–53. [Google Scholar] [CrossRef]
- Klobutcher, L.A.; Swanton, M.T.; Donini, P.; Prescott, D.M. All gene-sized DNA molecules in four species of hypotrichs have the same terminal sequence and an unusual 3’ terminus. Proc. Natl. Acad. Sci. USA 1981, 78, 3015–3019. [Google Scholar] [CrossRef] [Green Version]
- Szostak, J.W.; Blackburn, E.H. Cloning yeast telomeres on linear plasmid vectors. Cell 1982, 29, 245–255. [Google Scholar] [CrossRef]
- Shampay, J.; Szostak, J.W.; Blackburn, E.H. DNA sequences of telomeres maintained in yeast. Nature 1984, 310, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 1987, 51, 887–898. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 1989, 337, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.L.; Bradley, J.D.; Attardi, L.D.; Blackburn, E.H. In vivo alteration of telomere sequences and senescence caused by mutated Tetrahymena telomerase RNAs. Nature 1990, 344, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, V.; Szostak, J.W. A mutant with a defect in telomere elongation leads to senescence in yeast. Cell 1989, 57, 633–643. [Google Scholar] [CrossRef]
- Lendvay, T.S.; Morris, D.K.; Sah, J.; Balasubramanian, B.; Lundblad, V. Senescence mutants of Saccharomyces cerevisiae with a defect in telomere replication identify three additional EST genes. Genetics 1996, 144, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.S.; Gottschling, D.E. TLC1: Template RNA component of Saccharomyces cerevisiae telomerase. Science 1994, 266, 404–409. [Google Scholar] [CrossRef]
- Lingner, J.; Hughes, T.R.; Shevchenko, A.; Mann, M.; Lundblad, V.; Cech, T.R. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science 1997, 276, 561–567. [Google Scholar] [CrossRef]
- Feng, J.; Funk, W.D.; Wang, S.S.; Weinrich, S.L.; Avilion, A.A.; Chiu, C.P.; Adams, R.R.; Chang, E.; Allsopp, R.C.; Yu, J.; et al. The RNA component of human telomerase. Science 1995, 269, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.; McPhail, T.; Mar, V.; Zhou, W.; Oulton, R.; Bass, M.B.; Arruda, I.; Robinson, M.O. A mammalian telomerase-associated protein. Science 1997, 275, 973–977. [Google Scholar] [CrossRef]
- Waterman, D.P.; Haber, J.E.; Smolka, M.B. Checkpoint responses to DNA double-strand breaks. Annu. Rev. Biochem. 2020, 89, 103–133. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Mimitou, E.P.; Symington, L.S. Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 2010, 29, 3358–3369. [Google Scholar] [CrossRef] [Green Version]
- Shim, E.Y.; Chung, W.H.; Nicolette, M.L.; Zhang, Y.; Davis, M.; Zhu, Z.; Paull, T.T.; Ira, G.; Lee, S.E. Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 2010, 29, 3370–3380. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [Green Version]
- Cejka, P.; Symington, L.S. DNA end resection: Mechanism and control. Annu. Rev. Genet. 2021, 55, 285–307. [Google Scholar] [CrossRef] [PubMed]
- Syed, A.; Tainer, J.A. The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Gellert, M. The 3’ to 5’ exonuclease activity of Mre11 facilitates repair of DNA double-strand breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef]
- Trujillo, K.M.; Yuan, S.S.; Lee, E.Y.; Sung, P. Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J. Biol. Chem. 1998, 273, 21447–21450. [Google Scholar] [CrossRef] [Green Version]
- Cannavo, E.; Cejka, P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 2014, 514, 122–125. [Google Scholar] [CrossRef]
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 2008, 455, 770–774. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Chung, W.H.; Shim, E.Y.; Lee, S.E.; Ira, G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008, 134, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Cejka, P.; Cannavo, E.; Polaczek, P.; Masuda-Sasa, T.; Pokharel, S.; Campbell, J.L.; Kowalczykowski, S.C. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 2010, 467, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Niu, H.; Chung, W.H.; Zhu, Z.; Kwon, Y.; Zhao, W.; Chi, P.; Prakash, R.; Seong, C.; Liu, D.; Lu, L.; et al. Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature 2010, 467, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.; Phelps, S.E.; Gray, S.; Neale, M.J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 2011, 479, 241–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reginato, G.; Cannavo, E.; Cejka, P. Physiological protein blocks direct the Mre11-Rad50-Xrs2 and Sae2 nuclease complex to initiate DNA end resection. Genes Dev. 2017, 31, 2325–2330. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Daley, J.M.; Kwon, Y.; Krasner, D.S.; Sung, P. Plasticity of the Mre11-Rad50-Xrs2-Sae2 nuclease ensemble in the processing of DNA-bound obstacles. Genes Dev. 2017, 31, 2331–2336. [Google Scholar] [CrossRef] [Green Version]
- Lammens, K.; Bemeleit, D.J.; Möckel, C.; Clausing, E.; Schele, A.; Hartung, S.; Schiller, C.B.; Lucas, M.; Angermüller, C.; Söding, J.; et al. The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in DNA double-strand break repair. Cell 2011, 145, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.J.; Williams, R.S.; Williams, J.S.; Moncalian, G.; Arvai, A.S.; Limbo, O.; Guenther, G.; SilDas, S.; Hammel, M.; Russell, P.; et al. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat. Struct. Mol. Biol. 2011, 18, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Möckel, C.; Lammens, K.; Schele, A.; Hopfner, K.P. ATP driven structural changes of the bacterial Mre11:Rad50 catalytic head complex. Nucleic Acids Res. 2012, 40, 914–927. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, R.A.; Williams, G.J.; Limbo, O.; Williams, R.S.; Kuhnlein, J.; Lee, J.H.; Classen, S.; Guenther, G.; Russell, P.; Tainer, J.A.; et al. ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J. 2014, 33, 482–500. [Google Scholar] [CrossRef]
- Käshammer, L.; Saathoff, J.H.; Lammens, K.; Gut, F.; Bartho, J.; Alt, A.; Kessler, B.; Hopfner, K.P. Mechanism of DNA end sensing and processing by the Mre11-Rad50 complex. Mol. Cell 2019, 76, 382–394. [Google Scholar] [CrossRef]
- Lustig, A.J.; Petes, T.D. Identification of yeast mutants with altered telomere structure. Proc. Natl. Acad. Sci. USA 1986, 83, 1398–1402. [Google Scholar] [CrossRef] [PubMed]
- Nakada, D.; Matsumoto, K.; Sugimoto, K. ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 2003, 17, 1957–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Paull, T.T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef]
- You, Z.; Chahwan, C.; Bailis, J.; Hunter, T.; Russell, P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell Biol. 2005, 25, 5363–5379. [Google Scholar] [CrossRef] [Green Version]
- Cassani, C.; Gobbini, E.; Wang, W.; Niu, H.; Clerici, M.; Sung, P.; Longhese, M.P. Tel1 and Rif2 regulate MRX functions in end-tethering and repair of DNA double-strand breaks. PLoS Biol. 2016, 14, e1002387. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [Green Version]
- Rouse, J.; Jackson, S.P. Lcd1p recruits Mec1p to DNA lesions in vitro and in vivo. Mol. Cell 2002, 9, 857–869. [Google Scholar] [CrossRef]
- Sanchez, Y.; Bachant, J.; Wang, H.; Hu, F.; Liu, D.; Tetzlaff, M.; Elledge, S.J. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 1999, 286, 1166–1171. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, C.S.; Green, C.M.; Lowndes, N.F. Budding yeast Rad9 is an ATP-dependent Rad53 activating machine. Mol. Cell 2001, 8, 129–136. [Google Scholar] [CrossRef]
- Sweeney, F.D.; Yang, F.; Chi, A.; Shabanowitz, J.; Hunt, D.F.; Durocher, D. Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr. Biol. 2005, 15, 1364–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcasabas, A.A.; Osborn, A.J.; Bachant, J.; Hu, F.; Werler, P.J.; Bousset, K.; Furuya, K.; Diffley, J.F.; Carr, A.M.; Elledge, S.J. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 2001, 3, 958–965. [Google Scholar] [CrossRef]
- Tanaka, K.; Russell, P. Mrc1 channels the DNA replication arrest signal to checkpoint kinase Cds1. Nat. Cell Biol. 2001, 3, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Katou, Y.; Kanoh, Y.; Bando, M.; Noguchi, H.; Tanaka, H.; Ashikari, T.; Sugimoto, K.; Shirahige, K. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 2003, 424, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Osborn, A.J.; Elledge, S.J. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 2003, 17, 1755–1767. [Google Scholar] [CrossRef] [Green Version]
- Michelson, R.J.; Rosenstein, S.; Weinert, T. A telomeric repeat sequence adjacent to a DNA double-stranded break produces an anticheckpoint. Genes Dev. 2005, 19, 2546–2559. [Google Scholar] [CrossRef] [Green Version]
- Ribeyre, C.; Shore, D. Anticheckpoint pathways at telomeres in yeast. Nat. Struct. Mol. Biol. 2012, 19, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinert, T.A.; Hartwell, L.H. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 1988, 241, 317–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garvik, B.; Carson, M.; Hartwell, L. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol. Cell Biol. 1995, 15, 6128–6138. [Google Scholar] [CrossRef]
- Grandin, N.; Reed, S.I.; Charbonneau, M. Stn1, a new Saccharomyces cerevisiae protein, is implicated in telomere size regulation in association with Cdc13. Genes Dev. 1997, 11, 512–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandin, N.; Damon, C.; Charbonneau, M. Ten1 functions in telomere end protection and length regulation in association with Stn1 and Cdc13. EMBO J. 2001, 20, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Petreaca, R.C.; Gasparyan, H.J.; Vu, S.; Nugent, C.I. TEN1 is essential for CDC13-mediated telomere capping. Genetics 2009, 183, 793–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, C.M.; Boltz, K.A.; Chaiken, M.F.; Stewart, J.A.; Beilstein, M.A.; Shippen, D.E. Evolution of CST function in telomere maintenance. Cell Cycle 2010, 9, 3157–3165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Cervantes, R.B.; Mandell, E.K.; Otero, J.H.; Lundblad, V. RPA-like proteins mediate yeast telomere function. Nat. Struct. Mol. Biol. 2007, 14, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Hirano, Y.; Sugimoto, K. Cdc13 telomere capping decreases Mec1 association but does not affect Tel1 association with DNA ends. Mol. Biol. Cell 2007, 18, 2026–2036. [Google Scholar] [CrossRef] [PubMed]
- Vodenicharov, M.D.; Laterreur, N.; Wellinger, R.J. Telomere capping in non-dividing yeast cells requires Yku and Rap1. EMBO J. 2010, 29, 3007–3019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodenicharov, M.D.; Wellinger, R.J. DNA degradation at unprotected telomeres in yeast is regulated by the CDK1 (Cdc28/Clb) cell-cycle kinase. Mol. Cell 2006, 24, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Langston, R.E.; Palazzola, D.; Bonnell, E.; Wellinger, R.J.; Weinert, T. Loss of Cdc13 causes genome instability by a deficiency in replication-dependent telomere capping. PLoS Genet. 2020, 16, e1008733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diede, S.J.; Gottschling, D.E. Telomerase-mediated telomere addition in vivo requires DNA primase and DNA polymerases alpha and delta. Cell 1999, 99, 723–733. [Google Scholar] [CrossRef]
- Qi, H.; Zakian, V.A. The Saccharomyces telomere-binding protein Cdc13p interacts with both the catalytic subunit of DNA polymerase alpha and the telomerase-associated est1 protein. Genes Dev. 2000, 14, 1777–1788. [Google Scholar] [CrossRef]
- Grossi, S.; Puglisi, A.; Dmitriev, P.V.; Lopes, M.; Shore, D. Pol12, the B subunit of DNA polymerase alpha, functions in both telomere capping and length regulation. Genes Dev. 2004, 18, 992–1006. [Google Scholar] [CrossRef] [Green Version]
- Douglas, M.E.; Diffley, J.F.X. Budding yeast Rap1, but not telomeric DNA, is inhibitory for multiple stages of DNA replication in vitro. Nucleic Acids Res. 2021, 49, 5671–5683. [Google Scholar] [CrossRef] [PubMed]
- Makovets, S.; Herskowitz, I.; Blackburn, E.H. Anatomy and dynamics of DNA replication fork movement in yeast telomeric regions. Mol. Cell Biol. 2004, 24, 4019–4031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, P.; Min, J.N.; Wang, Y.; Huang, C.; Peng, T.; Chai, W.; Chang, S. CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J. 2012, 31, 2309–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Stewart, J.A.; Kasbek, C.; Zhao, Y.; Wright, W.E.; Price, C.M. Human CST has independent functions during telomere duplex replication and C-strand fill-in. Cell Rep. 2012, 2, 1096–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Hsu, S.J.; Kasbek, C.; Chaiken, M.; Price, C.M. CTC1-mediated C-strand fill-in is an essential step in telomere length maintenance. Nucleic Acids Res. 2017, 45, 4281–4293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Takai, H.; de Lange, T. Telomeric 3’ overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell 2012, 150, 39–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef]
- Lyu, X.; Sang, P.B.; Chai, W. CST in maintaining genome stability: Beyond telomeres. DNA Repair 2021, 102, 103104. [Google Scholar] [CrossRef] [PubMed]
- Gravel, S.; Larrivee, M.; Labrecque, P.; Wellinger, R.J. Yeast Ku as a regulator of chromosomal DNA end structure. Science 1998, 280, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Maringele, L.; Lydall, D. EXO1-dependent single-stranded DNA at telomeres activates subsets of DNA damage and spindle checkpoint pathways in budding yeast yku70Delta mutants. Genes Dev. 2002, 16, 1919–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetti, D.; Clerici, M.; Anbalagan, S.; Martina, M.; Lucchini, G.; Longhese, M.P. Shelterin-like proteins and Yku inhibit nucleolytic processing of Saccharomyces cerevisiae telomeres. PLoS Genet. 2010, 6, e1000966. [Google Scholar] [CrossRef] [Green Version]
- Teo, S.H.; Jackson, S.P. Telomerase subunit overexpression suppresses telomere-specific checkpoint activation in the yeast yku80 mutant. EMBO Rep. 2001, 2, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Holland, C.L.; Sanderson, B.A.; Titus, J.K.; Weis, M.F.; Riojas, A.M.; Malczewskyj, E.; Wasko, B.M.; Lewis, L.K. Suppression of telomere capping defects of Saccharomyces cerevisiae yku70 and yku80 mutants by telomerase. G3 2021, 11, jkab359. [Google Scholar] [CrossRef]
- Wang, Y.; Ghosh, G.; Hendrickson, E.A. Ku86 represses lethal telomere deletion events in human somatic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12430–12435. [Google Scholar] [CrossRef] [Green Version]
- Palm, W.; Hockemeyer, D.; Kibe, T.; de Lange, T. Functional dissection of human and mouse POT1 proteins. Mol. Cell Biol. 2009, 29, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Sfeir, A.; Kabir, S.; van Overbeek, M.; Celli, G.B.; de Lange, T. Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. Science 2010, 327, 1657–1661. [Google Scholar] [CrossRef] [Green Version]
- Lustig, A.J.; Kurtz, S.; Shore, D. Involvement of the silencer and UAS binding protein RAP1 in regulation of telomere length. Science 1990, 250, 549–553. [Google Scholar] [CrossRef]
- Wotton, D.; Shore, D. A novel Rap1p-interacting factor, Rif2p, cooperates with Rif1p to regulate telomere length in Saccharomyces cerevisiae. Genes Dev. 1997, 11, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Marcand, S.; Pardo, B.; Gratias, A.; Cahun, S.; Callebaut, I. Multiple pathways inhibit NHEJ at telomeres. Genes Dev. 2008, 22, 1153–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, K.B.; Petes, T.D. The Mre11p/Rad50p/Xrs2p complex and the Tel1p function in a single pathway for telomere maintenance in yeast. Genetics 2000, 155, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Hirano, Y.; Fukunaga, K.; Sugimoto, K. Rif1 and Rif2 inhibit localization of Tel1 to DNA ends. Mol. Cell 2009, 33, 312–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anbalagan, S.; Bonetti, D.; Lucchini, G.; Longhese, M.P. Rif1 supports the function of the CST complex in yeast telomere capping. PLoS Genet. 2011, 7, e1002024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lange, T. Shelterin-mediated telomere protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Denchi, E.L.; de Lange, T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 2007, 448, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- van Steensel, B.; Smogorzewska, A.; de Lange, T. TRF2 protects human telomeres from end-to-end fusions. Cell 1998, 92, 401–413. [Google Scholar] [CrossRef] [Green Version]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian telomeres end in a large duplex loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Stansel, R.M.; de Lange, T.; Griffith, J.D. T-loop assembly in vitro involves binding of TRF2 near the 3′ telomeric overhang. EMBO J. 2001, 20, 5532–5540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlseder, J.; Hoke, K.; Mirzoeva, O.K.; Bakkenist, C.; Kastan, M.B.; Petrini, J.H.; de Lange, T. The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response. PLoS Biol. 2004, 2, E240. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, D.; Riillo, C.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Alternative non-homologous end-joining: Error-prone DNA repair as cancer’s achilles’ heel. Cancers 2021, 13, 1392. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.E.; Oh, S.; Grimstead, J.W.; Zimbric, J.; Roger, L.; Heppel, N.H.; Ashelford, K.E.; Liddiard, K.; Hendrickson, E.A.; Baird, D.M. Escape from telomere-driven crisis is DNA ligase III dependent. Cell Rep. 2014, 8, 1063–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sfeir, A.; de Lange, T. Removal of shelterin reveals the telomere end-protection problem. Science 2012, 336, 593–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, I.R.; Chambers, A. Use of a selection technique to identify the diversity of binding sites for the yeast RAP1 transcription factor. Nucleic Acids Res. 1994, 22, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Konig, P.; Giraldo, R.; Chapman, L.; Rhodes, D. The crystal structure of the DNA-binding domain of yeast RAP1 in complex with telomeric DNA. Cell 1996, 85, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Taylor, H.O.; O’Reilly, M.; Leslie, A.G.; Rhodes, D. How the multifunctional yeast Rap1p discriminates between DNA target sites: A crystallographic analysis. J. Mol. Biol. 2000, 303, 693–707. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, E.A.; Galletto, R. The DNA-binding domain of yeast Rap1 interacts with double-stranded DNA in multiple binding modes. Biochemistry 2014, 53, 7471–7483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldmann, E.A.; De Bona, P.; Galletto, R. The wrapping loop and Rap1 C-terminal (RCT) domain of yeast Rap1 modulate access to different DNA binding modes. J. Biol. Chem. 2015, 290, 11455–11466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetti, D.; Rinaldi, C.; Vertemara, J.; Notaro, M.; Pizzul, P.; Tisi, R.; Zampella, G.; Longhese, M.P. DNA binding modes influence Rap1 activity in the regulation of telomere length and MRX functions at DNA ends. Nucleic Acids Res. 2020, 48, 2424–2441. [Google Scholar] [CrossRef] [PubMed]
- Hailemariam, S.; De Bona, P.; Galletto, R.; Hohl, M.; Petrini, J.H.; Burgers, P.M. The telomere-binding protein Rif2 and ATP-bound Rad50 have opposing roles in the activation of yeast Tel1ATM kinase. J. Biol. Chem. 2019, 294, 18846–18852. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sung, S.; Kim, Y.; Li, F.; Gwon, G.; Jo, A.; Kim, A.K.; Kim, T.; Song, O.K.; Lee, S.E.; et al. ATP-dependent DNA binding, unwinding, and resection by the Mre11/Rad50 complex. EMBO J. 2016, 35, 743–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, F.U.; Lammens, K.; Stoehr, G.; Kessler, B.; Hopfner, K.P. Structural mechanism of ATP-dependent DNA binding and DNA end bridging by eukaryotic Rad50. EMBO J. 2016, 35, 759–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hailemariam, S.; Kumar, S.; and Burgers, P.M. Activation of Tel1ATM kinase requires Rad50 ATPase and long nucleosome-free DNA but no DNA ends. J. Biol. Chem. 2019, 294, 10120–10130. [Google Scholar] [CrossRef] [PubMed]
- Khayat, F.; Cannavo, E.; Alshmery, M.; Foster, W.R.; Chahwan, C.; Maddalena, M.; Smith, C.; Oliver, A.W.; Watson, A.T.; Carr, A.M.; et al. Inhibition of MRN activity by a telomere protein motif. Nat. Commun. 2021, 12, 3856. [Google Scholar] [CrossRef] [PubMed]
- Marsella, A.; Gobbini, E.; Cassani, C.; Tisi, R.; Cannavo, E.; Reginato, G.; Cejka, P.; Longhese, M.P. Sae2 and Rif2 regulate MRX endonuclease activity at DNA double-strand breaks in opposite manners. Cell Rep. 2021, 34, 108906. [Google Scholar] [CrossRef] [PubMed]
- Roisné-Hamelin, F.; Pobiega, S.; Jézéquel, K.; Miron, S.; Dépagne, J.; Veaute, X.; Busso, D.; Du, M.L.; Callebaut, I.; Charbonnier, J.B.; et al. Mechanism of MRX inhibition by Rif2 at telomeres. Nat. Commun. 2021, 12, 2763. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, E.; Johnson, D.; Andres, S.N.; Kissling, V.M.; Reinert, J.K.; Garcia, V.; Erie, D.A.; Hess, D.; Thomä, N.H.; Enchev, R.I.; et al. Regulatory control of DNA end resection by Sae2 phosphorylation. Nat. Commun. 2018, 9, 4016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaizer, H.; Connelly, C.J.; Bettridge, K.; Viggiani, C.; Greider, C.W. Regulation of telomere length requires a conserved N-terminal domain of Rif2 in Saccharomyces cerevisiae. Genetics 2015, 201, 573–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Watson, J. Origin of concatemeric T7 DNA. Nat. New Biol. 1972, 239, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Olovnikov, A.M. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackett, J.A.; Feldser, D.M.; Greider, C.W. Telomere dysfunction increases mutation rate and genomic instability. Cell 2001, 106, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, S.; Glowczewski, L.; Berman, J. MEC3, MEC1, and DDC2 are essential components of a telomere checkpoint pathway required for cell cycle arrest during senescence in Saccharomyces cerevisiae. Mol. Biol. Cell 2002, 13, 2626–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IJpma, A.S.; Greider, C.W. Short telomeres induce a DNA damage response in Saccharomyces cerevisiae. Mol. Biol. Cell 2003, 14, 987–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Karlseder, J.; Smogorzewska, A.; de Lange, T. Senescence induced by altered telomere state, not telomere loss. Science 2002, 295, 2446–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesare, A.J.; Karlseder, J. A three-state model of telomere control over human proliferative boundaries. Curr. Opin. Cell Biol. 2012, 24, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Capper, R.; Britt-Compton, B.; Tankimanova, M.; Rowson, J.; Letsolo, B.; Man, S.; Haughton, M.; Baird, D.M. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev. 2007, 21, 2495–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letsolo, B.T.; Rowson, J.; Baird, D.M. Fusion of short telomeres in human cells is characterized by extensive deletion and microhomology, and can result in complex rearrangements. Nucleic Acids Res. 2010, 38, 1841–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, P.; Luciano, P.; Runge, K.W.; Lisby, M.; Géli, V.; Gilson, E.; Teixeira, M.T. A two-step model for senescence triggered by a single critically short telomere. Nat. Cell Biol. 2009, 11, 988–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khadaroo, B.; Teixeira, M.T.; Luciano, P.; Eckert-Boulet, N.; Germann, S.M.; Simon, M.N.; Gallina, I.; Abdallah, P.; Gilson, E.; Géli, V.; et al. The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nat. Cell Biol. 2009, 11, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Hemann, M.T.; Strong, M.A.; Hao, L.Y.; Greider, C.W. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 2001, 107, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Fallet, E.; Jolivet, P.; Soudet, J.; Lisby, M.; Gilson, E.; Teixeira, M.T. Length-dependent processing of telomeres in the absence of telomerase. Nucleic Acids Res. 2014, 42, 3648–3665. [Google Scholar] [CrossRef] [PubMed]
- Ballew, B.J.; Lundblad, V. Multiple genetic pathways regulate replicative senescence in telomerase-deficient yeast. Aging Cell 2013, 12, 719–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.; Rothstein, R. Rif1/2 and Tel1 function in separate pathways during replicative senescence. Cell Cycle 2011, 10, 3798–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantiero, D.; Clerici, M.; Lucchini, G.; Longhese, M.P. Dual role for Saccharomyces cerevisiae Tel1 in the checkpoint response to double-strand breaks. EMBO Rep. 2007, 8, 380–387. [Google Scholar] [CrossRef]
- Baldo, V.; Testoni, V.; Lucchini, G.; Longhese, M.P. Dominant TEL1-hy mutations compensate for Mec1 lack of functions in the DNA damage response. Mol. Cell Biol. 2008, 28, 358–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menin, L.; Colombo, C.V.; Maestrini, G.; Longhese, M.P.; Clerici, M. Tel1/ATM signaling to the checkpoint contributes to replicative senescence in the absence of telomerase. Genetics 2019, 213, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Artandi, S.E.; Chang, S.; Lee, S.L.; Alson, S.; Gottlieb, G.J.; Chin, L.; DePinho, R.A. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000, 406, 641–645. [Google Scholar] [CrossRef]
- Toczyski, D.P.; Galgoczy, D.J.; Hartwell, L.H. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 1997, 90, 1097–1106. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.E.; Moore, J.K.; Holmes, A.; Umezu, K.; Kolodner, R.D.; Haber, J.E. Saccharomyces Ku70, Mre11/Rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 1998, 94, 399–409. [Google Scholar] [CrossRef] [Green Version]
- Pellicioli, A.; Lee, S.E.; Lucca, C.; Foiani, M.; Haber, J.E. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol. Cell 2001, 7, 293–300. [Google Scholar] [CrossRef]
- Leroy, C.; Lee, S.E.; Vaze, M.B.; Ochsenbein, F.; Guerois, R.; Haber, J.E.; Marsolier-Kergoat, M.C. PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol. Cell 2003, 11, 827–835. [Google Scholar] [CrossRef]
- Lee, S.E.; Pellicioli, A.; Malkova, A.; Foiani, M.; Haber, J.E. The Saccharomyces recombination protein Tid1p is required for adaptation from G2/M arrest induced by a double-strand break. Curr. Biol. 2001, 11, 1053–1057. [Google Scholar] [CrossRef] [Green Version]
- Galgoczy, D.J.; Toczyski, D.P. Checkpoint adaptation precedes spontaneous and damage-induced genomic instability in yeast. Mol. Cell Biol. 2001, 21, 1710–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaye, J.A.; Melo, J.A.; Cheung, S.K.; Vaze, M.B.; Haber, J.E.; Toczyski, D.P. DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr. Biol. 2004, 14, 2096–2106. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.Y.; Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Adaptation of a DNA replication checkpoint response depends upon inactivation of Claspin by the Polo-like kinase. Cell 2004, 117, 575–588. [Google Scholar] [CrossRef] [Green Version]
- Syljuåsen, R.G.; Jensen, S.; Bartek, J.; Lukas, J. Adaptation to the ionizing radiation-induced G2 checkpoint occurs in human cells and depends on checkpoint kinase 1 and Polo-like kinase 1 kinases. Cancer Res. 2006, 66, 10253–10257. [Google Scholar] [CrossRef] [Green Version]
- Mersaoui, S.Y.; Gravel, S.; Karpov, V.; Wellinger, R.J. DNA damage checkpoint adaptation genes are required for division of cells harbouring eroded telomeres. Microb. Cell 2015, 2, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Coutelier, H.; Xu, Z.; Morisse, M.C.; Lhuillier-Akakpo, M.; Pelet, S.; Charvin, G.; Dubrana, K.; Teixeira, M.T. Adaptation to DNA damage checkpoint in senescent telomerase-negative cells promotes genome instability. Genes Dev. 2018, 32, 1499–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, T.; Weinert, T. Ontogeny of unstable chromosomes generated by telomere error in budding yeast. PLoS Genet. 2016, 12, e1006345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, W.C.; Counter, C.M.; Lundberg, A.S.; Beijersbergen, R.L.; Brooks, M.W.; Weinberg, R.A. Creation of human tumour cells with defined genetic elements. Nature 1999, 400, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, V.; Blackburn, E.H. An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell 1993, 73, 347–360. [Google Scholar] [CrossRef]
- Teng, S.C.; Zakian, V.A. Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol. Cell Biol. 1999, 19, 8083–8093. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Ijpma, A.; Greider, C.W. Two survivor pathways that allow growth in the absence of telomerase are generated by distinct telomere recombination events. Mol. Cell Biol. 2001, 21, 1819–1827. [Google Scholar] [CrossRef] [Green Version]
- Maringele, L.; Lydall, D. Telomerase- and recombination-independent immortalization of budding yeast. Genes Dev. 2004, 18, 2663–2675. [Google Scholar] [CrossRef] [PubMed]
- Grandin, N.; Charbonneau, M. Telomerase- and Rad52-independent immortalization of budding yeast by an inherited-long-telomere pathway of telomeric repeat amplification. Mol. Cell Biol. 2009, 29, 965–985. [Google Scholar] [CrossRef] [Green Version]
- Lebel, C.; Rosonina, E.; Sealey, D.C.; Pryde, F.; Lydall, D.; Maringele, L.; Harrington, L.A. Telomere maintenance and survival in Saccharomyces cerevisiae in the absence of telomerase and RAD52. Genetics 2009, 182, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Larrivée, M.; Wellinger, R.J. Telomerase- and capping-independent yeast survivors with alternate telomere states. Nat. Cell Biol. 2006, 8, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Wellinger, R.J.; Wolf, A.J.; Zakian, V.A. Saccharomyces telomeres acquire single-strand TG1-3 tails late in S phase. Cell 1993, 72, 51–60. [Google Scholar] [CrossRef]
- McEachern, M.J.; Blackburn, E.H. A conserved sequence motif within the exceptionally diverse telomeric sequences of budding yeasts. Proc. Natl. Acad. Sci. USA 1994, 91, 3453–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, S.; Moore, J.K.; Haber, J.E.; Greider, C.W. RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 1999, 152, 143–152. [Google Scholar] [CrossRef]
- Teng, S.C.; Chang, J.; McCowan, B.; Zakian, V.A. Telomerase-independent lengthening of yeast telomeres occurs by an abrupt Rad50p-dependent, Rif-inhibited recombinational process. Mol. Cell 2000, 6, 947–952. [Google Scholar] [CrossRef]
- Johnson, F.B.; Marciniak, R.A.; McVey, M.; Stewart, S.A.; Hahn, W.C.; Guarente, L. The Saccharomyces cerevisiae WRN homolog Sgs1p participates in telomere maintenance in cells lacking telomerase. EMBO J. 2001, 20, 905–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.; Pryde, F.E.; Lester, D.; Maddison, R.L.; Borts, R.H.; Hickson, I.D.; Louis, E.J. SGS1 is required for telomere elongation in the absence of telomerase. Curr. Biol. 2001, 11, 125–129. [Google Scholar] [CrossRef] [Green Version]
- Lydeard, J.R.; Jain, S.; Yamaguchi, M.; Haber, J.E. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 2007, 448, 820–823. [Google Scholar] [CrossRef]
- Kockler, Z.W.; Comeron, J.M.; Malkova, A. A unified alternative telomere-lengthening pathway in yeast survivor cells. Mol. Cell 2021, 81, 1816–1829. [Google Scholar] [CrossRef]
- Makovets, S.; Williams, T.L.; Blackburn, E.H. The telotype defines the telomere state in Saccharomyces cerevisiae and is inherited as a dominant non-Mendelian characteristic in cells lacking telomerase. Genetics 2008, 178, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Becerra, S.C.; Thambugala, H.T.; Erickson, A.R.; Lee, C.K.; Lewis, L.K. Reversibility of replicative senescence in Saccharomyces cerevisiae: Effect of homologous recombination and cell cycle checkpoints. DNA Repair 2012, 11, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.M.; Cooper, J.P.; Cech, T.R. Two modes of survival of fission yeast without telomerase. Science 1998, 282, 493–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Baumann, P. Chromosome fusions following telomere loss are mediated by single-strand annealing. Mol. Cell 2008, 31, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Almeida, H.; Godinho Ferreira, M. Spontaneous telomere to telomere fusions occur in unperturbed fission yeast cells. Nucleic Acids Res. 2013, 41, 3056–3067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, D.; Hebden, A.K.; Nakamura, T.M.; Miller, K.M.; Cooper, J.P. HAATI survivors replace canonical telomeres with blocks of generic heterochromatin. Nature 2010, 467, 223–227. [Google Scholar] [CrossRef]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Pickett, H.A. Targeting telomeres: Advances in telomere maintenance mechanism-specific cancer therapies. Nat. Rev. Cancer 2022, 22, 515–532. [Google Scholar] [CrossRef]
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere maintenance by recombination in human cells. Nat. Genet. 2000, 26, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, A.; Neumann, A.A.; Hills, M.; Reddel, R.R. Telomere elongation involves intra-molecular DNA replication in cells utilizing alternative lengthening of telomeres. Hum. Mol. Genet. 2009, 18, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.A.; Watson, C.M.; Noble, J.R.; Pickett, H.A.; Tam, P.P.; Reddel, R.R. Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev. 2013, 27, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Sfeir, A.; Kosiyatrakul, S.T.; Hockemeyer, D.; MacRae, S.L.; Karlseder, J.; Schildkraut, C.L.; de Lange, T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 2009, 138, 90–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandin, N.; Charbonneau, M. Mrc1, a non-essential DNA replication protein, is required for telomere end protection following loss of capping by Cdc13, Yku or telomerase. Mol. Genet. Genomics 2007, 277, 685–699. [Google Scholar] [CrossRef]
- Grandin, N.; Bailly, A.; Charbonneau, M. Activation of Mrc1, a mediator of the replication checkpoint, by telomere erosion. Biol. Cell 2005, 97, 799–814. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Kozak, M.; Martin, J.D.; Pennock, E.; Johnson, F.B. Evidence that a RecQ helicase slows senescence by resolving recombining telomeres. PLoS Biol. 2007, 5, e160. [Google Scholar] [CrossRef]
- Matmati, S.; Lambert, S.; Géli, V.; Coulon, S. Telomerase repairs collapsed replication forks at telomeres. Cell Rep. 2020, 30, 3312–3322. [Google Scholar] [CrossRef] [Green Version]
- Pickett, H.A.; Reddel, R.R. Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nat. Struct. Mol. Biol. 2015, 22, 875–880. [Google Scholar] [CrossRef]
- Sobinoff, A.P.; Pickett, H.A. Alternative lengthening of telomeres: DNA repair pathways converge. Trends Genet. 2017, 33, 921–932. [Google Scholar] [CrossRef]
- Kim, J.; Sun, C.; Tran, A.D.; Chin, P.J.; Ruiz, P.D.; Wang, K.; Gibbons, R.J.; Gamble, M.J.; Liu, Y.; Oberdoerffer, P. The macroH2A1.2 histone variant links ATRX loss to alternative telomere lengthening. Nat. Struct. Mol. Biol. 2019, 26, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Deng, Z.; Zhang, L.; Wu, C.; Jin, Y.; Hwang, I.; Vladimirova, O.; Xu, L.; Yang, L.; Lu, B.; et al. ATRX loss induces telomere dysfunction and necessitates induction of alternative lengthening of telomeres during human cell immortalization. EMBO J. 2019, 38, e96659. [Google Scholar] [CrossRef] [PubMed]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, 7538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.T.; Voon, H.P.J.; Xella, B.; Scott, C.; Clynes, D.; Babbs, C.; Ayyub, H.; Kerry, J.; Sharpe, J.A.; Sloane-Stanley, J.A.; et al. The chromatin remodelling factor ATRX suppresses R-loops in transcribed telomeric repeats. EMBO Rep. 2017, 18, 914–928. [Google Scholar] [CrossRef]
- O’Sullivan, R.J.; Almouzni, G. Assembly of telomeric chromatin to create ALTernative endings. Trends Cell Biol. 2014, 24, 675–685. [Google Scholar] [CrossRef]
- Maicher, A.; Kastner, L.; Dees, M.; Luke, B. Deregulated telomere transcription causes replication-dependent telomere shortening and promotes cellular senescence. Nucleic Acids Res. 2012, 40, 5966–6649. [Google Scholar] [CrossRef] [Green Version]
- Cusanelli, E.; Romero, C.A.; Chartrand, P. Telomeric noncoding RNA TERRA is induced by telomere shortening to nucleate telomerase molecules at short telomeres. Mol. Cell 2013, 51, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Moravec, M.; Wischnewski, H.; Bah, A.; Hu, Y.; Liu, N.; Lafranchi, L.; King, M.C.; Azzalin, C.M. TERRA promotes telomerase-mediated telomere elongation in Schizosaccharomyces pombe. EMBO Rep. 2016, 17, 999–1012. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Bennett, H.W.; Liu, N.; Moravec, M.; Williams, J.F.; Azzalin, C.M.; King, M.C. RNA-DNA hybrids support recombination-based telomere maintenance in fission yeast. Genetics 2019, 213, 431–447. [Google Scholar] [CrossRef]
- Balk, B.; Maicher, A.; Dees, M.; Klermund, J.; Luke-Glaser, S.; Bender, K.; Luke, B. Telomeric RNA-DNA hybrids affect telomere-length dynamics and senescence. Nat. Struct. Mol. Biol. 2013, 20, 1199–1205. [Google Scholar] [CrossRef]
- Yu, T.Y.; Kao, Y.W.; Lin, J.J. Telomeric transcripts stimulate telomere recombination to suppress senescence in cells lacking telomerase. Proc. Natl. Acad. Sci. USA 2014, 111, 3377–3382. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Bonetti, D.; Lockhart, A.; Serhal, K.; Kellner, V.; Maicher, A.; Jolivet, P.; Teixeira, M.T.; Luke, B. Telomere length determines TERRA and R-loop regulation through the cell cycle. Cell 2017, 170, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Silva, B.; Arora, R.; Bione, S.; Azzalin, C.M. TERRA transcription destabilizes telomere integrity to initiate break-induced replication in human ALT cells. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, K.L.; Millard, M.; Bosenberg, M.W.; DePinho, R.A. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat. Genet. 2001, 28, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wu, C.J.; Jaskelioff, M.; Ivanova, E.; Kost-Alimova, M.; Protopopov, A.; Chu, G.C.; Wang, G.; Lu, X.; Labrot, E.S.; et al. Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell 2012, 148, 896–907. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Hwang, S.S.; Liesa, M.; Gan, B.; Sahin, E.; Jaskelioff, M.; Ding, Z.; Ying, H.; Boutin, A.T.; Zhang, H.; et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 2012, 148, 651–663. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. cerevisiae | H. sapiens | Description |
|---|---|---|
| Mre11-Rad50-Xrs2 | MRE11-RAD50-NBS1 | DSB sensor; telomere length regulator |
| Ku70-Ku80 | KU70-KU80 | DSB sensor; telomere length regulator |
| Tel1 | ATM | Apical protein kinase; telomere length regulator |
| Sae2 | CtIP | Activator of MRX/MRN endonuclease |
| Exo1 | EXO1 | Exonuclease |
| Sgs1 | BLM | DNA helicase |
| Dna2 | DNA2 | DNA helicase and nuclease |
| Mec1-Ddc2 | ATR-ATRIP | Apical protein kinase and interacting factor |
| Rad9 | 53BP1 | Checkpoint adaptor/mediator |
| Mrc1 | Claspin | Replisome component; checkpoint activator |
| Rad53 | CHK2 | Downstream protein kinase |
| Chk1 | CHK1 | Downstream protein kinase |
| Cdc13-Stn1-Ten1 | CTC1-STN1-TEN1 | Telomere binding complex; telomere capping regulator |
| Rap1-Rif1-Rif2 | TRF1-TRF2-RAP1-TIN2-TPP1-POT1 | Telomere binding complex; telomere capping and length regulator |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casari, E.; Gnugnoli, M.; Rinaldi, C.; Pizzul, P.; Colombo, C.V.; Bonetti, D.; Longhese, M.P. To Fix or Not to Fix: Maintenance of Chromosome Ends Versus Repair of DNA Double-Strand Breaks. Cells 2022, 11, 3224. https://doi.org/10.3390/cells11203224
Casari E, Gnugnoli M, Rinaldi C, Pizzul P, Colombo CV, Bonetti D, Longhese MP. To Fix or Not to Fix: Maintenance of Chromosome Ends Versus Repair of DNA Double-Strand Breaks. Cells. 2022; 11(20):3224. https://doi.org/10.3390/cells11203224
Chicago/Turabian StyleCasari, Erika, Marco Gnugnoli, Carlo Rinaldi, Paolo Pizzul, Chiara Vittoria Colombo, Diego Bonetti, and Maria Pia Longhese. 2022. "To Fix or Not to Fix: Maintenance of Chromosome Ends Versus Repair of DNA Double-Strand Breaks" Cells 11, no. 20: 3224. https://doi.org/10.3390/cells11203224