
Long-Term IGF1 Stimulation Leads to Cellular Senescence via Functional Interaction with the Thioredoxin-Interacting Protein, TXNIP





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Generation of TXNIP Knock-Out Cell Lines Using CRISPR/Cas9
2.3. Overexpression Studies
2.4. Real Time-Quantitative Polymerase Chain Reactions (RT-QPCR)
2.5. Western Blot Analyses
2.6. Cell Treatments
2.7. Cell Viability Assays
2.8. Senescence-Associated β-Galactosidase Assays
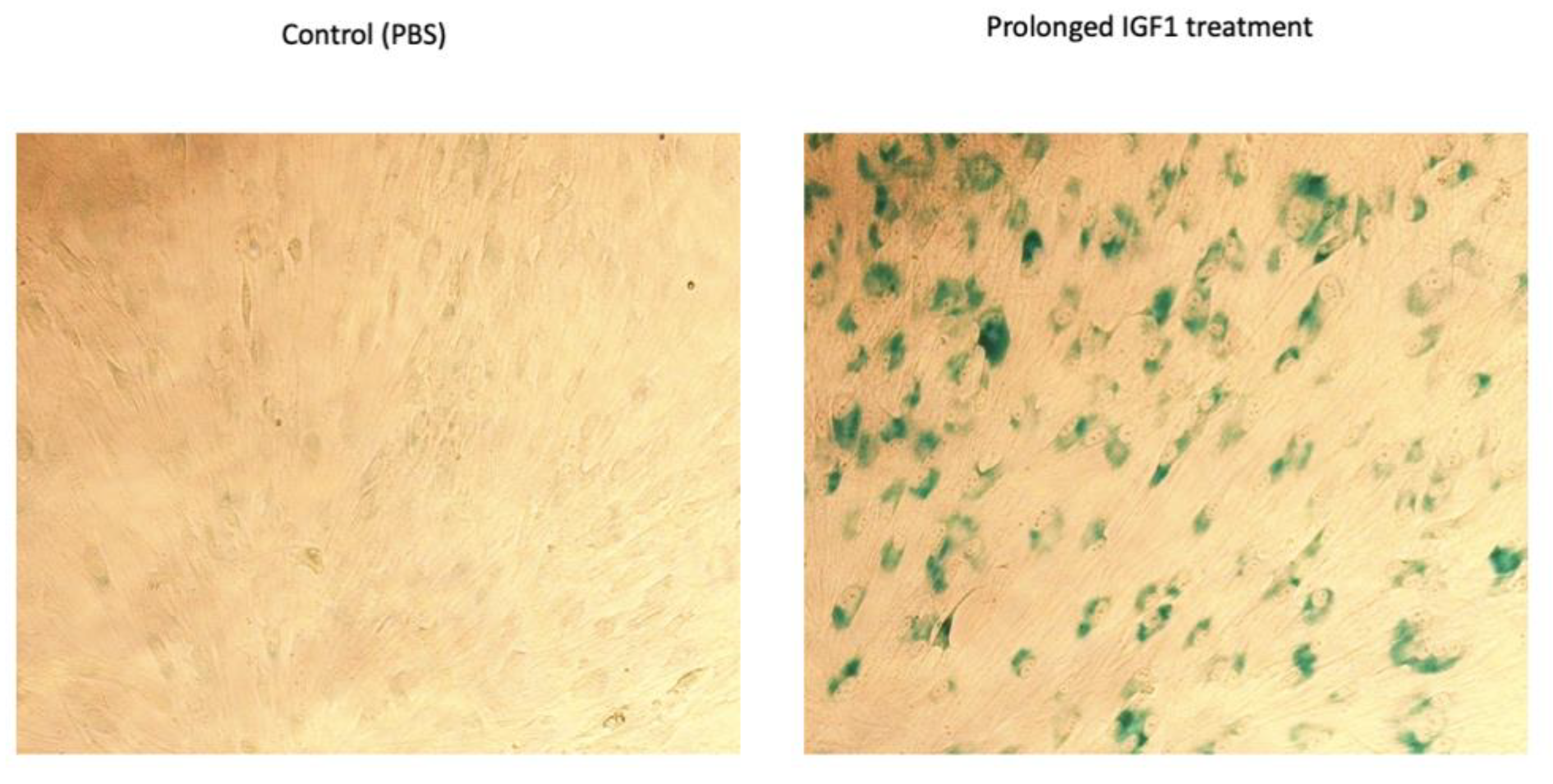
2.9. Prolonged IGF1-Mediated Senescence Induction
2.10. Flow Cytometry Analysis for Apoptotic Cell Death
2.11. Flow Cytometry-Based Cell Cycle Analysis
2.12. LC-MS/MS-Based Proteomic Analyses
2.13. Data Analyses
2.14. Statistical Analyses
3. Results
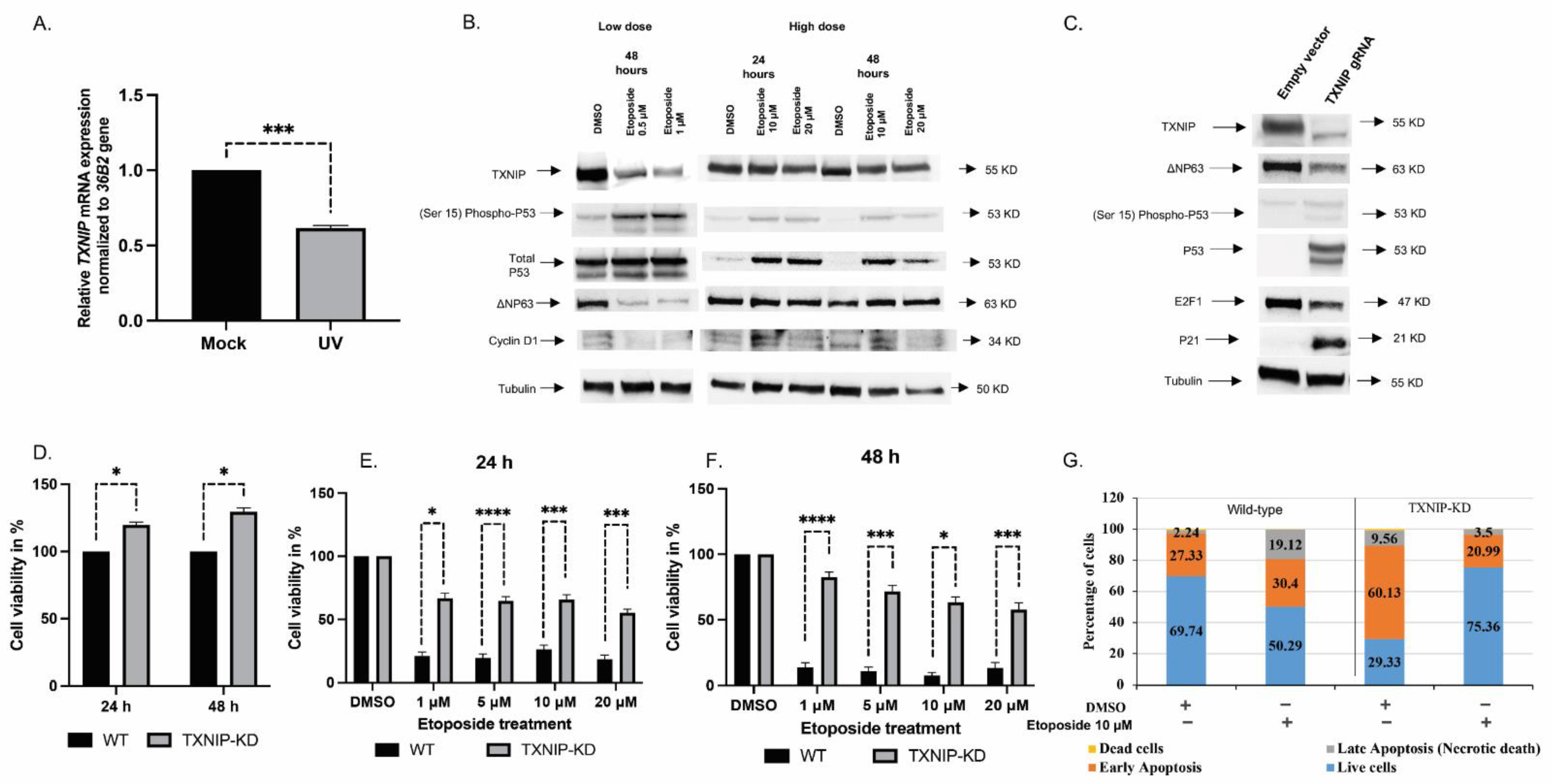
3.1. TXNIP Maintains Cellular Homeostasis upon DNA Damage Stress
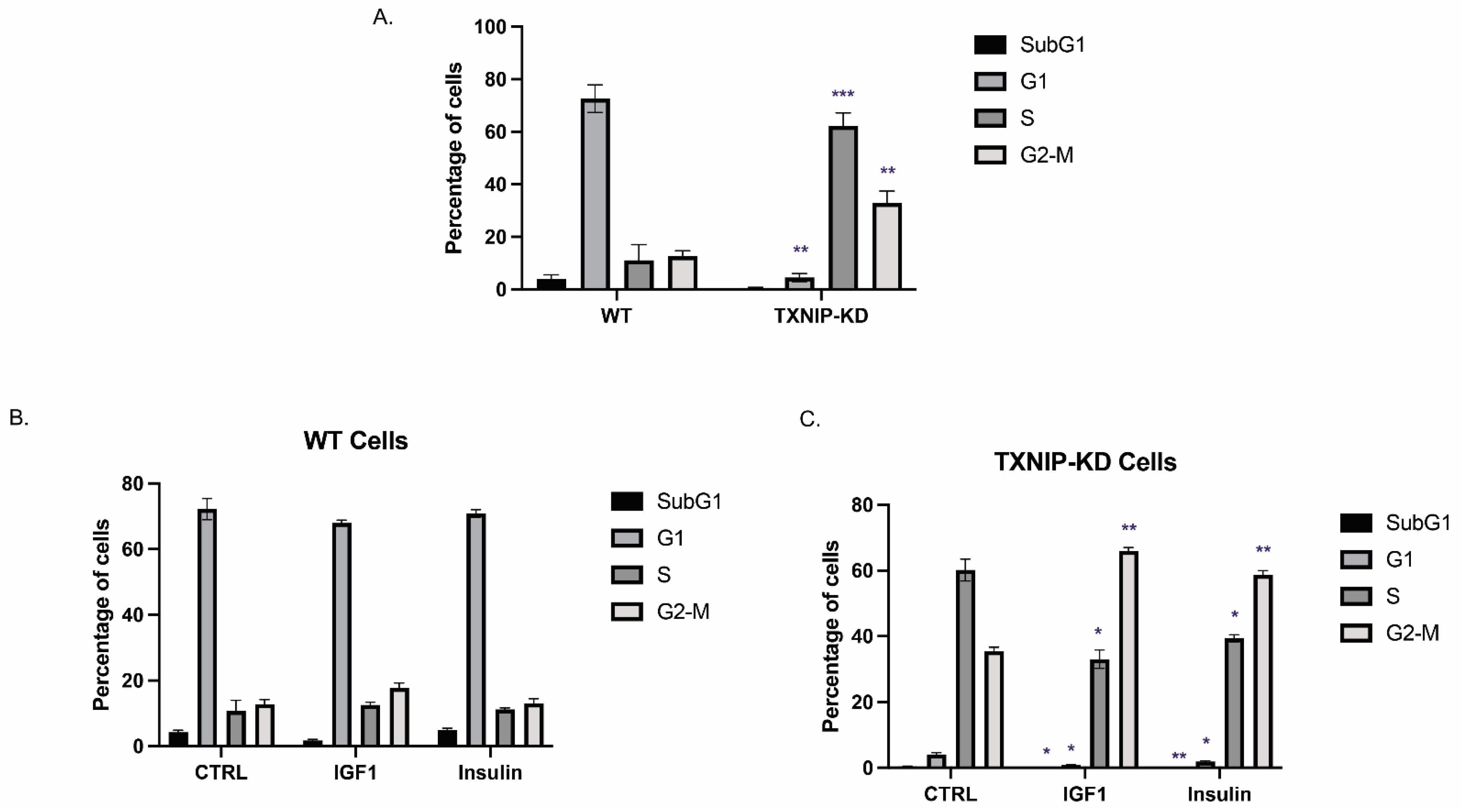
3.2. The Mitogenic Effects of IGF1 and Insulin Are Amplified in TXNIP-Deficient Cells
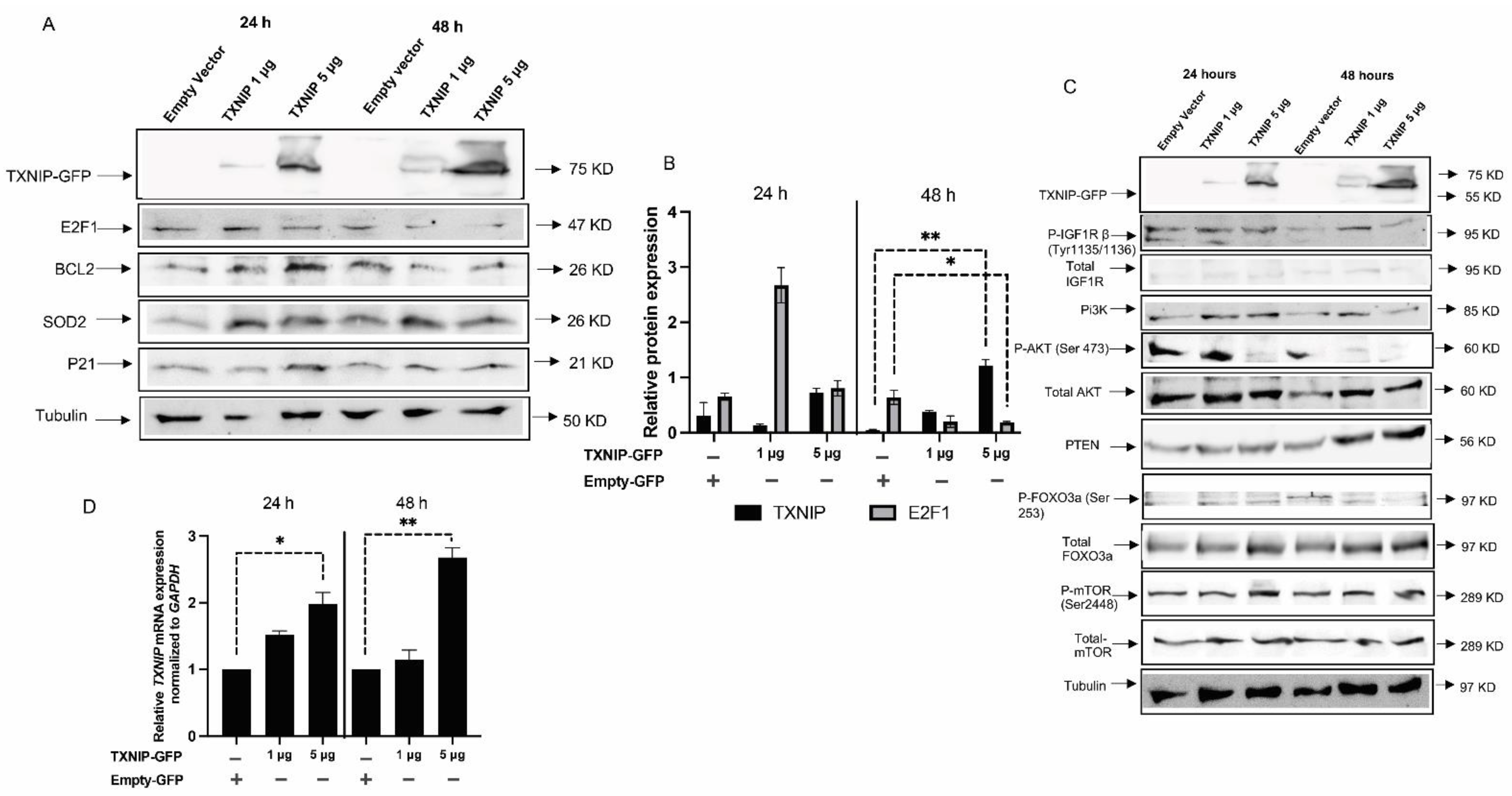
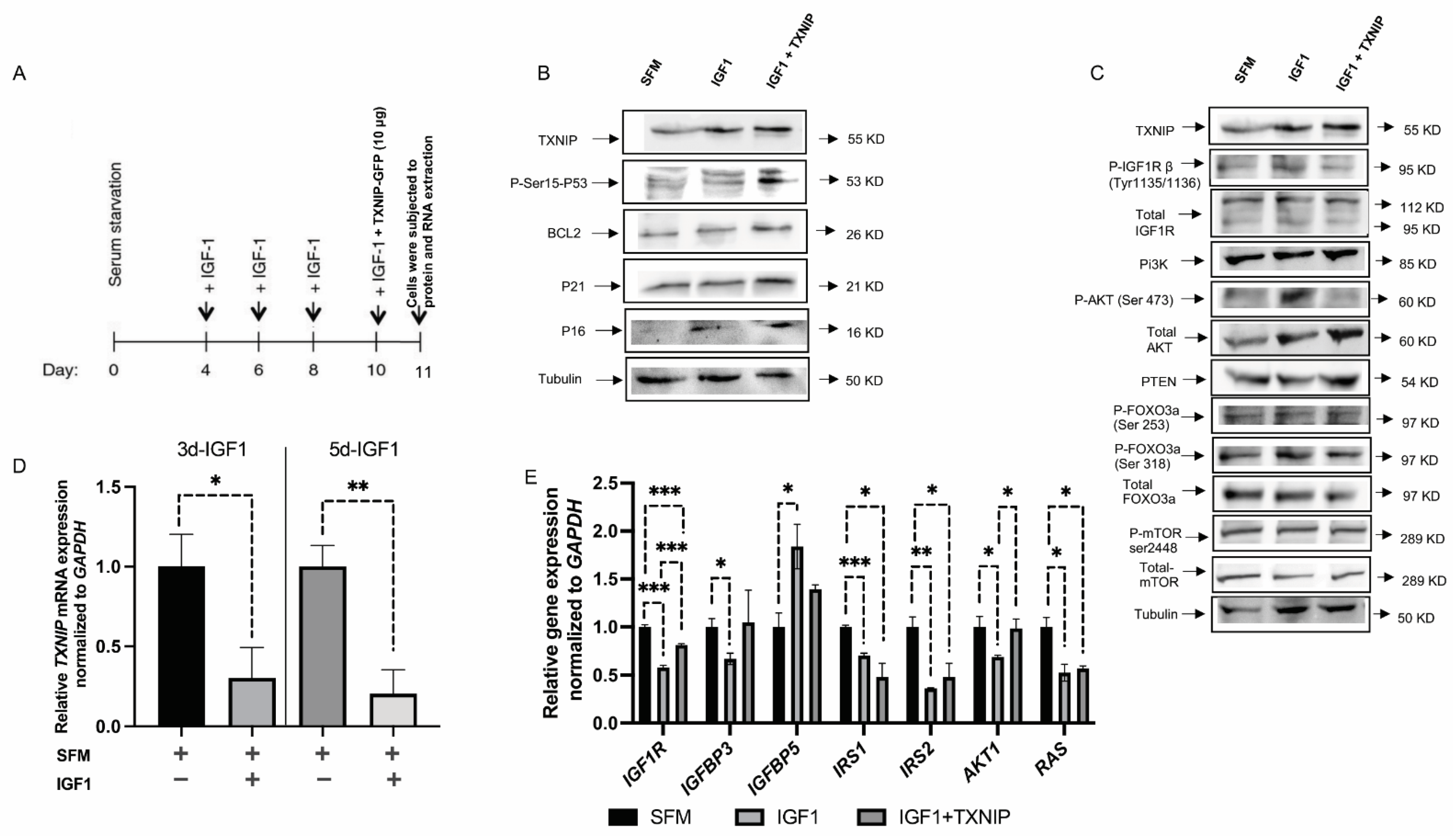
3.3. TXNIP Reactivation Upregulates E2F1 and Inhibits the IGF1R Signaling Pathway
3.4. TXNIP Augments the Effect of Prolonged IGF1-Induced Premature Senescence
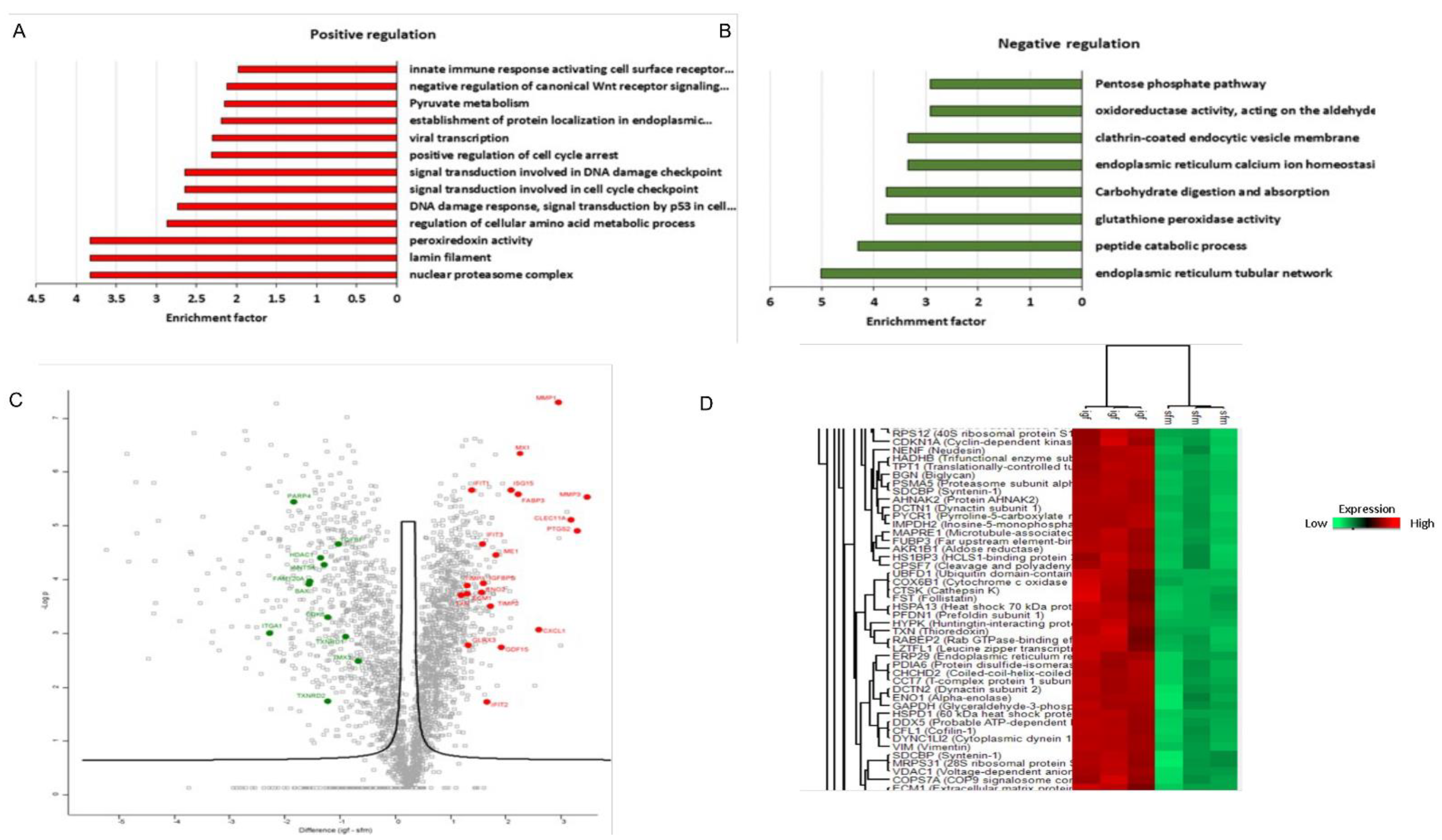
3.5. Proteomic Analysis Revealed a Unique Senescence Network Signature in IGF1-Induced Premature Senescence
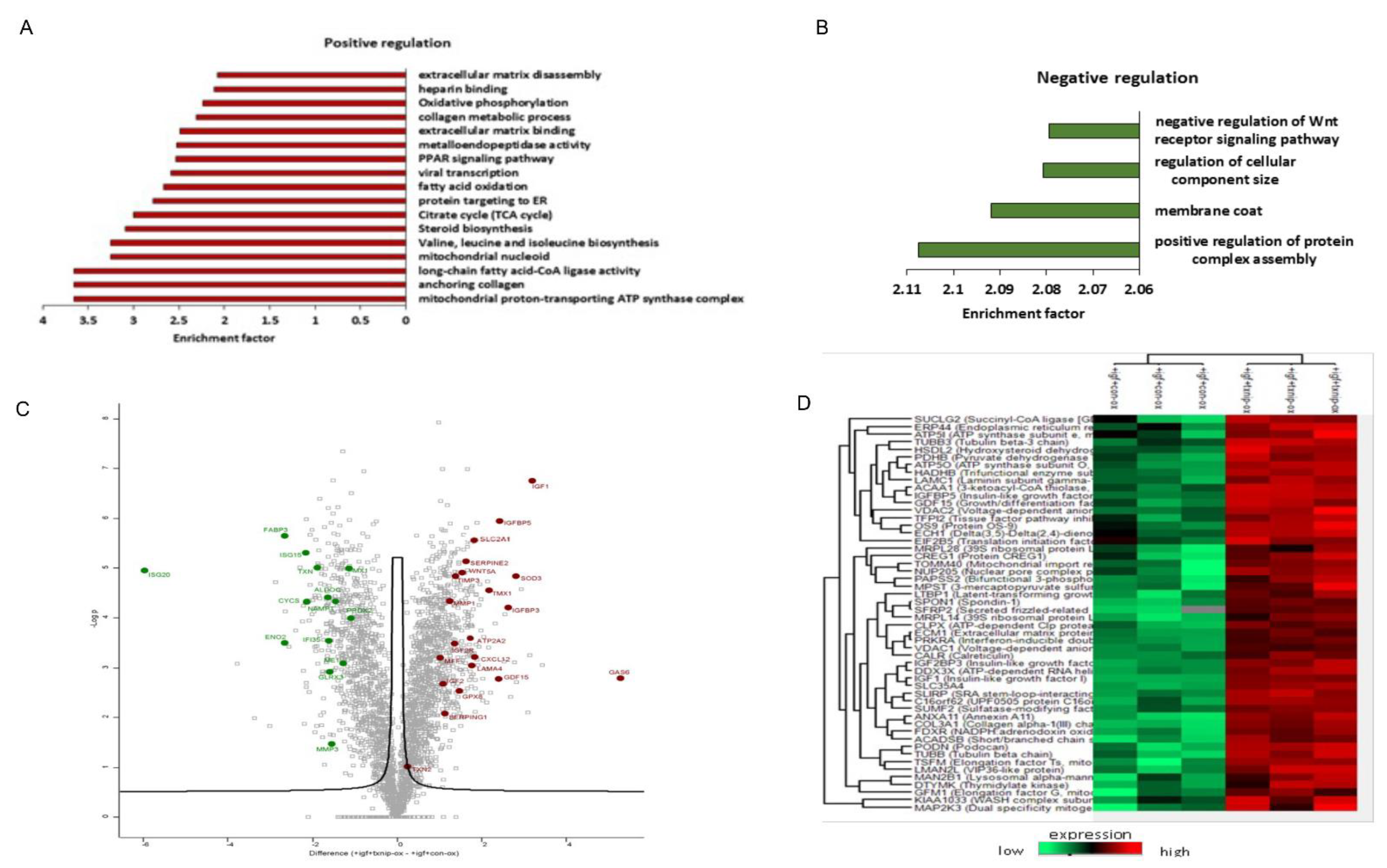
3.6. TXNIP Induction Drives a Distinct Senescence Phenotype in Prolonged IGF1-Induced Senescence
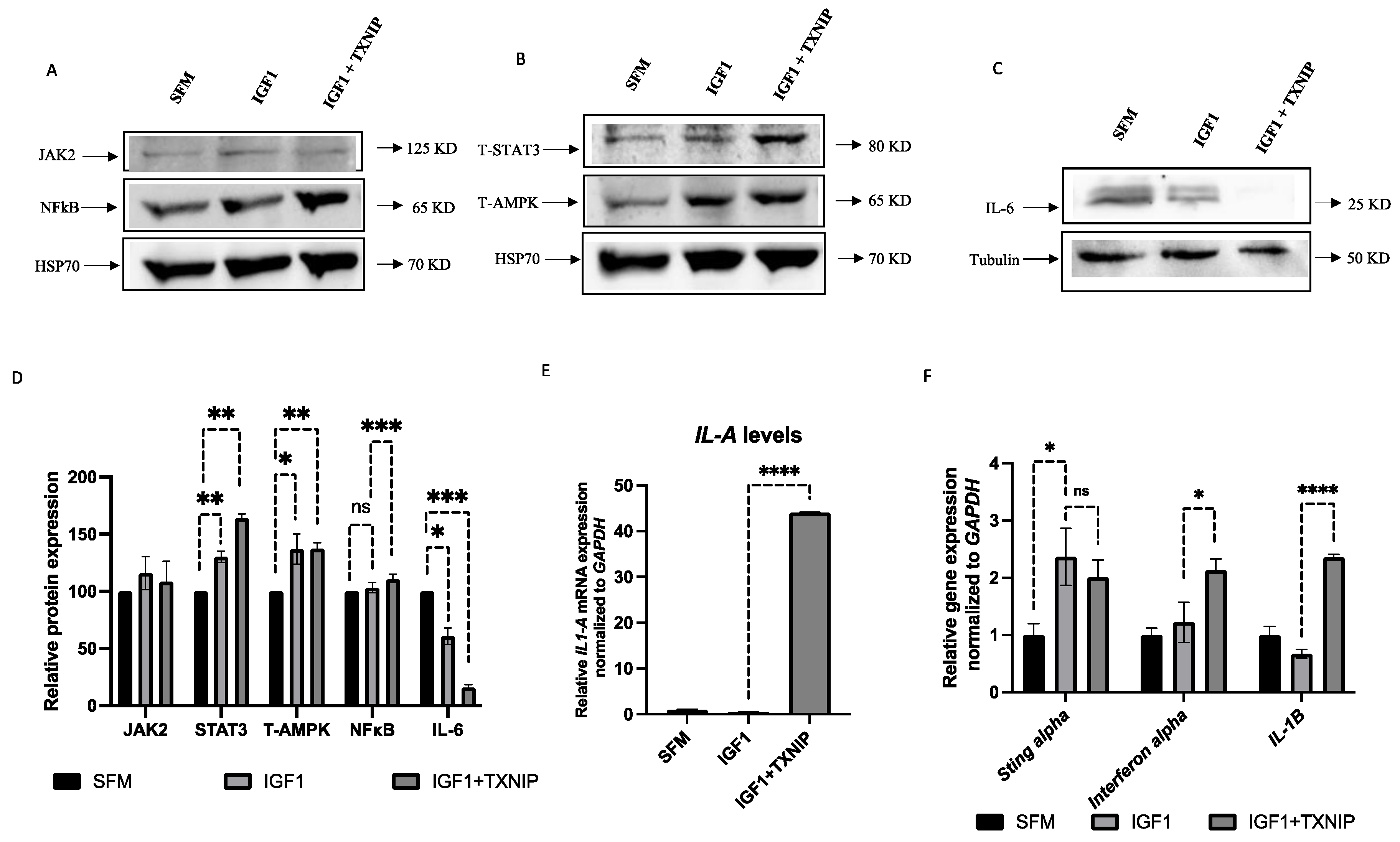
3.7. TXNIP Induces a STAT3/IL-1A-Mediated Senescence-Associated Secretory Phenotype (SASP) in IGF1-Induced Senescence
4. Discussion
4.1. Identification of Functional Interactions between TXNIP and the IGF1 Pathway
4.2. Prolonged IGF1-Induced Cellular Senescence Upregulates TXNIP Levels
4.3. Proteomic Analyses Reveal Activation of a Pro-Inflammatory Network in IGF1/TXNIP-Induced Premature Senescence
4.4. Proteins Involved in IGF1 Signaling Are Upregulated upon TXNIP Induction
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- LeRoith, D.; Yakar, S. Mechanisms of disease: Metabolic effects of growth hormone and insulin-like growth factor-1. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Yakar, S.; Adamo, M.L. Insulin-like growth factor 1 physiology: Lessons from mouse models. Endocrinol. Metab. Clin. N. Am. 2012, 41, 231–247. [Google Scholar] [CrossRef]
- Hawkes, C.P.; Grimberg, A. Insulin-like growth factor-1 is a marker for the nutrition state. Pediatr. Endocr. Rev. 2015, 13, 499–511. [Google Scholar]
- Klammt, J.; Pfaffle, R.; Werner, H.; Kiess, W. IGF signaling defects as causes of growth failure and IUGR. Trends Endocrinol. Metab. 2008, 19, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.; Stampfer, M.J.; Giovannucci, E.; Gann, P.H.; Ma, J.; Wilkinson, P.; Hennekens, C.H.; Pollak, M. Plasma insulin-like growth factor-I and prostate cancer risk: A prospective study. Science 1998, 279, 563–566. [Google Scholar] [CrossRef]
- Hankinson, S.E.; Willett, W.C.; Colditz, G.A.; Hunter, D.J.; Michaud, D.S.; Deroo, B.; Rosner, B.; Speizer, F.E.; Pollak, M. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet 1998, 351, 1393–1396. [Google Scholar] [CrossRef]
- Renehan, A.G.; Zwahlen, M.; Minder, C.; O’Dwyer, S.T.; Shalet, S.M.; Egger, M. Insulin-like growth factor-I, IGF binding protein-3, and cancer risk: Systematic review and meta-regression analysis. Lancet 2004, 363, 1346–1353. [Google Scholar] [CrossRef]
- Wang, S.; Wang, N.; Yu, B.; Cao, M.; Wang, Y.; Guo, Y.; Zhang, Y.; Zhang, P.; Yu, X.; Wang, S.; et al. Circulating IGF-1 promotes prostate adenocarcinoma via FOXO3A/BIM signaling in a double-transgenic mouse model. Oncogene 2019, 38, 6338–6353. [Google Scholar] [CrossRef]
- Ostermann, A.L.; Wunderlich, C.M.; Schneiders, L.; Vogt, M.C.; Woeste, M.A.; Belgardt, B.F.; Niessen, C.M.; Martiny, B.; Schauss, A.C.; Frommolt, P.; et al. Intestinal insulin/IGF1 signalling through FoxO1 regulates epithelial integrity and susceptibility to colon cancer. Nat. Metab. 2019, 1, 371–389. [Google Scholar] [CrossRef]
- Werner, H. Tumor suppressors govern insulin-like growth factor signaling pathways: Implications in metabolism and cancer. Oncogene 2012, 31, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Werner, H. The pathophysiological significance of IGF-I receptor overexpression: New insights. Ped. Endocrinol. Rev. 2009, 7, 2–5. [Google Scholar]
- Larsson, O.; Girnita, A.; Girnita, L. Role of insulin-like growth factor-I receptor signalling in cancer. British J. Cancer 2005, 92, 2097–2101. [Google Scholar] [CrossRef] [PubMed]
- Sarfstein, R.; Maor, S.; Reizner, N.; Abramovitch, S.; Werner, H. Transcriptional regulation of the insulin-like growth factor-1 receptor in breast cancer. Mol. Cell. Endocrinol. 2006, 252, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Gubbi, S.; Quipildor, G.F.; Barzilai, N.; Huffman, D.M.; Milman, S. 40 years of IGF1: IGF1: The Jekyll and Hyde of the aging brain. J. Mol. Endocrinol. 2018, 61, T171–T185. [Google Scholar] [CrossRef]
- Vitale, G.; Pellegrino, G.; Vollery, M.; Hofland, L.J. Role of IGF-1 system in the modulation of longevity: Controversies and new insights from a centenarian’s perspective. Front. Endocrinol. 2019, 10, 27. [Google Scholar] [CrossRef]
- Hinkal, G.; Donehower, L.A. How does suppression of IGF-1 signaling by DNA damage affect aging and longevity? Mech. Ageing Dev. 2008, 129, 243–253. [Google Scholar] [CrossRef][Green Version]
- Milman, S.; Huffman, D.M.; Barzilai, N. The somatotropic axis in human aging: Framework for the current state of knowledge and future research. Cell Metab. 2016, 23, 980–989. [Google Scholar] [CrossRef]
- Mao, K.; Quipildor, G.F.; Tabrizian, T.; Novaj, A.; Guan, F.; Walters, R.O.; Delahaye, F.; Hubbard, G.B.; Ikeno, Y.; Ejima, K.; et al. Late-life targeting of the IGF-1 receptor improves healthspan and lifespan in female mice. Nat. Commun. 2018, 9, 2394. [Google Scholar] [CrossRef]
- Laron, Z.; Pertzelan, A.; Mannheimer, S. Genetic pituitary dwarfism with high serum concentration of growth hormone-a new inborn error of metabolism? Isr. J. Med. Sci. 1966, 2, 152–155. [Google Scholar]
- Amselem, S.; Duquesnoy, P.; Attree, O.; Novelli, G.; Bousnina, S.; Postel-Vinay, M.C.; Goossens, M. Laron dwarfism and mutations of the growth hormone-receptor gene. N. Engl. J. Med. 1989, 321, 989–995. [Google Scholar] [CrossRef]
- Laron, Z. Extensive personal experience. Laron syndrome (primary growth hormone resistance or insensitivity): The personal experience 1958-2003. J. Clin. Endocrinol. Metab. 2004, 89, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z.; Kopchik, J.J. Laron Syndrome—From Man to Mouse; Springer: Heidelberg, Germany, 2011. [Google Scholar]
- Shevah, O.; Laron, Z. Patients with congenital deficiency of IGF-I seem protected from the development of malignancies: A preliminary report. Growth Horm. IGF Res. 2007, 17, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Steuerman, R.; Shevah, O.; Laron, Z. Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur. J. Endocrinol. 2011, 164, 485–489. [Google Scholar] [CrossRef]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef] [PubMed]
- Lapkina-Gendler, L.; Rotem, I.; Pasmanik-Chor, M.; Gurwitz, D.; Sarfstein, R.; Laron, Z.; Werner, H. Identification of signaling pathways associated with cancer protection in Laron syndrome. Endocr. Relat. Cancer 2016, 23, 399–410. [Google Scholar] [CrossRef]
- Werner, H.; Lapkina-Gendler, L.; Achlaug, L.; Nagaraj, K.; Somri, L.; Yaron-Saminsky, D.; Pasmanik-Chor, M.; Sarfstein, R.; Laron, Z.; Yakar, S. Genome-wide profiling of Laron Syndrome patients identifies novel cancer protection pathways. Cells 2019, 8, E596. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Laron, Z. Role of the GH-IGF1 system in progression of cancer. Mol. Cell. Endocrinol. 2020, 518, 111003. [Google Scholar] [CrossRef]
- Zhou, J.; Yu, Q.; Chng, W.J. TXNIP (VDUP-1, TBP-2): A major redox regulator commonly suppressed in cancer by epigenetic mechanisms. Int. J. Biochem. Cell Biol. 2011, 43, 1668–1673. [Google Scholar] [CrossRef]
- Chen, K.S.; DeLuca, H.F. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochim. Biophys. Acta—Gene Struct. Expr. 1994, 1219, 26–32. [Google Scholar] [CrossRef]
- Nishiyama, A.; Matsui, M.; Iwata, S.; Hirota, K.; Masutani, H.; Nakamura, H.; Takagi, Y.; Sono, H.; Gon, Y.; Yodoi, J. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 1999, 274, 21645–21650. [Google Scholar] [CrossRef]
- Patwari, P.; Higgins, L.J.; Chutkow, W.A.; Yoshioka, J.; Lee, R.T. The interaction of thioredoxin with Txnip: Evidence for formation of a mixed disulfide by disulfide exchange. J. Biol. Chem. 2006, 281, 21884–21891. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, J.; Hirota, K.; Itoh, T.; Shinkura, R.; Kitada, K.; Yodoi, J.; Namba, T.; Fukuda, K. Thioredoxin inhibits tumor necrosis factor- or interleukin-1-induced NF-kappaB activation at a level upstream of NF-kappaB-inducing kinase. Antioxid. Redox Signal. 2000, 2, 83–92. [Google Scholar] [CrossRef]
- Aitken, C.J.; Hodge, J.M.; Nishinaka, Y.; Vaughan, T.; Yodoi, J.; Day, C.J.; Morrison, N.A.; Nicholson, G.C. Regulation of human osteoclast differentiation by thioredoxin binding protein-2 and redox-sensitive signaling. J. Bone Min. Res. 2004, 19, 2057–2064. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.F.; Miele, M.E.; Hatta, N.; Takata, M.; Paquette-Straub, C.; Freedman, L.P.; Welch, D.R. Melanoma metastasis suppression by chromosome 6: Evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Res. 2003, 63, 432–440. [Google Scholar] [PubMed]
- Zhang, X.; Fu, T.; He, Q.; Gao, X.; Luo, Y. Glucose-6-phosphate upregulates Txnip expression by interacting with MondoA. Front. Mol. Biosci. 2020, 6, 147. [Google Scholar] [CrossRef] [PubMed]
- Chutkow, W.A.; Patwari, P.; Yoshioka, J.; Lee, R.T. Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J. Biol. Chem. 2008, 283, 2397–2406. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.H.; Lee, K.N.; Hwang, C.Y.; Kwon, K.S.; You, K.H.; Choi, I. Tumor suppressor VDUP1 increases p27(kip1) stability by inhibiting JAB1. Cancer Res. 2005, 65, 4485–4489. [Google Scholar]
- Elgort, M.G.; O’Shea, J.M.; Jiang, Y.; Ayer, D.E. Transcriptional and translational downregulation of thioredoxin interacting protein is required for metabolic reprogramming during G(1). Genes Cancer 2010, 1, 893–907. [Google Scholar]
- Chutkow, W.A.; Lee, R.T. Thioredoxin regulates adipogenesis through thioredoxin-interacting protein (Txnip) protein stability. J. Biol. Chem. 2011, 286, 29139–29145. [Google Scholar] [CrossRef]
- Masutani, H.; Yoshihara, E.; Masaki, S.; Chen, Z.; Yodoi, J. Thioredoxin binding protein (TBP)-2/Txnip and α-arrestin proteins in cancer and diabetes mellitus. J. Clin. Biochem. Nutr. 2012, 50, 23–34. [Google Scholar] [CrossRef]
- Butler, L.M.; Zhou, X.; Xu, W.S.; Scher, H.I.; Rifkind, R.A.; Marks, P.A.; Richon, V.M. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc. Natl. Acad. Sci. USA 2002, 99, 11700–11705. [Google Scholar] [CrossRef]
- Park, J.W.; Lee, S.H.; Woo, G.H.; Kwon, H.J.; Kim, D.Y. Downregulation of TXNIP leads to high proliferative activity and estrogen-dependent cell growth in breast cancer. Biochem. Biophys. Res. Commun. 2018, 498, 566–572. [Google Scholar] [CrossRef]
- Jung, H.; Kim, D.O.; Byun, J.E.; Kim, W.S.; Kim, M.J.; Song, H.Y.; Kim, Y.K.; Kang, D.K.; Park, Y.J.; Kim, T.D.; et al. Thioredoxin-interacting protein regulates haematopoietic stem cell ageing and rejuvenation by inhibiting p38 kinase activity. Nat. Commun. 2016, 7, 13674. [Google Scholar] [CrossRef]
- Muri, J.; Heer, S.; Matsushita, M.; Pohlmeier, L.; Tortola, L.; Fuhrer, T.; Conrad, M.; Zamboni, N.; Kisielow, J.; Kopf, M. The thioredoxin-1 system is essential for fueling DNA synthesis during T-cell metabolic reprogramming and proliferation. Nat. Commun. 2018, 9, 1851. [Google Scholar] [CrossRef]
- Katsu-Jiménez, Y.; Vázquez-Calvo, C.; Maffezzini, C.; Halldin, M.; Peng, X.; Freyer, C.; Wredenberg, A.; Giménez-Cassina, A.; Wedell, A.; Arnér, E.S.J. Absence of TXNIP in humans leads to lactic acidosis and low serum methionine linked to deficient respiration on pyruvate. Diabetes 2019, 68, 709–723. [Google Scholar] [CrossRef]
- Zhou, J.; Chng, W.J. Roles of thioredoxin binding protein (TXNIP) in oxidative stress, apoptosis and cancer. Mitochondrion 2013, 13, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, K.; Lapkina-Gendler, L.; Sarfstein, R.; Gurwitz, D.; Pasmanik-Chor, M.; Laron, Z.; Yakar, S.; Werner, H. Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc. Natl. Acad. Sci. USA 2018, 115, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Bergholz, J.; Zhang, H.; He, H.; Wang, Y.; Zhang, Y.; Li, Q.; Kirkland, J.L.; Xiao, Z.X. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell 2014, 13, 669–678. [Google Scholar] [CrossRef]
- Bae, V.L.; Jackson-Cook, C.K.; Maygarden, S.J.; Plymate, S.R.; Chen, J.; Ware, J.L. Metastatic sublines of an SV40 large T antigen immortalized human prostate epithelial cell line. Prostate 1998, 34, 275–282. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Nahor, I.; Abramovitch, S.; Engeland, K.; Werner, H. The p53-family members p63 and p73 inhibit insulin-like growth factor-I receptor gene expression in colon cancer cells. Growth Horm. IGF Res. 2005, 15, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.T.; Andres, A.M.; Miller, A.K.; Spann, N.J.; Potter, D.W.; Post, N.M.; Chen, A.Z.; Sachithanantham, S.; Jung, D.Y.; Kim, J.K.; et al. Txnip balances metabolic and growth signaling via PTEN disulfide reduction. Proc. Natl. Acad. Sci. USA 2008, 105, 3921–3926. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Bang, E.; Arulkumar, R.; Ha, S.; Chung, K.W.; Park, M.H.; Choi, Y.J.; Yu, B.P.; Chung, H.Y. Senoinflammation: A major mediator underlying age-related metabolic dysregulation. Exp. Gerontol. 2020, 134, 110891. [Google Scholar] [CrossRef]
- Pillai, P.S.; Molony, R.D.; Martinod, K.; Dong, H.; Pang, I.K.; Tal, M.C.; Solis, A.G.; Bielecki, P.; Mohanty, S.; Trentalange, M.; et al. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science 2016, 352, 463–466. [Google Scholar] [CrossRef]
- Yamagami, M.; Otsuka, M.; Kishikawa, T.; Sekiba, K.; Seimiya, T.; Tanaka, E.; Suzuki, T.; Ishibashi, R.; Ohno, M.; Koike, K. ISGF3 with reduced phosphorylation is associated with constitutive expression of interferon-induced genes in aging cells. NPJ. Aging Mech. Dis. 2018, 4, 11. [Google Scholar] [CrossRef]
- Piñeiro-Hermida, S.; López, I.P.; Alfaro-Arnedo, E.; Torrens, R.; Iñiguez, M.; Alvarez-Erviti, L.; Ruíz-Martínez, C.; Pichel, J.G. IGF1R deficiency attenuates acute inflammatory response in a bleomycin-induced lung injury mouse model. Sci. Rep. 2017, 7, 4290. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.; Porciuncula, A.; Yu, A.; Iwakura, Y.; David, G. Uncoupling the senescence-associated secretory phenotype from cell cycle exit via interleukin-1 inactivation unveils Its protumorigenic role. Mol. Cell Biol. 2019, 39, e00586. [Google Scholar] [CrossRef]
- Kenyon, C. A conserved regulatory system for aging. Cell 2001, 105, 165–168. [Google Scholar] [CrossRef]
- Wrigley, S.; Arafa, D.; Tropea, D. Insulin-Like Growth Factor 1: At the Crossroads of Brain Development and Aging. Front. Cell. Neurosci. 2017, 11, 14. [Google Scholar] [CrossRef]
- Marais, T.L.D.; Kluz, T.; Xu, D.; Zhang, X.; Gesumaria, L.; Matsui, M.S.; Costa, M.; Sun, H. Transcription factors and stress response gene alterations in human keratinocytes following Solar Simulated Ultra Violet Radiation. Sci. Rep. 2017, 7, 13622. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sun, B.; Clewell, R.A.; Adeleye, Y.; Andersen, M.E.; Zhang, Q. Dose-response modeling of etoposide-induced DNA damage response. Toxicol. Sci. 2014, 137, 371–384. [Google Scholar] [CrossRef]
- Zhao, Q.; Che, X.; Zhang, H.; Fan, P.; Tan, G.; Liu, L.; Jiang, D.; Zhao, J.; Xiang, X.; Liang, Y.; et al. Thioredoxin-interacting protein links endoplasmic reticulum stress to inflammatory brain injury and apoptosis after subarachnoid haemorrhage. J. Neuroinflammation 2017, 14, 104. [Google Scholar] [CrossRef] [PubMed]
- Demidenko, Z.N.; Blagosklonny, M.V. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle 2008, 7, 3355–3361. [Google Scholar] [CrossRef]
- Kamitori, K.; Yamaguchi, F.; Dong, Y.; Hossain, A.; Katagi, A.; Noguchi, C.; Hirata, Y.; Tsukamoto, I.; Hatano, N.; Tokuda, M. Both Ser361 phosphorylation and the C-arrestin domain of thioredoxin interacting protein are important for cell cycle blockade at the G1/S checkpoint. FEBS Open Biol. 2018, 8, 1804–1819. [Google Scholar] [CrossRef]
- Hodge, R.D.; D’Ercole, A.J.; O’Kusky, J.R. Insulin-like growth factor-I accelerates the cell cycle by decreasing G1 phase length and increases cell cycle reentry in the embryonic cerebral cortex. J. Neurosci. 2004, 24, 10201–20210. [Google Scholar] [CrossRef]
- Ireland, L.; Santos, A.; Campbell, F.; Figueiredo, C.; Hammond, D.; Ellies, L.G.; Weyer-Czernilofsky, U.; Bogenrieder, T.; Schmid, M.; Mielgo, A. Blockade of insulin-like growth factors increases efficacy of paclitaxel in metastatic breast cancer. Oncogene 2018, 37, 2022–2036. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Itahana, K.; Acosta, M.; Campisi, J. Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Mol. Cell Biol. 2000, 20, 273–285. [Google Scholar] [CrossRef]
- Marinkovic, D.; Zhang, X.; Yalcin, S.; Luciano, J.P.; Brugnara, C.; Huber, T.; Ghaffari, S. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J. Clin. Investig. 2007, 117, 2133–2144. [Google Scholar] [CrossRef]
- Klotz, L.O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef]
- Crudden, C.; Girnita, A.; Girnita, L. Targeting the IGF-1R: The tale of the tortoise and the hare. Front. Endocrinol. 2015, 6, 64. [Google Scholar] [CrossRef]
- Paez-Ribes, M.; González-Gualda, E.; Doherty, G.J.; Muñoz-Espín, D. Targeting senescent cells in translational medicine. EMBO Mol. Med. 2019, 11, e10234. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Karin, O.; Agrawal, A.; Porat, Z.; Krizhanovsky, V.; Alon, U. Senescent cell turnover slows with age providing an explanation for the Gompertz law. Nat. Commun. 2019, 10, 5495. [Google Scholar] [CrossRef]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar]
- Chesnokova, V.; Melmed, S. GH and senescence: A new understanding of adult GH action. J. Endocr. Soc. 2022, 6, bvab177. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef]
- Pan, C.; Locasale, J.W. Targeting metabolism to influence aging. Science 2021, 371, 234–235. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef]
- Chan, L.N.; Chen, Z.; Braas, D.; Lee, J.W.; Xiao, G.; Geng, H.; Cosgun, K.N.; Hurtz, C.; Shojaee, S.; Cazzaniga, V.; et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature 2017, 542, 479–483. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Quax, W.J.; Demaria, M. Regulation of survival networks in senescent cells: From mechanisms to interventions. J. Mol. Biol. 2019, 431, 2629–2643. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 2017, 27, 2652–2660. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Moiseeva, O.; Bourdeau, V.; Roux, A.; Deschênes-Simard, X.; Ferbeyre, G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol. Cell Biol. 2009, 29, 4495–4507. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef]
- Perng, Y.C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439. [Google Scholar] [CrossRef]
- Freitas-Rodríguez, S.; Folgueras, A.R.; López-Otín, C. The role of matrix metalloproteinases in aging: Tissue remodeling and beyond. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2015–2025. [Google Scholar] [CrossRef]
- Quan, T.; Qin, Z.; Xia, W.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Matrix-degrading metalloproteinases in photoaging. J. Inv. Dermatol. Symp. Proc. 2009, 14, 20–24. [Google Scholar] [CrossRef]
- Gonzalez-Meljem, J.M.; Apps, J.R.; Fraser, H.C.; Martinez-Barbera, J.P. Paracrine roles of cellular senescence in promoting tumourigenesis. Br. J. Cancer 2018, 118, 1283–1288. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic potential of NAD-boosting molecules: The in vivo evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. The genetic/metabolic transformation concept of carcinogenesis. Cancer Metastasis Rev. 2012, 31, 123–130. [Google Scholar] [CrossRef]
- Weinberg, R.A. How cancer arises. Sci. Am. 1996, 275, 62–70. [Google Scholar] [CrossRef]
- Zhu, J.; Woods, D.; McMahon, M.; Bishop, J.M. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998, 12, 2997–3007. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, N.K.; Lim, H.J.; Ji, S.T.; Kim, Y.J.; Jang, W.B.; Kim, D.Y.; Kang, S.; Yun, J.; Ha, J.S.; et al. Pharmacological inhibition of mTOR attenuates replicative cell senescence and improves cellular function via regulating the STAT3-PIM1 axis in human cardiac progenitor cells. Exp. Mol. Med. 2020, 52, 615–628. [Google Scholar] [CrossRef]
- Xu, S.; Cai, Y.; Wei, Y. mTOR signaling from cellular senescence to organismal aging. Aging Dis. 2013, 5, 263–273. [Google Scholar] [CrossRef]
- Ye, Z.; Ayer, D.E. Ras suppresses TXNIP expression by restricting ribosome translocation. Mol. Cell Biol. 2018, 38, e00178. [Google Scholar] [CrossRef]
- Shen, L.; O’Shea, J.M.; Kaadige, M.R.; Cunha, S.; Wilde, B.R.; Cohen, A.L.; Welm, A.L.; Ayer, D.E. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc. Natl. Acad. Sci. USA 2015, 112, 5425–5430. [Google Scholar] [CrossRef]
- Kaadige, M.R.; Yang, J.; Wilde, B.R.; Ayer, D.E. MondoA-Mlx transcriptional activity is limited by mTOR-MondoA interaction. Mol. Cell Biol. 2015, 35, 101–110. [Google Scholar] [CrossRef]
- Nassar, M.; Tabib, Y.; Capucha, T.; Mizraji, G.; Nir, T.; Pevsner-Fischer, M.; Zilberman-Schapira, G.; Heyman, O.; Nussbaum, G.; Bercovier, H.; et al. GAS6 is a key homeostatic immunological regulator of host-commensal interactions in the oral mucosa. Proc. Natl. Acad. Sci. USA 2017, 114, E337–E346. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Zhang, Y.; Chen, Q.; Yan, K.; Han, D. Toll-like receptor-mediated inhibition of Gas6 and ProS expression facilitates inflammatory cytokine production in mouse macrophages. Immunology 2012, 135, 40–50. [Google Scholar] [CrossRef]
- Sanada, F.; Taniyama, Y.; Muratsu, J.; Otsu, R.; Shimizu, H.; Rakugi, H.; Morishita, R. IGF binding protein-5 induces cell senescence. Front. Endocrinol. 2018, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Isik, M.; Moroz, N.; Steinbaugh, M.J.; Zhang, P.; Blackwell, T.K. Dietary restriction extends lifespan through metabolic regulation of innate immunity. Cell Metab. 2019, 29, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Allard, J.B.; Duan, C. IGF-binding proteins: Why do they exist and why are there so many? Front. Endocrinol. 2018, 9, 117. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagaraj, K.; Sarfstein, R.; Laron, Z.; Werner, H. Long-Term IGF1 Stimulation Leads to Cellular Senescence via Functional Interaction with the Thioredoxin-Interacting Protein, TXNIP. Cells 2022, 11, 3260. https://doi.org/10.3390/cells11203260
Nagaraj K, Sarfstein R, Laron Z, Werner H. Long-Term IGF1 Stimulation Leads to Cellular Senescence via Functional Interaction with the Thioredoxin-Interacting Protein, TXNIP. Cells. 2022; 11(20):3260. https://doi.org/10.3390/cells11203260
Chicago/Turabian StyleNagaraj, Karthik, Rive Sarfstein, Zvi Laron, and Haim Werner. 2022. "Long-Term IGF1 Stimulation Leads to Cellular Senescence via Functional Interaction with the Thioredoxin-Interacting Protein, TXNIP" Cells 11, no. 20: 3260. https://doi.org/10.3390/cells11203260
APA StyleNagaraj, K., Sarfstein, R., Laron, Z., & Werner, H. (2022). Long-Term IGF1 Stimulation Leads to Cellular Senescence via Functional Interaction with the Thioredoxin-Interacting Protein, TXNIP. Cells, 11(20), 3260. https://doi.org/10.3390/cells11203260