
Neutral CB1 Receptor Antagonists as Pharmacotherapies for Substance Use Disorders: Rationale, Evidence, and Challenge


Abstract
:1. Introduction
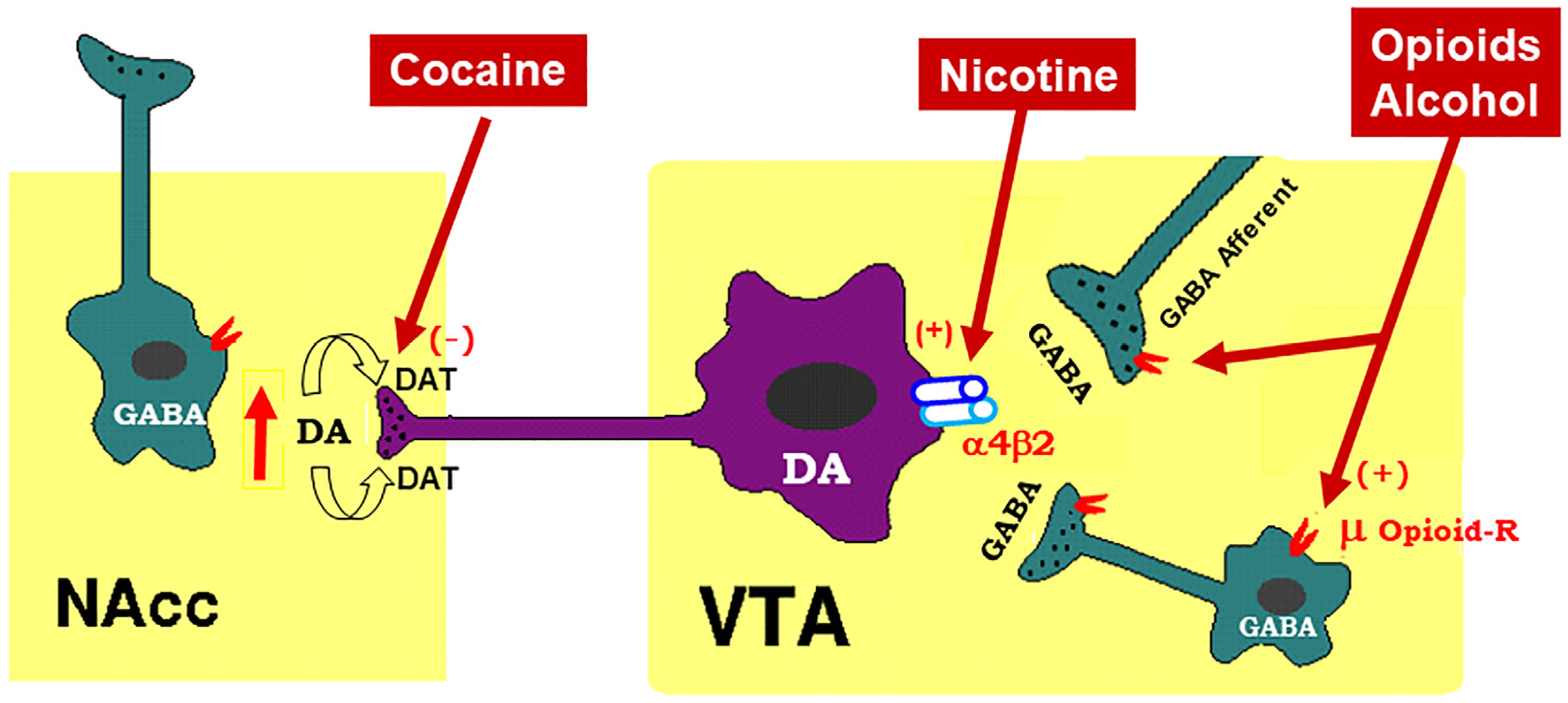
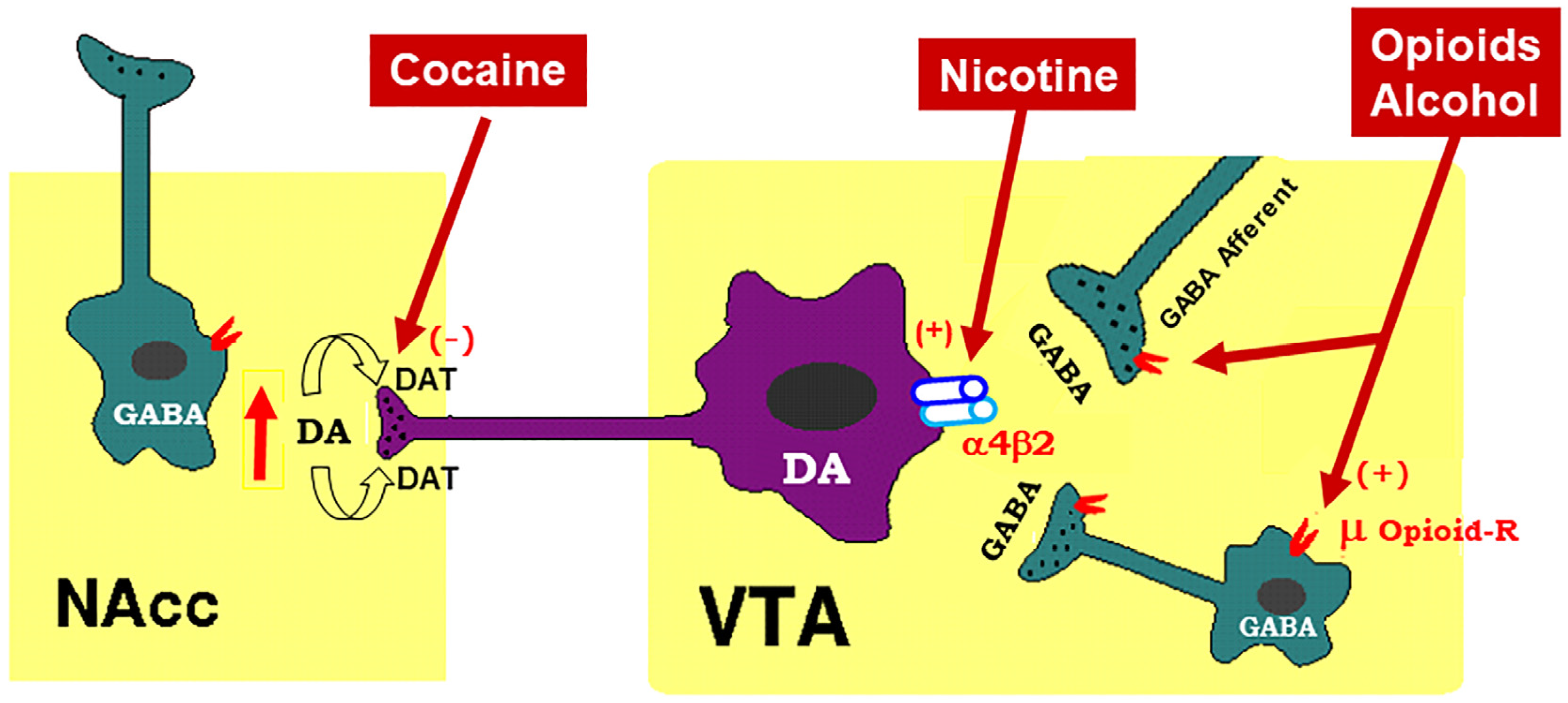
2. Mesocorticolimbic Dopamine System
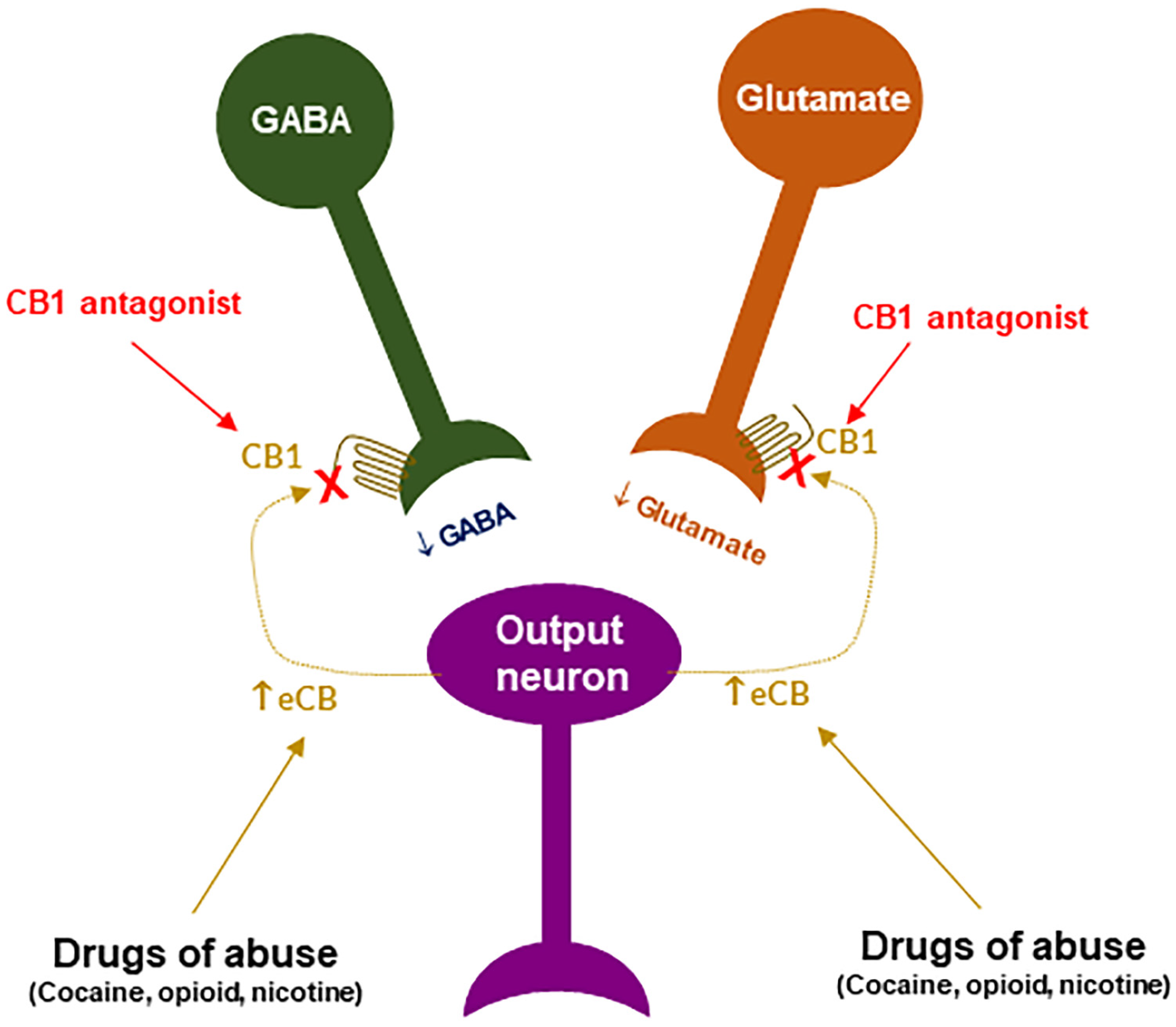
3. Endocannabinoid System
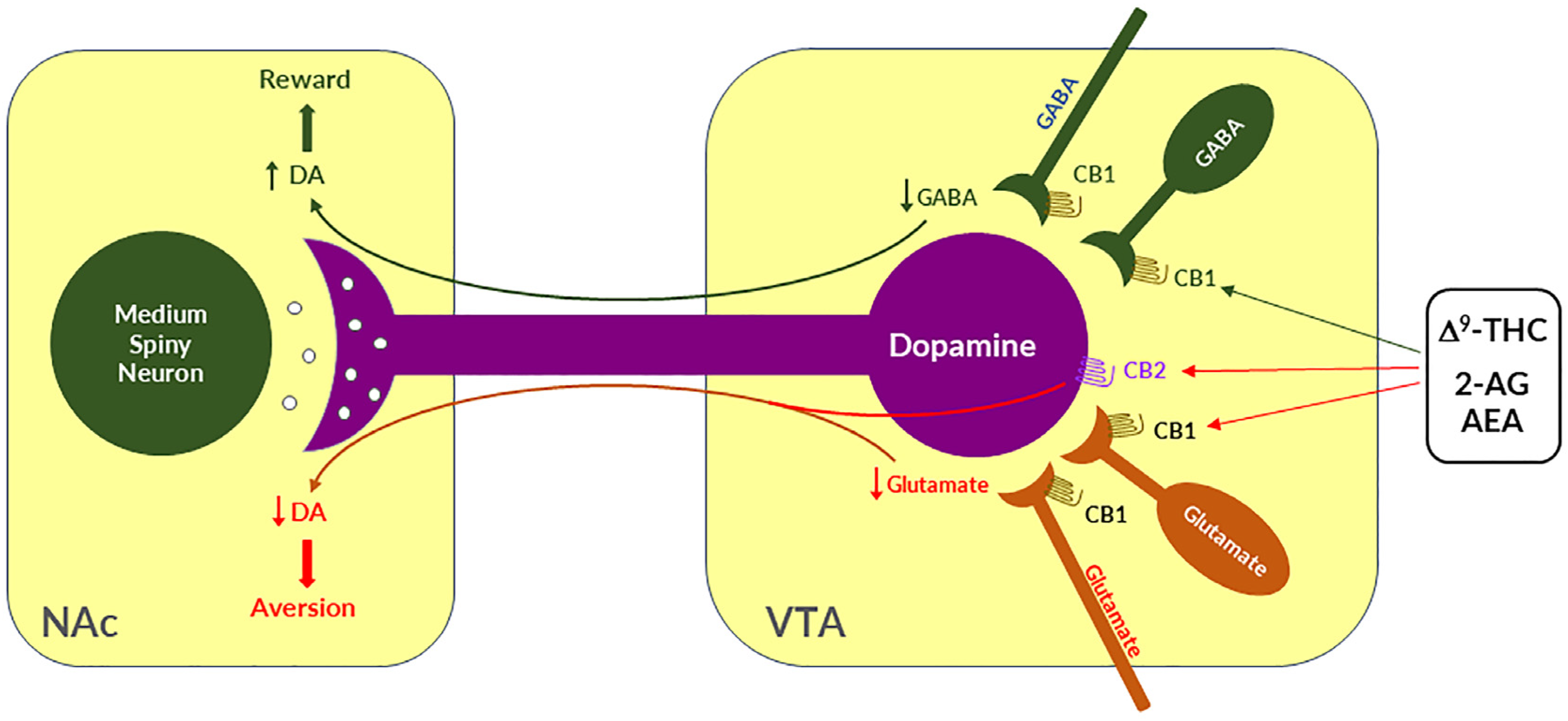
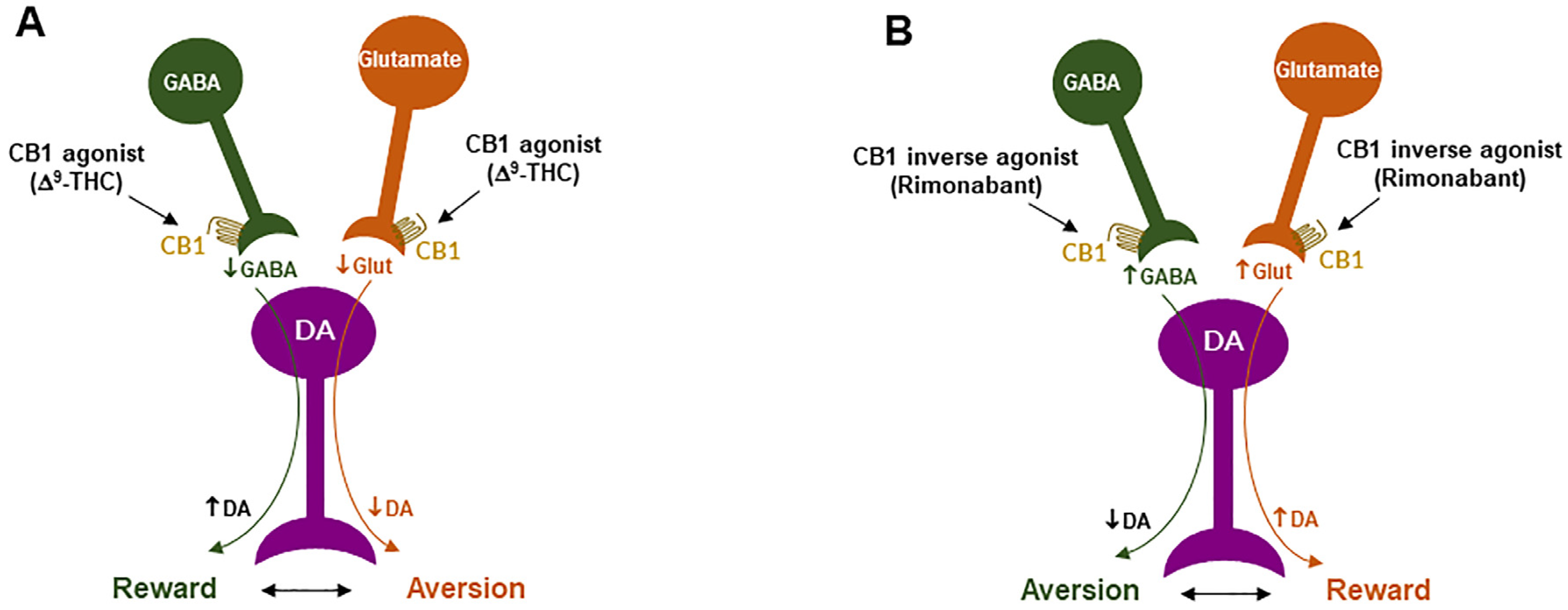
4. Cannabinoid Reward versus Aversion
4.1. GABAergic CB1R Hypothesis of Cannabis Reward
4.2. Glutamatergic CB1 Hypothesis of Cannabinoid Aversion
4.3. Dopaminergic CB2 Hypothesis of Cannabinoid Aversion
5. Rimonabant: The First CB1R Antagonist Approved for the Treatment of Obesity
6. CB1R Antagonists Are Promising for the Treatments of SUDs
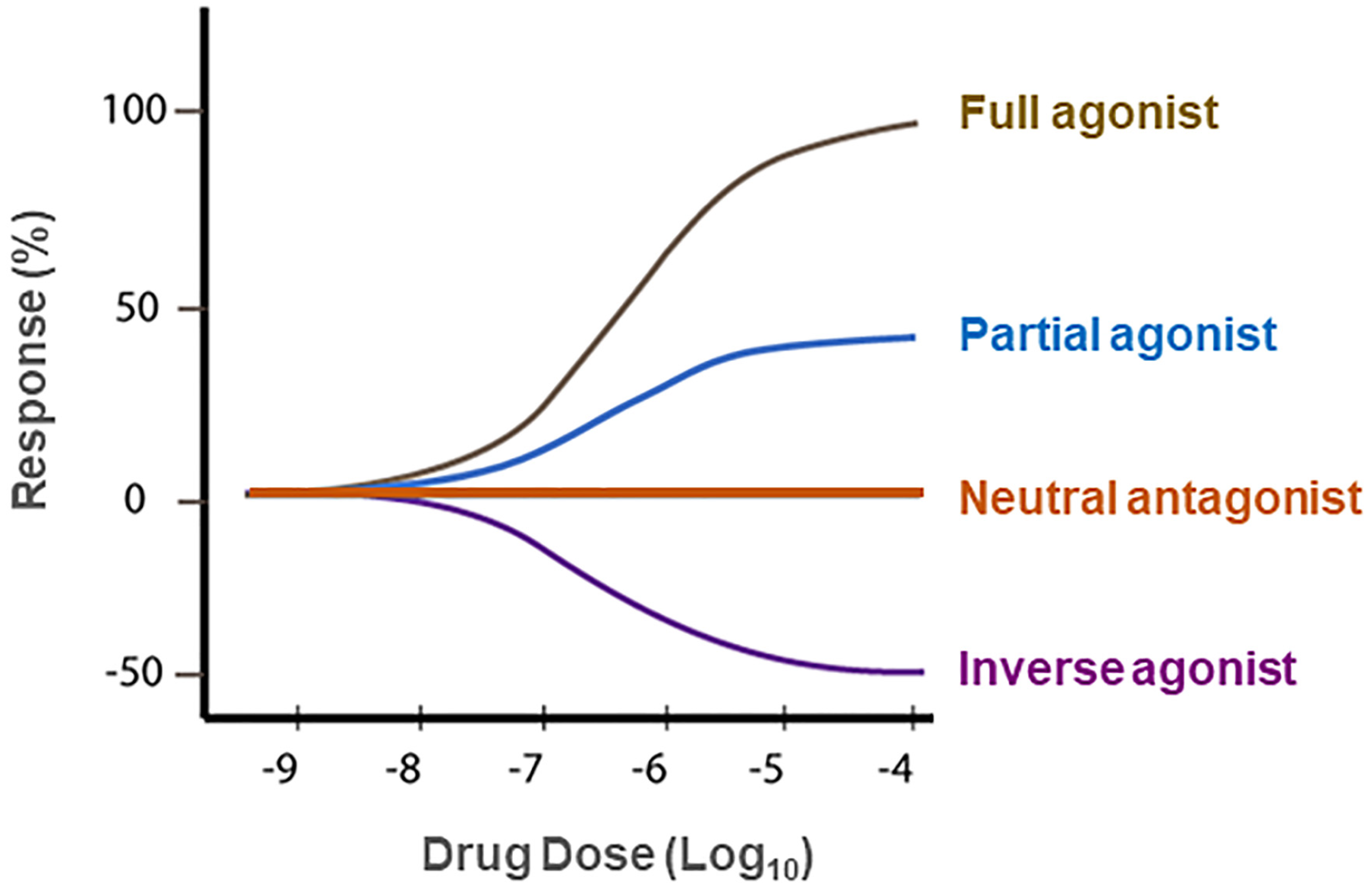
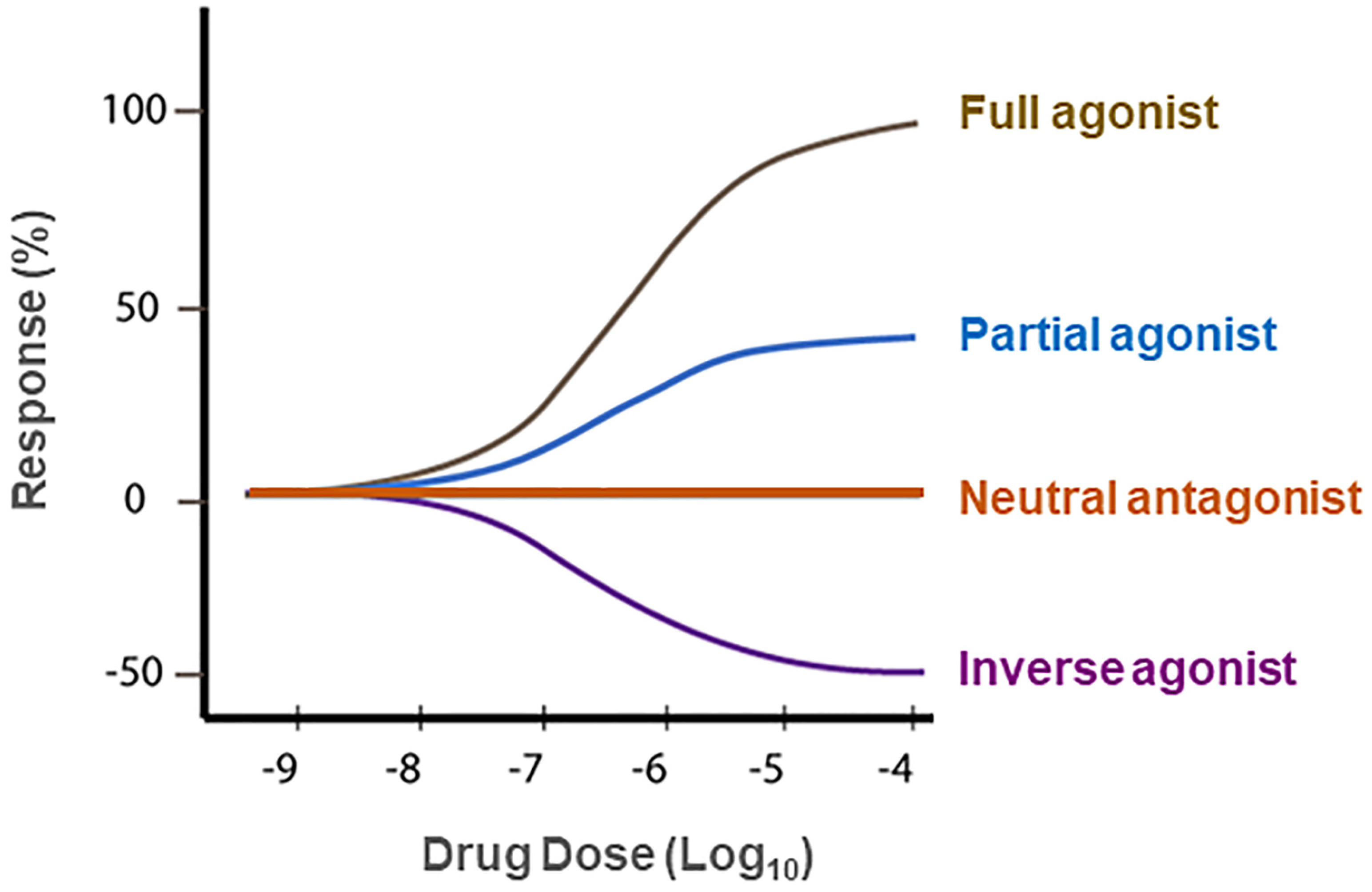
7. Neutral CB1R Antagonists as New Promising Therapies for SUDs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Doses | Species | Results | References |
|---|---|---|---|---|
| PIMSR | 10, 30 mg/kg | Rat | ↓ Cocaine self-administration (FR2, FR5, PR) | [13] |
| PIMSR | 3, 10, 30 mg/kg, i.p. | Rat | ↓ Cocaine-cue-induced reinstatement | [13] |
| PIMSR | 3, 10, 30 mg/kg, i.p. | Rat | ↓ Cocaine-enhanced electrical brain-stimulation reward | [13] |
| PIMSR | 10, 30 mg/kg | Mouse | Not produce CPP or CPA by itself | [13] |
| PIMSR | 3, 10, 30 mg/kg, i.p. | Rat | Not alter electrical brain-stimulation reward by itself | [13] |
| AM4113 | 3, 10 mg/kg, i.p. | Rat | Not alter cocaine self-administration | [139] |
| AM4113 | 0.3–3 mg/kg, i.m. | Monkey | Not alter cocaine self-administration | [160] |
| AM4113 | 0.3, 1, 3, 10 mg/kg, i.p. | Rat | ↓ PR cocaine self-administration | [158] |
| AM4113 | 0.3–3 mg/kg, i.m. | Monkey | ↓ Cue-induced cocaine seeking | [160] |
| AM4113 | 0.3–3 mg/kg, i.m. | Monkey | ↓ Cocaine-primed drug seeking | [160] |
| AM4113 | 3, 10 mg/kg, i.p. | Rat | ↓ Methamphetamine self-administration | [139] |
| AM4113 | 0.3, 1, 3, 10 mg/kg, i.p. | Rat | ↓ Nicotine self-administration | [158] |
| AM4113 | 0.3–3 mg/kg, i.m. | Monkey | ↓ Nicotine self-administration | [160] |
| AM4113 | 0.3, 1, 3, 10 mg/kg, i.p.; | Rat | ↓ Cue-induced nicotine seeking | [158] |
| AM4113 | 0.3–3 mg/kg, i.m. | Monkey | ↓ Cue-induced nicotine seeking | [160] |
| AM4113 | 0.3–3 mg/kg, i.m. | Monkey | ↓ Drug priming-induced nicotine seeking | [160] |
| AM4113 | 0.3–3 mg/kg, i.m. | Monkey | ↓ Δ9-THC self-administration | [160] |
| AM4113 | 0.3–3 mg/kg, i.m. | Monkey | ↓ Cue-induced Δ9-THC seeking | [160] |
| AM4113 | 0.3–3 mg/kg, i.m. | Monkey | ↓ Drug priming-induced Δ9-THC seeking | [160] |
| AM4113 | 3, 10 mg/kg, i.p. | Rat | ↓ Heroin self-administration | [139] |
| AM4113 | 1, 2.5 mg/kg, i.p. | Rat | ↓ Naloxone-precipitated CPA | [166] |
| AM4113 | 3, 10 mg/kg, i.p. | Rat | No effect on electrical brain-stimulation reward by itself | [139] |
| AM6527 | 1, 2.5 mg/kg, i.p. | Rat | ↓ Naloxone-precipitated CPA | [166] |
| Δ8-THCV | 10, 20 mg/kg, i.p. | Rat | ↓ Nicotine self-administration | [174] |
| Δ8-THCV | 0.03~3 mg/kg, i.p. | Mouse | ↓ Nicotine-induced CPP | [174] |
| Δ8-THCV | 10, 20 mg/kg, i.p. | Rat | ↓ Nicotine- or cue-induced nicotine seeking | [174] |
| Δ8-THCV | 0.3 mg/kg, i.p. | Mouse | ↓ Nicotine withdrawal-induced somatic signs | [174] |
8. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Rudd, R.A.; Aleshire, N.; Zibbell, J.E.; Gladden, R.M. Increases in drug and opioid overdose deaths—United States, 2000–2014. MMWR Morb. Mortal Wkly. Rep. 2016, 64, 1378–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CDC (Center for Disease Control and Prevention). Drug over Dose Deaths; CDC: Atlanta, GA, USA, 2017.
- Jordan, C.J.; Xi, Z.X. Progress in brain cannabinoid CB2 receptor research: From genes to behavior. Neurosci. Biobehav. Rev. 2019, 98, 208–220. [Google Scholar] [CrossRef]
- Jordan, C.J.; Xi, Z.X. Discovery and development of varenicline for smoking cessation. Expert Opin. Drug Discov. 2018, 13, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Galaj, E.; Newman, A.H.; Xi, Z.X. Dopamine D3 receptor-based medication development for the treatment of opioid use disorder: Rationale, progress, and challenges. Neurosci. Biobehav. Rev. 2020, 114, 38–52. [Google Scholar] [CrossRef]
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773. [Google Scholar] [CrossRef]
- Galaj, E.; Xi, Z.X. Potential of Cannabinoid Receptor Ligands as Treatment for Substance Use Disorders. CNS Drugs 2019, 33, 1001–1030. [Google Scholar] [CrossRef] [PubMed]
- Le Foll, B.; Goldberg, S.R. Cannabinoid CB1 receptor antagonists as promising new medications for drug dependence. J. Pharmacol. Exp. Ther. 2005, 312, 875–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, M.E.; Gowin, J.L.; Ramchandani, V.A.; Hurd, Y.L.; Le Foll, B. The endocannabinoid system as a target for addiction treatment: Trials and tribulations. Neuropharmacology 2017, 124, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.; Le Foll, B. Novel therapeutic and drug development strategies for tobacco use disorder: Endocannabinoid modulation. Expert. Opin. Drug Discov. 2020, 15, 1065–1080. [Google Scholar] [CrossRef]
- Le Foll, B.; Gorelick, D.A.; Goldberg, S.R. The future of endocannabinoid-oriented clinical research after CB1 antagonists. Psychopharmacology 2009, 205, 171–174. [Google Scholar] [CrossRef]
- Compton, W.M.; Wargo, E.M.; Volkow, N.D. Neuropsychiatric Model of Addiction Simplified. Psychiatr. Clin. N. Am. 2022, 45, 321–334. [Google Scholar] [CrossRef]
- Galaj, E.; Hempel, B.; Moore, A.; Klein, B.; Bi, G.H.; Gardner, E.L.; Seltzman, H.H.; Xi, Z.X. Therapeutic potential of PIMSR, a novel CB1 receptor neutral antagonist, for cocaine use disorder: Evidence from preclinical research. Transl. Psychiatry 2022, 12, 286. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Wise, R.A.; Baler, R. The dopamine motive system: Implications for drug and food addiction. Nat. Rev. Neurosci. 2017, 18, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.X.; Gardner, E.L. Hypothesis-driven medication discovery for the treatment of psychostimulant addiction. Curr. Drug Abuse Rev. 2008, 1, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.X.; Spiller, K.; Gardner, E.L. Mechanism-based medication development for the treatment of nicotine dependence. Acta Pharmacol. Sin. 2009, 30, 723–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, C.J.; Cao, J.; Newman, A.H.; Xi, Z.X. Progress in agonist therapy for substance use disorders: Lessons learned from methadone and buprenorphine. Neuropharmacology 2019, 158, 107609. [Google Scholar] [CrossRef] [PubMed]
- Wise, R.A. Brain reward circuitry: Insights from unsensed incentives. Neuron 2002, 36, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Ron, D.; Wang, J. The NMDA Receptor and Alcohol Addiction. In Biology of the NMDA Receptor; Van Dongen, A.M., Ed.; Frontiers in Neuroscience: Boca Raton, FL, USA, 2009; Chapter 4. [Google Scholar]
- Berrettini, W. Alcohol addiction and the mu-opioid receptor. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 65, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Di Chiara, G. Alcohol and dopamine. Alcohol Health Res. World 1997, 21, 108–114. [Google Scholar] [PubMed]
- Galaj, E.; Xi, Z.X. Progress in opioid reward research: From a canonical two-neuron hypothesis to two neural circuits. Pharmacol. Biochem. Behav. 2021, 200, 173072. [Google Scholar] [CrossRef] [PubMed]
- Hempel, B.; Xi, Z.X. Receptor mechanisms underlying the CNS effects of cannabinoids: CB1 receptor and beyond. Adv. Pharmacol. 2022, 93, 275–333. [Google Scholar] [CrossRef] [PubMed]
- Mackie, K. Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb. Exp. Pharmacol. 2005, 168, 299–325. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- McPartland, J.M.; Glass, M.; Pertwee, R.G. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: Interspecies differences. Br. J. Pharmacol. 2007, 152, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Hillard, C.J.; Edgemond, W.S.; Jarrahian, A.; Campbell, W.B. Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J. Neurochem. 1997, 69, 631–638. [Google Scholar] [CrossRef]
- Sugiura, T.; Waku, K. 2-Arachidonoylglycerol and the cannabinoid receptors. Chem. Phys. Lipids 2000, 108, 89–106. [Google Scholar] [CrossRef]
- Trangenstein, P.J.; Whitehill, J.M.; Jenkins, M.C.; Jernigan, D.H.; Moreno, M.A. Cannabis Marketing and Problematic Cannabis Use Among Adolescents. J. Stud. Alcohol Drugs 2021, 82, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Fattore, L.; Fadda, P.; Spano, M.S.; Pistis, M.; Fratta, W. Neurobiological mechanisms of cannabinoid addiction. Mol. Cell Endocrinol. 2008, 286, S97–S107. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, R.; Valverde, O.; Berrendero, F. Involvement of the endocannabinoid system in drug addiction. Trends Neurosci. 2006, 29, 225–232. [Google Scholar] [CrossRef]
- NSDUH. National Survey on Drug Use and Health. 2019. Available online: https://datafiles.samhsa.gov/ (accessed on 20 August 2022).
- D’Souza, D.C.; Perry, E.; MacDougall, L.; Ammerman, Y.; Cooper, T.; Wu, Y.T.; Braley, G.; Gueorguieva, R.; Krystal, J.H. The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: Implications for psychosis. Neuropsychopharmacology 2004, 29, 1558–1572. [Google Scholar] [CrossRef]
- Raft, D.; Gregg, J.; Ghia, J.; Harris, L. Effects of intravenous tetrahydrocannabinol on experimental and surgical pain. Psychological correlates of the analgesic response. Clin. Pharmacol. Ther. 1977, 21, 26–33. [Google Scholar] [CrossRef]
- Lac, A.; Luk, J.W. Testing the Amotivational Syndrome: Marijuana Use Longitudinally Predicts Lower Self-Efficacy Even After Controlling for Demographics, Personality, and Alcohol and Cigarette Use. Prev. Sci. 2018, 19, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Petrucci, A.S.; LaFrance, E.M.; Cuttler, C. A Comprehensive Examination of the Links between Cannabis Use and Motivation. Subst. Use Misuse 2020, 55, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Barch, D.M.; Dowd, E.C. Goal representations and motivational drive in schizophrenia: The role of prefrontal-striatal interactions. Schizophr. Bull. 2010, 36, 919–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, C.H.; Glazer, J.E.; Lee, R.; Nusslock, R.; de Wit, H. Delta9-THC reduces reward-related brain activity in healthy adults. Psychopharmacology 2022, 239, 2829–2840. [Google Scholar] [CrossRef] [PubMed]
- van Hell, H.H.; Jager, G.; Bossong, M.G.; Brouwer, A.; Jansma, J.M.; Zuurman, L.; van Gerven, J.; Kahn, R.S.; Ramsey, N.F. Involvement of the endocannabinoid system in reward processing in the human brain. Psychopharmacology 2012, 219, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Jansma, J.M.; van Hell, H.H.; Vanderschuren, L.J.; Bossong, M.G.; Jager, G.; Kahn, R.S.; Ramsey, N.F. THC reduces the anticipatory nucleus accumbens response to reward in subjects with a nicotine addiction. Transl. Psychiatry 2013, 3, e234. [Google Scholar] [CrossRef] [Green Version]
- Lawn, W.; Freeman, T.P.; Pope, R.A.; Joye, A.; Harvey, L.; Hindocha, C.; Mokrysz, C.; Moss, A.; Wall, M.B.; Bloomfield, M.A.; et al. Acute and chronic effects of cannabinoids on effort-related decision-making and reward learning: An evaluation of the cannabis ‘amotivational’ hypotheses. Psychopharmacology 2016, 233, 3537–3552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, T.P.; Pope, R.A.; Wall, M.B.; Bisby, J.A.; Luijten, M.; Hindocha, C.; Mokrysz, C.; Lawn, W.; Moss, A.; Bloomfield, M.A.P.; et al. Cannabis Dampens the Effects of Music in Brain Regions Sensitive to Reward and Emotion. Int. J. Neuropsychopharmacol. 2018, 21, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Bloomfield, M.A.P.; Hindocha, C.; Green, S.F.; Wall, M.B.; Lees, R.; Petrilli, K.; Costello, H.; Ogunbiyi, M.O.; Bossong, M.G.; Freeman, T.P. The neuropsychopharmacology of cannabis: A review of human imaging studies. Pharmacol. Ther. 2019, 195, 132–161. [Google Scholar] [CrossRef]
- Justinova, Z.; Tanda, G.; Redhi, G.H.; Goldberg, S.R. Self-administration of delta9-tetrahydrocannabinol (THC) by drug naive squirrel monkeys. Psychopharmacology 2003, 169, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Tanda, G.; Munzar, P.; Goldberg, S.R. Self-administration behavior is maintained by the psychoactive ingredient of marijuana in squirrel monkeys. Nat. Neurosci. 2000, 3, 1073–1074. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.T.; Waters, W.; McLendon, D. Evaluation of reinforcing capability of delta-9-tetrahydrocannabinol in rhesus monkeys. Psychopharmacologia 1974, 37, 23–29. [Google Scholar] [CrossRef] [PubMed]
- John, W.S.; Martin, T.J.; Nader, M.A. Behavioral Determinants of Cannabinoid Self-Administration in Old World Monkeys. Neuropsychopharmacology 2017, 42, 1522–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansbach, R.S.; Nicholson, K.L.; Martin, B.R.; Balster, R.L. Failure of Delta(9)-tetrahydrocannabinol and CP 55,940 to maintain intravenous self-administration under a fixed-interval schedule in rhesus monkeys. Behav. Pharmacol. 1994, 5, 219–225. [Google Scholar] [CrossRef]
- Panagis, G.; Vlachou, S.; Nomikos, G.G. Behavioral pharmacology of cannabinoids with a focus on preclinical models for studying reinforcing and dependence-producing properties. Curr. Drug Abuse Rev. 2008, 1, 350–374. [Google Scholar] [CrossRef]
- Vlachou, S.; Panagis, G. Regulation of brain reward by the endocannabinoid system: A critical review of behavioral studies in animals. Curr. Pharm. Des. 2014, 20, 2072–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, S.; Neuhofer, D.; Chioma, V.C.; Garcia-Keller, C.; Schwartz, D.J.; Allen, N.; Scofield, M.D.; Ortiz-Ithier, T.; Kalivas, P.W. A Model of Delta(9)-Tetrahydrocannabinol Self-administration and Reinstatement That Alters Synaptic Plasticity in Nucleus Accumbens. Biol. Psychiatry 2018, 84, 601–610. [Google Scholar] [CrossRef]
- Neuhofer, D.; Spencer, S.M.; Chioma, V.C.; Beloate, L.N.; Schwartz, D.; Kalivas, P.W. The loss of NMDAR-dependent LTD following cannabinoid self-administration is restored by positive allosteric modulation of CB1 receptors. Addict. Biol. 2020, 25, e12843. [Google Scholar] [CrossRef]
- Cheer, J.F.; Kendall, D.A.; Marsden, C.A. Cannabinoid receptors and reward in the rat: A conditioned place preference study. Psychopharmacology 2000, 151, 25–30. [Google Scholar] [CrossRef] [PubMed]
- DeVuono, M.V.; Wills, K.L.; MacPherson, D.V.; Hrelja, K.M.; Parker, L.A. Effect of footshock stress on place conditioning produced by Delta(9)-tetrahydrocannabinol and the fatty acid amide hydrolase (FAAH) inhibitor, URB597, in Sprague-Dawley rats. Psychopharmacology 2017, 234, 3229–3240. [Google Scholar] [CrossRef] [PubMed]
- Braida, D.; Iosue, S.; Pegorini, S.; Sala, M. Delta9-tetrahydrocannabinol-induced conditioned place preference and intracerebroventricular self-administration in rats. Eur. J. Pharmacol. 2004, 506, 63–69. [Google Scholar] [CrossRef]
- Lepore, M.; Liu, X.; Savage, V.; Matalon, D.; Gardner, E.L. Genetic differences in delta 9-tetrahydrocannabinol-induced facilitation of brain stimulation reward as measured by a rate-frequency curve-shift electrical brain stimulation paradigm in three different rat strains. Life Sci. 1996, 58, PL365–PL372. [Google Scholar] [CrossRef]
- Gardner, E.L.; Paredes, W.; Smith, D.; Donner, A.; Milling, C.; Cohen, D.; Morrison, D. Facilitation of brain stimulation reward by delta 9-tetrahydrocannabinol. Psychopharmacology 1988, 96, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Katsidoni, V.; Kastellakis, A.; Panagis, G. Biphasic effects of Delta9-tetrahydrocannabinol on brain stimulation reward and motor activity. Int. J. Neuropsychopharmacol. 2013, 16, 2273–2284. [Google Scholar] [CrossRef] [Green Version]
- Kwilasz, A.J.; Negus, S.S. Dissociable effects of the cannabinoid receptor agonists Delta9-tetrahydrocannabinol and CP55940 on pain-stimulated versus pain-depressed behavior in rats. J. Pharmacol. Exp. Ther. 2012, 343, 389–400. [Google Scholar] [CrossRef] [Green Version]
- Negus, S.S.; Miller, L.L. Intracranial self-stimulation to evaluate abuse potential of drugs. Pharmacol. Rev. 2014, 66, 869–917. [Google Scholar] [CrossRef] [Green Version]
- Vlachou, S.; Nomikos, G.G.; Stephens, D.N.; Panagis, G. Lack of evidence for appetitive effects of Delta 9-tetrahydrocannabinol in the intracranial self-stimulation and conditioned place preference procedures in rodents. Behav. Pharmacol. 2007, 18, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Wiebelhaus, J.M.; Grim, T.W.; Owens, R.A.; Lazenka, M.F.; Sim-Selley, L.J.; Abdullah, R.A.; Niphakis, M.J.; Vann, R.E.; Cravatt, B.F.; Wiley, J.L.; et al. Delta9-tetrahydrocannabinol and endocannabinoid degradative enzyme inhibitors attenuate intracranial self-stimulation in mice. J. Pharmacol. Exp. Ther. 2015, 352, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Spiller, K.J.; Bi, G.H.; He, Y.; Galaj, E.; Gardner, E.L.; Xi, Z.X. Cannabinoid CB1 and CB2 receptor mechanisms underlie cannabis reward and aversion in rats. Br. J. Pharmacol. 2019, 176, 1268–1281. [Google Scholar] [CrossRef]
- Han, X.; He, Y.; Bi, G.H.; Zhang, H.Y.; Song, R.; Liu, Q.R.; Egan, J.M.; Gardner, E.L.; Li, J.; Xi, Z.X. CB1 Receptor Activation on VgluT2-Expressing Glutamatergic Neurons Underlies Delta(9)-Tetrahydrocannabinol (Delta(9)-THC)-Induced Aversive Effects in Mice. Sci. Rep. 2017, 7, 12315. [Google Scholar] [CrossRef] [Green Version]
- Humburg, B.A.; Jordan, C.J.; Zhang, H.Y.; Shen, H.; Han, X.; Bi, G.H.; Hempel, B.; Galaj, E.; Baumann, M.H.; Xi, Z.X. Optogenetic brain-stimulation reward: A new procedure to re-evaluate the rewarding versus aversive effects of cannabinoids in dopamine transporter-Cre mice. Addict. Biol. 2021, 26, e13005. [Google Scholar] [CrossRef] [PubMed]
- Cheer, J.F.; Kendall, D.A.; Mason, R.; Marsden, C.A. Differential cannabinoid-induced electrophysiological effects in rat ventral tegmentum. Neuropharmacology 2003, 44, 633–641. [Google Scholar] [CrossRef]
- Cheer, J.F.; Wassum, K.M.; Heien, M.L.; Phillips, P.E.; Wightman, R.M. Cannabinoids enhance subsecond dopamine release in the nucleus accumbens of awake rats. J. Neurosci. 2004, 24, 4393–4400. [Google Scholar] [CrossRef] [Green Version]
- Tanda, G.; Pontieri, F.E.; Di Chiara, G. Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science 1997, 276, 2048–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covey, D.P.; Mateo, Y.; Sulzer, D.; Cheer, J.F.; Lovinger, D.M. Endocannabinoid modulation of dopamine neurotransmission. Neuropharmacology 2017, 124, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.L.; Shobin, E.; Pickel, V.M. Cannabinoid modulation of the dopaminergic circuitry: Implications for limbic and striatal output. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 38, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paladini, C.A.T. Neurophysiology of substantia nigra dopamine neurons: Modulation by GABA and glutamate. In Handbook of Basal Ganglia Structure and Function; Elsevier: Amsterdam, The Netherlands, 2017; pp. 335–360. [Google Scholar]
- Szabo, B.; Muller, T.; Koch, H. Effects of cannabinoids on dopamine release in the corpus striatum and the nucleus accumbens in vitro. J. Neurochem. 1999, 73, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Castañeda, E.M.; Oddie, S.D.; Whishaw, Q. THC does not affect striatal dopamine release: Microdialysis in freely moving rats. Pharmacol. Biochem. Behav. 1991, 40, 587–591. [Google Scholar] [CrossRef]
- Pillolla, G.; Melis, M.; Perra, S.; Muntoni, A.L.; Gessa, G.L.; Pistis, M. Medial forebrain bundle stimulation evokes endocannabinoid-mediated modulation of ventral tegmental area dopamine neuron firing in vivo. Psychopharmacology 2007, 191, 843–853. [Google Scholar] [CrossRef]
- Sidlo, Z.; Reggio, P.H.; Rice, M.E. Inhibition of striatal dopamine release by CB1 receptor activation requires nonsynaptic communication involving GABA, H2O2, and KATP channels. Neurochem. Int. 2008, 52, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Hempel, B.J.; Yang, H.J.; Han, X.; Bi, G.H.; Gardner, E.L.; Xi, Z.X. Dissecting the role of CB1 and CB2 receptors in cannabinoid reward versus aversion using transgenic CB1- and CB2-knockout mice. Eur. Neuropsychopharmacol. 2021, 43, 38–51. [Google Scholar] [CrossRef]
- Lupica, C.R.; Riegel, A.C. Endocannabinoid release from midbrain dopamine neurons: A potential substrate for cannabinoid receptor antagonist treatment of addiction. Neuropharmacology 2005, 48, 1105–1116. [Google Scholar] [CrossRef]
- Szabo, B.; Siemes, S.; Wallmichrath, I. Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Eur. J. Neurosci. 2002, 15, 2057–2061. [Google Scholar] [CrossRef]
- Melis, M.; Pistis, M. Endocannabinoid signaling in midbrain dopamine neurons: More than physiology? Curr. Neuropharmacol. 2007, 5, 268–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melis, M.; Sagheddu, C.; De Felice, M.; Casti, A.; Madeddu, C.; Spiga, S.; Muntoni, A.L.; Mackie, K.; Marsicano, G.; Colombo, G.; et al. Enhanced endocannabinoid-mediated modulation of rostromedial tegmental nucleus drive onto dopamine neurons in Sardinian alcohol-preferring rats. J. Neurosci. 2014, 34, 12716–12724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.F.; Galaj, E.; Bi, G.H.; Zhang, C.; He, Y.; Zhan, J.; Bauman, M.H.; Gardner, E.L.; Xi, Z.X. Different receptor mechanisms underlying phytocannabinoid- versus synthetic cannabinoid-induced tetrad effects: Opposite roles of CB1 /CB2 versus GPR55 receptors. Br. J. Pharmacol. 2020, 177, 1865–1880. [Google Scholar] [CrossRef] [PubMed]
- Manzanares, J.; Cabanero, D.; Puente, N.; Garcia-Gutierrez, M.S.; Grandes, P.; Maldonado, R. Role of the endocannabinoid system in drug addiction. Biochem. Pharmacol. 2018, 157, 108–121. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.Y.; Gao, M.; Liu, Q.R.; Bi, G.H.; Li, X.; Yang, H.J.; Gardner, E.L.; Wu, J.; Xi, Z.X. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E5007–E5015. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.Y.; Gao, M.; Shen, H.; Bi, G.H.; Yang, H.J.; Liu, Q.R.; Wu, J.; Gardner, E.L.; Bonci, A.; Xi, Z.X. Expression of functional cannabinoid CB2 receptor in VTA dopamine neurons in rats. Addict. Biol. 2017, 22, 752–765. [Google Scholar] [CrossRef]
- Xi, Z.X.; Peng, X.Q.; Li, X.; Song, R.; Zhang, H.Y.; Liu, Q.R.; Yang, H.J.; Bi, G.H.; Li, J.; Gardner, E.L. Brain cannabinoid CB(2) receptors modulate cocaine’s actions in mice. Nat. Neurosci. 2011, 14, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, D.J.; Wilson, J.M.; Remke, D.H.; Mahmood, M.S.; Uddin, M.J.; Wess, J.; Patel, S.; Marnett, L.J.; Niswender, C.M.; Jones, C.K.; et al. Antipsychotic-like Effects of M4 Positive Allosteric Modulators Are Mediated by CB2 Receptor-Dependent Inhibition of Dopamine Release. Neuron 2016, 91, 1244–1252. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.Y.; Bi, G.H.; Li, X.; Li, J.; Qu, H.; Zhang, S.J.; Li, C.Y.; Onaivi, E.S.; Gardner, E.L.; Xi, Z.X.; et al. Species differences in cannabinoid receptor 2 and receptor responses to cocaine self-administration in mice and rats. Neuropsychopharmacology 2015, 40, 1037–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aracil-Fernandez, A.; Trigo, J.M.; Garcia-Gutierrez, M.S.; Ortega-Alvaro, A.; Ternianov, A.; Navarro, D.; Robledo, P.; Berbel, P.; Maldonado, R.; Manzanares, J. Decreased cocaine motor sensitization and self-administration in mice overexpressing cannabinoid CB(2) receptors. Neuropsychopharmacology 2012, 37, 1749–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delis, F.; Polissidis, A.; Poulia, N.; Justinova, Z.; Nomikos, G.G.; Goldberg, S.R.; Antoniou, K. Attenuation of Cocaine-Induced Conditioned Place Preference and Motor Activity via Cannabinoid CB2 Receptor Agonism and CB1 Receptor Antagonism in Rats. Int. J. Neuropsychopharmacol. 2017, 20, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azar, S.; Sherf-Dagan, S.; Nemirovski, A.; Webb, M.; Raziel, A.; Keidar, A.; Goitein, D.; Sakran, N.; Shibolet, O.; Tam, J.; et al. Circulating Endocannabinoids Are Reduced Following Bariatric Surgery and Associated with Improved Metabolic Homeostasis in Humans. Obes. Surg. 2019, 29, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, A.M.; Piscitelli, F.; Dalle Grave, R.; El Ghoch, M.; Di Marzo, V.; Maj, M.; Monteleone, P. Peripheral Endocannabinoid Responses to Hedonic Eating in Binge-Eating Disorder. Nutrients 2017, 9, 1377. [Google Scholar] [CrossRef] [Green Version]
- van Eyk, H.J.; van Schinkel, L.D.; Kantae, V.; Dronkers, C.E.A.; Westenberg, J.J.M.; de Roos, A.; Lamb, H.J.; Jukema, J.W.; Harms, A.C.; Hankemeier, T.; et al. Caloric restriction lowers endocannabinoid tonus and improves cardiac function in type 2 diabetes. Nutr. Diabetes 2018, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, J.M.; Davis, S.N. Endocannabinoid system overactivity and the metabolic syndrome: Prospects for treatment. Curr. Diab. Rep. 2008, 8, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Duffy, D.; Rader, D. Endocannabinoid antagonism: Blocking the excess in the treatment of high-risk abdominal obesity. Trends Cardiovasc. Med. 2007, 17, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi-Carmona, M.; Barth, F.; Heaulme, M.; Shire, D.; Calandra, B.; Congy, C.; Martinez, S.; Maruani, J.; Neliat, G.; Caput, D.; et al. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994, 350, 240–244. [Google Scholar] [CrossRef] [Green Version]
- EMA. European Public Assessment Report (EPAR) Acomplia: EPAR Summary for the Public; EMA: London, UK, 2007. [Google Scholar]
- Di Marzo, V.; Despres, J.P. CB1 antagonists for obesity—What lessons have we learned from rimonabant? Nat. Rev. Endocrinol. 2009, 5, 633–638. [Google Scholar] [CrossRef]
- Christensen, R.; Kristensen, P.K.; Bartels, E.M.; Bliddal, H.; Astrup, A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet 2007, 370, 1706–1713. [Google Scholar] [CrossRef]
- Sam, A.H.; Salem, V.; Ghatei, M.A. Rimonabant: From RIO to Ban. J. Obes. 2011, 2011, 432607. [Google Scholar] [CrossRef] [Green Version]
- EMA. European Medicines Agency: Public Statement on Acomplia (Rimonabant) Withdrawal of the Marketing Authorisation in the European Union; EMA: London, UK, 2009. [Google Scholar]
- Saul, S. F.D.A. Panel Rejects Drug for Obesity. The New York Times. 2007. Available online: https://www.nytimes.com/2007/06/14/business/14drugs.html (accessed on 20 August 2022).
- Nguyen, T.; Thomas, B.F.; Zhang, Y. Overcoming the Psychiatric Side Effects of the Cannabinoid CB1 Receptor Antagonists: Current Approaches for Therapeutics Development. Curr. Top. Med. Chem. 2019, 19, 1418–1435. [Google Scholar] [CrossRef] [PubMed]
- Despres, J.P.; Ross, R.; Boka, G.; Almeras, N.; Lemieux, I.; Investigators, A.D.-L. Effect of rimonabant on the high-triglyceride/low-HDL-cholesterol dyslipidemia, intraabdominal adiposity, and liver fat: The ADAGIO-Lipids trial. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 416–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenstock, J.; Hollander, P.; Chevalier, S.; Iranmanesh, A.; Group, S.S. SERENADE: The Study Evaluating Rimonabant Efficacy in Drug-naive Diabetic Patients: Effects of monotherapy with rimonabant, the first selective CB1 receptor antagonist, on glycemic control, body weight, and lipid profile in drug-naive type 2 diabetes. Diabetes Care 2008, 31, 2169–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pi-Sunyer, F.X.; Aronne, L.J.; Heshmati, H.M.; Devin, J.; Rosenstock, J.; Rio-North America Study Group. Effect of rimonabant, a cannabinoid-1 receptor blocker, on weight and cardiometabolic risk factors in overweight or obese patients: RIO-North America: A randomized controlled trial. JAMA 2006, 295, 761–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bluher, M. Efficacy and safety of the weight-loss drug rimonabant. Lancet 2008, 371, 1706. [Google Scholar] [CrossRef]
- Despres, J.P.; Van Gaal, L.; Pi-Sunyer, X.; Scheen, A. Efficacy and safety of the weight-loss drug rimonabant. Lancet 2008, 371, 555; author reply 556–557. [Google Scholar] [CrossRef]
- Rinaldi-Carmona, M.; Barth, F.; Heaulme, M.; Alonso, R.; Shire, D.; Congy, C.; Soubrie, P.; Breliere, J.C.; Le Fur, G. Biochemical and pharmacological characterisation of SR141716A, the first potent and selective brain cannabinoid receptor antagonist. Life Sci. 1995, 56, 1941–1947. [Google Scholar] [CrossRef]
- Compton, D.R.; Aceto, M.D.; Lowe, J.; Martin, B.R. In vivo characterization of a specific cannabinoid receptor antagonist (SR141716A): Inhibition of delta 9-tetrahydrocannabinol-induced responses and apparent agonist activity. J. Pharmacol. Exp. Ther. 1996, 277, 586–594. [Google Scholar]
- Richardson, J.D.; Aanonsen, L.; Hargreaves, K.M. SR 141716A, a cannabinoid receptor antagonist, produces hyperalgesia in untreated mice. Eur. J. Pharmacol. 1997, 319, R3–R4. [Google Scholar] [CrossRef]
- Landsman, R.S.; Burkey, T.H.; Consroe, P.; Roeske, W.R.; Yamamura, H.I. SR141716A is an inverse agonist at the human cannabinoid CB1 receptor. Eur. J. Pharmacol. 1997, 334, R1–R2. [Google Scholar] [CrossRef]
- Cahill, K.; Ussher, M.H. Cannabinoid type 1 receptor antagonists for smoking cessation. Cochrane Database Syst. Rev. 2011, CD005353. [Google Scholar] [CrossRef] [PubMed]
- Elrashidi, M.Y.; Ebbert, J.O. Emerging drugs for the treatment of tobacco dependence: 2014 update. Expert Opin. Emerg. Drugs 2014, 19, 243–260. [Google Scholar] [CrossRef]
- Steinberg, M.B.; Foulds, J. Rimonabant for treating tobacco dependence. Vasc. Health Risk Manag. 2007, 3, 307–311. [Google Scholar]
- Huestis, M.A.; Boyd, S.J.; Heishman, S.J.; Preston, K.L.; Bonnet, D.; Le Fur, G.; Gorelick, D.A. Single and multiple doses of rimonabant antagonize acute effects of smoked cannabis in male cannabis users. Psychopharmacology 2007, 194, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Solinas, M.; Panlilio, L.V.; Goldberg, S.R. Exposure to delta-9-tetrahydrocannabinol (THC) increases subsequent heroin taking but not heroin’s reinforcing efficacy: A self-administration study in rats. Neuropsychopharmacology 2004, 29, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Gamaleddin, I.; Wertheim, C.; Zhu, A.Z.; Coen, K.M.; Vemuri, K.; Makryannis, A.; Goldberg, S.R.; Le Foll, B. Cannabinoid receptor stimulation increases motivation for nicotine and nicotine seeking. Addict. Biol. 2012, 17, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Colombo, G.; Serra, S.; Brunetti, G.; Gomez, R.; Melis, S.; Vacca, G.; Carai, M.M.; Gessa, L. Stimulation of voluntary ethanol intake by cannabinoid receptor agonists in ethanol-preferring sP rats. Psychopharmacology 2002, 159, 181–187. [Google Scholar] [CrossRef]
- Linsenbardt, D.N.; Boehm, S.L., 2nd. Agonism of the endocannabinoid system modulates binge-like alcohol intake in male C57BL/6J mice: Involvement of the posterior ventral tegmental area. Neuroscience 2009, 164, 424–434. [Google Scholar] [CrossRef]
- Gamaleddin, I.H.; Trigo, J.M.; Gueye, A.B.; Zvonok, A.; Makriyannis, A.; Goldberg, S.R.; Le Foll, B. Role of the endogenous cannabinoid system in nicotine addiction: Novel insights. Front. Psychiatry 2015, 6, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maccioni, P.; Colombo, G.; Carai, M.A. Blockade of the cannabinoid CB1 receptor and alcohol dependence: Preclinical evidence and preliminary clinical data. CNS Neurol. Disord. Drug Targets 2010, 9, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Solinas, M.; Panlilio, L.V.; Antoniou, K.; Pappas, L.A.; Goldberg, S.R. The cannabinoid CB1 antagonist N-piperidinyl-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl) -4-methylpyrazole-3-carboxamide (SR-141716A) differentially alters the reinforcing effects of heroin under continuous reinforcement, fixed ratio, and progressive ratio schedules of drug self-administration in rats. J. Pharmacol. Exp. Ther. 2003, 306, 93–102. [Google Scholar] [CrossRef] [PubMed]
- De Vries, T.J.; Homberg, J.R.; Binnekade, R.; Raaso, H.; Schoffelmeer, A.N.M. Cannabinoid modulation of the reinforcing and motivational properties of heroin and heroin-associated cues in rats. Psychopharmacology 2003, 168, 164–169. [Google Scholar] [CrossRef]
- Cohen, C.; Perrault, G.; Voltz, C.; Steinberg, R.; Soubrie, P. SR141716, a central cannabinoid (CB(1)) receptor antagonist, blocks the motivational and dopamine-releasing effects of nicotine in rats. Behav. Pharmacol. 2002, 13, 451–463. [Google Scholar] [CrossRef]
- Cohen, C.; Perrault, G.; Griebel, G.; Soubrie, P. Nicotine-associated cues maintain nicotine-seeking behavior in rats several weeks after nicotine withdrawal: Reversal by the cannabinoid (CB1) receptor antagonist, rimonabant (SR141716). Neuropsychopharmacology 2005, 30, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Foll, B.; Goldberg, S.R. Rimonabant, a CB1 antagonist, blocks nicotine-conditioned place preferences. Neuroreport. 2004, 15, 2139–2143. [Google Scholar] [CrossRef]
- Chaperon, F.; Soubrie, P.; Puech, A.J.; Thiebot, M.H. Involvement of central cannabinoid (CB1) receptors in the establishment of place conditioning in rats. Psychopharmacology 1998, 135, 324–332. [Google Scholar] [CrossRef]
- De Vries, T.J.; Shaham, Y.; Homberg, J.R.; Crombag, H.; Schuurman, K.; Dieben, J.; Vanderschuren, L.J.; Schoffelmeer, A.N. A cannabinoid mechanism in relapse to cocaine seeking. Nat. Med. 2001, 7, 1151–1154. [Google Scholar] [CrossRef] [Green Version]
- Anggadiredja, K.; Nakamichi, M.; Hiranita, T.; Tanaka, H.; Shoyama, Y.; Watanabe, S.; Yamamoto, T. Endocannabinoid system modulates relapse to methamphetamine seeking: Possible mediation by the arachidonic acid cascade. Neuropsychopharmacology 2004, 29, 1470–1478. [Google Scholar] [CrossRef] [PubMed]
- Henderson-Redmond, A.N.; Guindon, J.; Morgan, D.J. Roles for the endocannabinoid system in ethanol-motivated behavior. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 65, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, S.; Schmid, P.C.; Fernandez-Ruiz, J.; Krebsbach, R.; Schmid, H.H.; Ramos, J.A. Region-dependent changes in endocannabinoid transmission in the brain of morphine-dependent rats. Addict. Biol. 2003, 8, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Vigano, D.; Grazia Cascio, M.; Rubino, T.; Fezza, F.; Vaccani, A.; Di Marzo, V.; Parolaro, D. Chronic morphine modulates the contents of the endocannabinoid, 2-arachidonoyl glycerol, in rat brain. Neuropsychopharmacology 2003, 28, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Vigano, D.; Valenti, M.; Cascio, M.G.; Di Marzo, V.; Parolaro, D.; Rubino, T. Changes in endocannabinoid levels in a rat model of behavioural sensitization to morphine. Eur. J. Neurosci. 2004, 20, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.; Cascio, M.G.; Fernandez-Ruiz, J.; Fezza, F.; Di Marzo, V.; Ramos, J.A. Changes in endocannabinoid contents in the brain of rats chronically exposed to nicotine, ethanol or cocaine. Brain Res. 2002, 954, 73–81. [Google Scholar] [CrossRef]
- Wang, H.; Treadway, T.; Covey, D.P.; Cheer, J.F.; Lupica, C.R. Cocaine-Induced Endocannabinoid Mobilization in the Ventral Tegmental Area. Cell Rep. 2015, 12, 1997–2008. [Google Scholar] [CrossRef] [Green Version]
- Caille, S.; Alvarez-Jaimes, L.; Polis, I.; Stouffer, D.G.; Parsons, L.H. Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. J. Neurosci. 2007, 27, 3695–3702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Lipinski, A.A.; Liktor-Busa, E.; Smith, A.F.; Moutal, A.; Khanna, R.; Langlais, P.R.; Largent-Milnes, T.M.; Vanderah, T.W. The Effects of Repeated Morphine Treatment on the Endogenous Cannabinoid System in the Ventral Tegmental Area. Front. Pharmacol. 2021, 12, 632757. [Google Scholar] [CrossRef] [PubMed]
- Buczynski, M.W.; Polis, I.Y.; Parsons, L.H. The volitional nature of nicotine exposure alters anandamide and oleoylethanolamide levels in the ventral tegmental area. Neuropsychopharmacology 2013, 38, 574–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.H.; Jordan, C.J.; Vemuri, K.; Bi, G.H.; Zhan, J.; Gardner, E.L.; Makriyannis, A.; Wang, Y.L.; Xi, Z.X. Cannabinoid CB1 receptor neutral antagonist AM4113 inhibits heroin self-administration without depressive side effects in rats. Acta Pharmacol. Sin. 2019, 40, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Anisman, H.; Matheson, K. Stress, depression, and anhedonia: Caveats concerning animal models. Neurosci. Biobehav. Rev. 2005, 29, 525–546. [Google Scholar] [CrossRef] [PubMed]
- Scheggi, S.; De Montis, M.G.; Gambarana, C. Making Sense of Rodent Models of Anhedonia. Int. J. Neuropsychopharmacol. 2018, 21, 1049–1065. [Google Scholar] [CrossRef]
- Arnold, J.C.; Hunt, G.E.; McGregor, I.S. Effects of the cannabinoid receptor agonist CP 55,940 and the cannabinoid receptor antagonist SR 141716 on intracranial self-stimulation in Lewis rats. Life Sci. 2001, 70, 97–108. [Google Scholar] [CrossRef]
- Grim, T.W.; Wiebelhaus, J.M.; Morales, A.J.; Negus, S.S.; Lichtman, A.H. Effects of acute and repeated dosing of the synthetic cannabinoid CP55,940 on intracranial self-stimulation in mice. Drug Alcohol Depend. 2015, 150, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Vlachou, S.; Nomikos, G.G.; Panagis, G. Effects of endocannabinoid neurotransmission modulators on brain stimulation reward. Psychopharmacology 2006, 188, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Vlachou, S.; Stamatopoulou, F.; Nomikos, G.G.; Panagis, G. Enhancement of endocannabinoid neurotransmission through CB1 cannabinoid receptors counteracts the reinforcing and psychostimulant effects of cocaine. Int. J. Neuropsychopharmacol. 2008, 11, 905–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Hoffman, A.F.; Peng, X.Q.; Lupica, C.R.; Gardner, E.L.; Xi, Z.X. Attenuation of basal and cocaine-enhanced locomotion and nucleus accumbens dopamine in cannabinoid CB1-receptor-knockout mice. Psychopharmacology 2009, 204, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Murillo-Rodriguez, E.; Machado, S.; Rocha, N.B.; Budde, H.; Yuan, T.F.; Arias-Carrion, O. Revealing the role of the endocannabinoid system modulators, SR141716A, URB597 and VDM-11, in sleep homeostasis. Neuroscience 2016, 339, 433–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Fang, Q.; Liu, Y.; Zhao, M.; Li, D.; Wang, J.; Lu, L. Cannabinoid CB(1) receptor antagonist rimonabant attenuates reinstatement of ketamine conditioned place preference in rats. Eur. J. Pharmacol. 2008, 589, 122–126. [Google Scholar] [CrossRef]
- Biala, G.; Budzynska, B.; Staniak, N. Effects of rimonabant on the reinstatement of nicotine-conditioned place preference by drug priming in rats. Behav. Brain. Res. 2009, 202, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.E.; Verty, A.N.; McGregor, I.S.; Mallet, P.E. A cannabinoid receptor antagonist attenuates conditioned place preference but not behavioural sensitization to morphine. Brain Res. 2004, 1026, 244–253. [Google Scholar] [CrossRef]
- Fang, Q.; Li, F.Q.; Li, Y.Q.; Xue, Y.X.; He, Y.Y.; Liu, J.F.; Lu, L.; Wang, J.S. Cannabinoid CB1 receptor antagonist rimonabant disrupts nicotine reward-associated memory in rats. Pharmacol. Biochem. Behav. 2011, 99, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Hurst, D.; Umejiego, U.; Lynch, D.; Seltzman, H.; Hyatt, S.; Roche, M.; McAllister, S.; Fleischer, D.; Kapur, A.; Abood, M.; et al. Biarylpyrazole inverse agonists at the cannabinoid CB1 receptor: Importance of the C-3 carboxamide oxygen/lysine3.28(192) interaction. J. Med. Chem. 2006, 49, 5969–5987. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.F.; Lycas, M.D.; Kaczmarzyk, J.R.; Spivak, C.E.; Baumann, M.H.; Lupica, C.R. Disruption of hippocampal synaptic transmission and long-term potentiation by psychoactive synthetic cannabinoid ‘Spice’ compounds: Comparison with Delta(9) -tetrahydrocannabinol. Addict Biol. 2017, 22, 390–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seltzman, H.H.; Maitra, R.; Bortoff, K.; Henson, J.; Reggio, P.H.; Wesley, D.; Tam, J. Metabolic Profiling of CB1 Neutral Antagonists. Methods Enzymol. 2017, 593, 199–215. [Google Scholar] [CrossRef] [Green Version]
- Salamone, J.D.; McLaughlin, P.J.; Sink, K.; Makriyannis, A.; Parker, L.A. Cannabinoid CB1 receptor inverse agonists and neutral antagonists: Effects on food intake, food-reinforced behavior and food aversions. Physiol. Behav. 2007, 91, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sink, K.S.; McLaughlin, P.J.; Wood, J.A.; Brown, C.; Fan, P.; Vemuri, V.K.; Peng, Y.; Olszewska, T.; Thakur, G.A.; Makriyannis, A.; et al. The novel cannabinoid CB1 receptor neutral antagonist AM4113 suppresses food intake and food-reinforced behavior but does not induce signs of nausea in rats. Neuropsychopharmacology 2008, 33, 946–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, A.P.; Vemuri, V.K.; Peng, Y.; Wood, J.T.; Olszewska, T.; Pittman, Q.J.; Makriyannis, A.; Sharkey, K.A. A neutral CB1 receptor antagonist reduces weight gain in rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R2185–R2193. [Google Scholar] [CrossRef]
- Gueye, A.B.; Pryslawsky, Y.; Trigo, J.M.; Poulia, N.; Delis, F.; Antoniou, K.; Loureiro, M.; Laviolette, S.R.; Vemuri, K.; Makriyannis, A.; et al. The CB1 Neutral Antagonist AM4113 Retains the Therapeutic Efficacy of the Inverse Agonist Rimonabant for Nicotine Dependence and Weight Loss with Better Psychiatric Tolerability. Int. J. Neuropsychopharmacol. 2016, 19, pyw068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cluny, N.L.; Vemuri, V.K.; Chambers, A.P.; Limebeer, C.L.; Bedard, H.; Wood, J.T.; Lutz, B.; Zimmer, A.; Parker, L.A.; Makriyannis, A.; et al. A novel peripherally restricted cannabinoid receptor antagonist, AM6545, reduces food intake and body weight, but does not cause malaise, in rodents. Br. J. Pharmacol. 2010, 161, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.W.; Redhi, G.H.; Vemuri, K.; Makriyannis, A.; Le Foll, B.; Bergman, J.; Goldberg, S.R.; Justinova, Z. Blockade of Nicotine and Cannabinoid Reinforcement and Relapse by a Cannabinoid CB1-Receptor Neutral Antagonist AM4113 and Inverse Agonist Rimonabant in Squirrel Monkeys. Neuropsychopharmacology 2016, 41, 2283–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balla, A.; Dong, B.; Shilpa, B.M.; Vemuri, K.; Makriyannis, A.; Pandey, S.C.; Sershen, H.; Suckow, R.F.; Vinod, K.Y. Cannabinoid-1 receptor neutral antagonist reduces binge-like alcohol consumption and alcohol-induced accumbal dopaminergic signaling. Neuropharmacology 2018, 131, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Kangas, B.D.; Delatte, M.S.; Vemuri, V.K.; Thakur, G.A.; Nikas, S.P.; Subramanian, K.V.; Shukla, V.G.; Makriyannis, A.; Bergman, J. Cannabinoid discrimination and antagonism by CB(1) neutral and inverse agonist antagonists. J. Pharmacol. Exp. Ther. 2013, 344, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Tai, S.; Nikas, S.P.; Shukla, V.G.; Vemuri, K.; Makriyannis, A.; Jarbe, T.U. Cannabinoid withdrawal in mice: Inverse agonist vs neutral antagonist. Psychopharmacology 2015, 232, 2751–2761. [Google Scholar] [CrossRef] [Green Version]
- Jarbe, T.U.; LeMay, B.J.; Olszewska, T.; Vemuri, V.K.; Wood, J.T.; Makriyannis, A. Intrinsic effects of AM4113, a putative neutral CB1 receptor selective antagonist, on open-field behaviors in rats. Pharmacol. Biochem. Behav. 2008, 91, 84–90. [Google Scholar] [CrossRef] [Green Version]
- Sink, K.S.; Vemuri, V.K.; Wood, J.; Makriyannis, A.; Salamone, J.D. Oral bioavailability of the novel cannabinoid CB1 antagonist AM6527: Effects on food-reinforced behavior and comparisons with AM4113. Pharmacol. Biochem. Behav. 2009, 91, 303–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wills, K.L.; Vemuri, K.; Kalmar, A.; Lee, A.; Limebeer, C.L.; Makriyannis, A.; Parker, L.A. CB1 antagonism: Interference with affective properties of acute naloxone-precipitated morphine withdrawal in rats. Psychopharmacology 2014, 231, 4291–4300. [Google Scholar] [CrossRef] [Green Version]
- Kangas, B.D.; Zakarian, A.S.; Vemuri, K.; Alapafuja, S.O.; Jiang, S.; Nikas, S.P.; Makriyannis, A.; Bergman, J. Cannabinoid Antagonist Drug Discrimination in Nonhuman Primates. J. Pharmacol. Exp. Ther. 2020, 372, 119–127. [Google Scholar] [CrossRef]
- Stewart, J.L.; McMahon, L.R. Rimonabant-induced Delta9-tetrahydrocannabinol withdrawal in rhesus monkeys: Discriminative stimulus effects and other withdrawal signs. J. Pharmacol. Exp. Ther. 2010, 334, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G. Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci. 2005, 76, 1307–1324. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Thomas, A.; Stevenson, L.A.; Ross, R.A.; Varvel, S.A.; Lichtman, A.H.; Martin, B.R.; Razdan, R.K. The psychoactive plant cannabinoid, Delta9-tetrahydrocannabinol, is antagonized by Delta8- and Delta9-tetrahydrocannabivarin in mice in vivo. Br. J. Pharmacol. 2007, 150, 586–594. [Google Scholar] [CrossRef] [Green Version]
- Batkai, S.; Mukhopadhyay, P.; Horvath, B.; Rajesh, M.; Gao, R.Y.; Mahadevan, A.; Amere, M.; Battista, N.; Lichtman, A.H.; Gauson, L.A.; et al. Delta8-Tetrahydrocannabivarin prevents hepatic ischaemia/reperfusion injury by decreasing oxidative stress and inflammatory responses through cannabinoid CB2 receptors. Br. J. Pharmacol. 2012, 165, 2450–2461. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Stevenson, L.A.; Wease, K.N.; Price, M.R.; Baillie, G.; Ross, R.A.; Pertwee, R.G. Evidence that the plant cannabinoid Delta9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor antagonist. Br. J. Pharmacol. 2005, 146, 917–926. [Google Scholar] [CrossRef] [Green Version]
- Riedel, G.; Fadda, P.; McKillop-Smith, S.; Pertwee, R.G.; Platt, B.; Robinson, L. Synthetic and plant-derived cannabinoid receptor antagonists show hypophagic properties in fasted and non-fasted mice. Br. J. Pharmacol. 2009, 156, 1154–1166. [Google Scholar] [CrossRef] [Green Version]
- Xi, Z.X.; Muldoon, P.; Wang, X.F.; Bi, G.H.; Damaj, M.I.; Lichtman, A.H.; Pertwee, R.G.; Gardner, E.L. Delta(8)—Tetrahydrocannabivarin has potent anti-nicotine effects in several rodent models of nicotine dependence. Br. J. Pharmacol. 2019, 176, 4773–4784. [Google Scholar] [CrossRef]
- Tudge, L.; Williams, C.; Cowen, P.J.; McCabe, C. Neural effects of cannabinoid CB1 neutral antagonist tetrahydrocannabivarin on food reward and aversion in healthy volunteers. Int. J. Neuropsychopharmacol. 2014, 18, pyu094. [Google Scholar] [CrossRef] [Green Version]
- Rzepa, E.; Tudge, L.; McCabe, C. The CB1 Neutral Antagonist Tetrahydrocannabivarin Reduces Default Mode Network and Increases Executive Control Network Resting State Functional Connectivity in Healthy Volunteers. Int. J. Neuropsychopharmacol. 2015, 19, pyv092. [Google Scholar] [CrossRef]



| Compound | Doses | Species | Results | References |
|---|---|---|---|---|
| Rimonabant | 20 mg/kg, i.p. | Rat | ↓ Electrical brain-stimulation reward | [142] |
| Rimonabant | 0.3, 1, 3, 10 mg/kg, i.p. | Rat | ↓ Electrical brain-stimulation reward | [139] |
| Rimonabant | 0.02, 0.3, 1.0 mg/kg, i.p. | Rat | No effect on electrical brain-stimulation reward | [144] |
| Rimonabant | 0.02 mg/kg, i.p. | Rat | No effect on electrical brain-stimulation reward | [145] |
| Rimonabant | 3, 10 mg/kg, i.p., | Mouse | No effect on electrical brain-stimulation reward | [143] |
| Rimonabant | 0.3, 1, 3 mg/kg, i.p. | Rat | Not produce CPP or CPA | [127] |
| Rimonabant | 0.1, 0.5, 3.0 mg/kg, i.p. | Rat | Not produces CPP or CPA | [148] |
| Rimonabant | 0.5, 1, 2 mg/kg, i.p. | Rat | Not produce CPP or CPA | [149] |
| Rimonabant | 3 mg/kg, i.p. | Rat | Not produce CPP or CPA | [89] |
| Rimonabant | 0.25, 0.5, 1 mg/kg, i.p. | Rat | Not produce CPP or CPA | [55] |
| Rimonabant | 0.1, 0.5, 3 mg/kg | Rat | Not produce CPP or CPA | [150] |
| Rimonabant | 0.3, 3 mg/kg, i.p. | Rat | Not produce CPP or CPA | [151] |
| Rimonabant | 0.25, 0.5, 2, 3 mg/kg | Rat | Produces CPP | [53] |
| Rimonabant | 3 mg/kg | Mouse | ↓ Accumbens DA | [146] |
| Rimonabant | 2, 10 mg/kg, i.p. | Rat | No effect on accumbens DA | [76] |
| Rimonabant | 1, 10, 30, 100 mM, intra-NAc | Rat | ↑ Accumbens DA | [76] |
| Rimonabant | 5, 10, 20 mg/kg, i.p. | Rat | ↑ Accumbens DA | [147] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soler-Cedeno, O.; Xi, Z.-X. Neutral CB1 Receptor Antagonists as Pharmacotherapies for Substance Use Disorders: Rationale, Evidence, and Challenge. Cells 2022, 11, 3262. https://doi.org/10.3390/cells11203262
Soler-Cedeno O, Xi Z-X. Neutral CB1 Receptor Antagonists as Pharmacotherapies for Substance Use Disorders: Rationale, Evidence, and Challenge. Cells. 2022; 11(20):3262. https://doi.org/10.3390/cells11203262
Chicago/Turabian StyleSoler-Cedeno, Omar, and Zheng-Xiong Xi. 2022. "Neutral CB1 Receptor Antagonists as Pharmacotherapies for Substance Use Disorders: Rationale, Evidence, and Challenge" Cells 11, no. 20: 3262. https://doi.org/10.3390/cells11203262
APA StyleSoler-Cedeno, O., & Xi, Z.-X. (2022). Neutral CB1 Receptor Antagonists as Pharmacotherapies for Substance Use Disorders: Rationale, Evidence, and Challenge. Cells, 11(20), 3262. https://doi.org/10.3390/cells11203262