

Research Progress on the Role of Pyroptosis in Myocardial Ischemia-Reperfusion Injury


Abstract
:1. Introduction
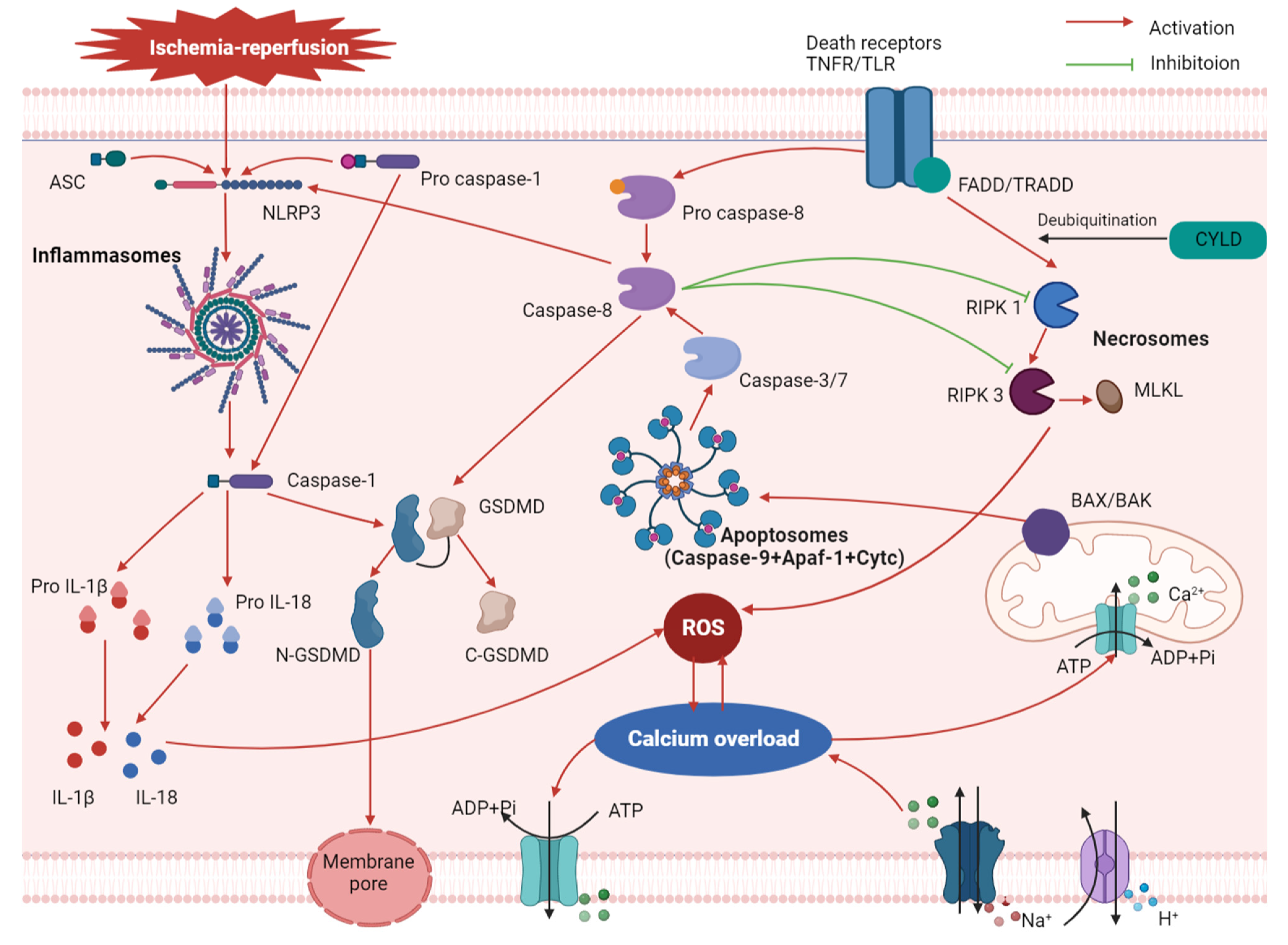
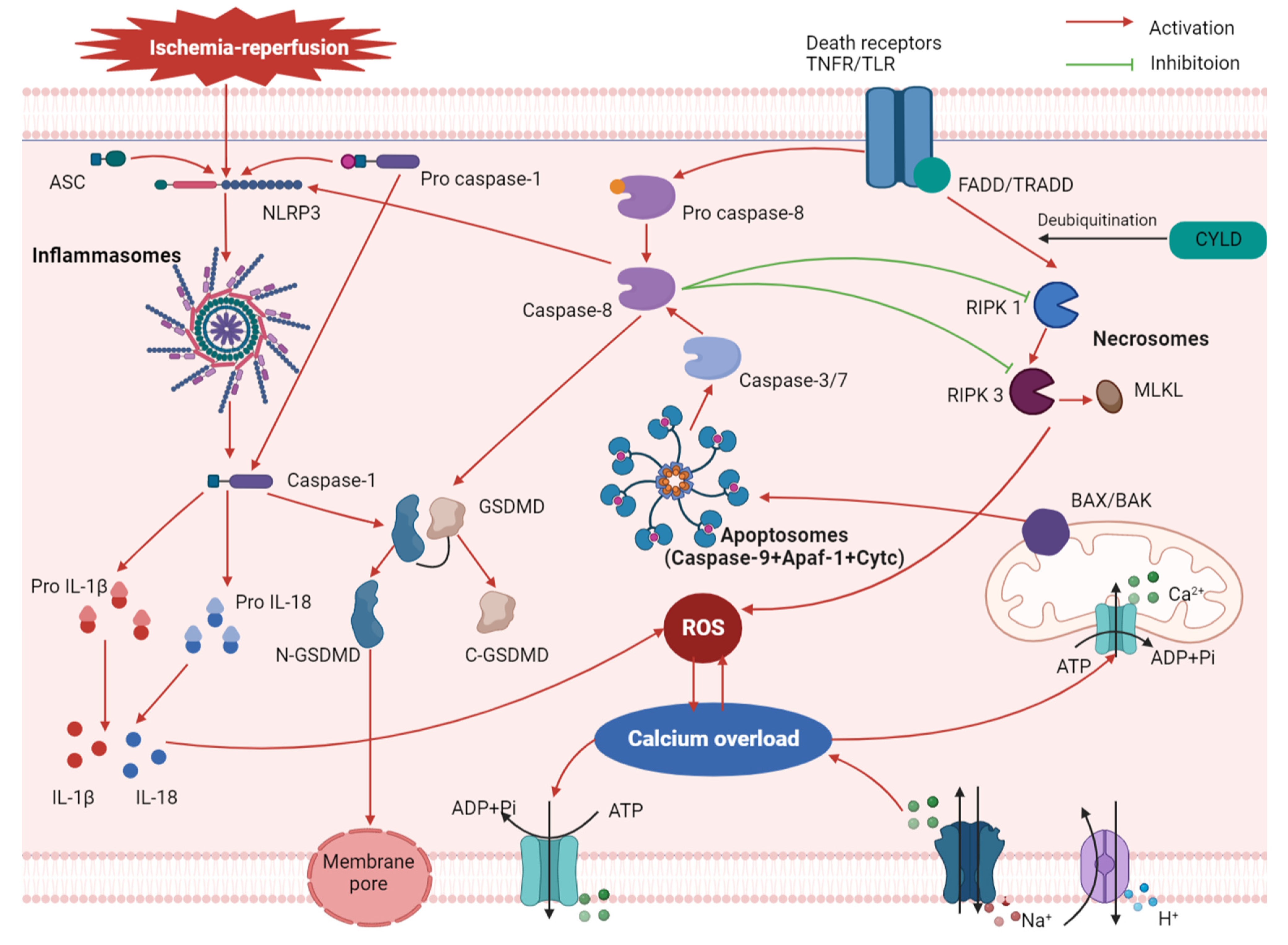
2. Mechanism of Pyroptosis in MIRI
3. Relationship between Pyroptosis and Other Associated Mechanisms of MIRI
3.1. Oxidative Stress
3.2. Calcium Overload
3.3. Apoptosis and Necroptosis
4. Effect of Pyroptosis on Non-Cardiomyocytes and Its Role in MIRI
4.1. Fibroblasts
4.2. Vascular Endothelial Cells
4.3. Macrophages
5. Regulation of Pyroptosis by Non-Coding RNAs in MIRI
5.1. MicroRNA
5.2. LncRNA
5.3. CircRNA
6. Inhibition of Pyroptosis by Drugs and Improved MIRI
6.1. Small Molecular Substances
6.2. Clinical Drugs
6.3. Natural Substances
6.4. Gases
7. Thinking and Improvement
8. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ASC | Apoptosis-associated speck-like protein containing a carboxy-terminal CARD |
| Circ | Circular RNAs |
| DAMPs | Danger-associated molecular patterns |
| EVs | Extracellular vesicles |
| FADD | Fas-associated death domain |
| GSDMD | Gasdermin D |
| IL-1β | Interleukin 1β |
| LncRNAs | Long noncoding RNAs |
| MIRI | Myocardial ischemia-reperfusion injury |
| MiRNAs | MicroRNAs |
| NcRNAs | Noncoding RNAs |
| NLRP3 | Nod-like receptor protein 3 |
| ROS | Reactive oxygen species |
| TNF | Tumor necrosis factor |
References
- Anderson, J.L.; Morrow, D.A. Acute Myocardial Infarction. N. Engl. J. Med. 2017, 376, 2053–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algoet, M.; Janssens, S.; Himmelreich, U.; Gsell, W.; Pusovnik, M.; Van den Eynde, J.; Oosterlinck, W. Myocardial ischemia-reperfusion injury and the influence of inflammation. Trends Cardiovasc. Med. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.F. Clinical manifestations and basic mechanisms of myocardial ischemia/reperfusion injury. Tzu-Chi Med. J. 2018, 30, 209–215. [Google Scholar] [CrossRef]
- Bell, R.M.; Bøtker, H.E.; Carr, R.D.; Davidson, S.M.; Downey, J.M.; Dutka, D.P.; Heusch, G.; Ibanez, B.; Macallister, R.; Stoppe, C.; et al. 9th Hatter Biannual Meeting: Position document on ischaemia/reperfusion injury, conditioning and the ten commandments of cardioprotection. Basic Res. Cardiol. 2016, 111, 41. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, B.; Heusch, G.; Ovize, M.; Van de Werf, F. Evolving therapies for myocardial ischemia/reperfusion injury. J. Am. Coll. Cardiol. 2015, 65, 1454–1471. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S. Perspectives on the therapeutic modulation of an alternative cell death, programmed necrosis (review). Int. J. Mol. Med. 2014, 33, 1401–1406. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G. CIRCUS: A kiss of death for cardioprotection? Cardiovasc. Res. 2015, 108, 215–216. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef]
- Zhaolin, Z.; Guohua, L.; Shiyuan, W.; Zuo, W. Role of pyroptosis in cardiovascular disease. Cell Prolif. 2019, 52, e12563. [Google Scholar] [CrossRef]
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Lan, Z.; Xin, Z.; He, C.; Guo, Z.; Xia, X.; Hu, T. Emerging insights into molecular mechanisms underlying pyroptosis and functions of inflammasomes in diseases. J. Cell. Physiol. 2020, 235, 3207–3221. [Google Scholar] [CrossRef]
- Yabal, M.; Calleja, D.J.; Simpson, D.S.; Lawlor, K.E. Stressing out the mitochondria: Mechanistic insights into NLRP3 inflammasome activation. J. Leukoc. Biol. 2019, 105, 377–399. [Google Scholar] [CrossRef]
- Tang, Y.S.; Zhao, Y.H.; Zhong, Y.; Li, X.Z.; Pu, J.X.; Luo, Y.C.; Zhou, Q.L. Neferine inhibits LPS-ATP-induced endothelial cell pyroptosis via regulation of ROS/NLRP3/Caspase-1 signaling pathway. Inflamm. Res 2019, 68, 727–738. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Gong, W.; Shi, Y.; Ren, J. Research progresses of molecular mechanism of pyroptosis and its related diseases. Immunobiology 2020, 225, 151884. [Google Scholar] [CrossRef]
- Broz, P.; Pelegrín, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Zhan, K.Y.; Yu, P.L.; Liu, C.H.; Luo, J.H.; Yang, W. Detrimental or beneficial: The role of TRPM2 in ischemia/reperfusion injury. Acta Pharmacol. Sin. 2016, 37, 4–12. [Google Scholar] [CrossRef] [Green Version]
- González-Montero, J.; Brito, R.; Gajardo, A.I.; Rodrigo, R. Myocardial reperfusion injury and oxidative stress: Therapeutic opportunities. World J. Cardiol. 2018, 10, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Kurian, G.A.; Rajagopal, R.; Vedantham, S.; Rajesh, M. The Role of Oxidative Stress in Myocardial Ischemia and Reperfusion Injury and Remodeling: Revisited. Oxidative Med. Cell. Longev. 2016, 2016, 1656450. [Google Scholar] [CrossRef] [Green Version]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef]
- Minutoli, L.; Puzzolo, D.; Rinaldi, M.; Irrera, N.; Marini, H.; Arcoraci, V.; Bitto, A.; Crea, G.; Pisani, A.; Squadrito, F.; et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxidative Med. Cell. Longev. 2016, 2016, 2183026. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, K.; Prabhu, S.D.; Reddy, V.S.; Boylston, W.H.; Valente, A.J.; Chandrasekar, B. Neutralization of interleukin-18 ameliorates ischemia/reperfusion-induced myocardial injury. J. Biol. Chem. 2009, 284, 7853–7865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Markel, T.A.; Meldrum, D.R. Interleukin 18 in the heart. Shock 2008, 30, 3–10. [Google Scholar] [CrossRef]
- Wu, X.; Ren, G.; Zhou, R.; Ge, J.; Chen, F.H. The role of Ca2+ in acid-sensing ion channel 1a-mediated chondrocyte pyroptosis in rat adjuvant arthritis. Lab. Investig. 2019, 99, 499–513. [Google Scholar] [CrossRef]
- Zhou, B.; Lin, Y.; Chen, S.; Cai, J.; Luo, Z.; Yu, S.; Lu, J. Activation of Ca2+/Calmodulin-Dependent Protein Kinase II (CaMKII) with Lidocaine Provokes Pyroptosis of Glioblastoma Cells. Bull. Exp. Biol. Med. 2021, 171, 297–304. [Google Scholar] [CrossRef]
- Mo, G.; Liu, X.; Zhong, Y.; Mo, J.; Li, Z.; Li, D.; Zhang, L.; Liu, Y. IP3R1 regulates Ca2+ transport and pyroptosis through the NLRP3/Caspase-1 pathway in myocardial ischemia/reperfusion injury. Cell Death Discov. 2021, 7, 31. [Google Scholar] [CrossRef]
- Wang, M.; Wang, R.; Sun, H.; Sun, G.; Sun, X. Ginsenoside Rb1 ameliorates cardiotoxicity triggered by aconitine via inhibiting calcium overload and pyroptosis. Phytomed. Int. J. Phytother. Phytopharm. 2021, 83, 153468. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef]
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 2019, 38, e101638. [Google Scholar] [CrossRef] [PubMed]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.E.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal. 2007, 19, 2056–2067. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Wickliffe, K.E.; Dugger, D.L.; Maltzman, A.; Roose-Girma, M.; Dohse, M.; Kőműves, L.; Webster, J.D.; Dixit, V.M. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 2019, 574, 428–431. [Google Scholar] [CrossRef]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef]
- Newton, K.; Wickliffe, K.E.; Maltzman, A.; Dugger, D.L.; Reja, R.; Zhang, Y.; Roose-Girma, M.; Modrusan, Z.; Sagolla, M.S.; Webster, J.D.; et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature 2019, 575, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vince, J.E.; De Nardo, D.; Gao, W.; Vince, A.J.; Hall, C.; McArthur, K.; Simpson, D.; Vijayaraj, S.; Lindqvist, L.M.; Bouillet, P.; et al. The Mitochondrial Apoptotic Effectors BAX/BAK Activate Caspase-3 and -7 to Trigger NLRP3 Inflammasome and Caspase-8 Driven IL-1β Activation. Cell Rep. 2018, 25, 2339–2353. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.E.; Wong, W.W.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015, 6, 7515. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandanger, Ø.; Ranheim, T.; Vinge, L.E.; Bliksøen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2013, 99, 164–174. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M. NLRP3 in myocardial ischaemia-reperfusion injury: Inflammasome-dependent or -independent role in different cell types. Cardiovasc. Res. 2013, 99, 4–5. [Google Scholar] [CrossRef]
- Hou, M.; Wu, X.; Zhao, Z.; Deng, Q.; Chen, Y.; Yin, L. Endothelial cell-targeting, ROS-ultrasensitive drug/siRNA co-delivery nanocomplexes mitigate early-stage neutrophil recruitment for the anti-inflammatory treatment of myocardial ischemia reperfusion injury. Acta Biomater. 2022, 143, 344–355. [Google Scholar] [CrossRef]
- Korayem, A.H.; Mujica, P.E.; Aramoto, H.; Durán, R.G.; Nepali, P.R.; Kim, D.D.; Harris, A.L.; Sánchez, F.A.; Durán, W.N. Endothelial cAMP deactivates ischemia-reperfusion-induced microvascular hyperpermeability via Rap1-mediated mechanisms. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H179–H189. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, L.; Ma, H.; Sun, F.; Wen, F. microRNA-129 overexpression in endothelial cell-derived extracellular vesicle influences inflammatory response caused by myocardial ischemia/reperfusion injury. Cell Biol. Int. 2021, 45, 1743–1756. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Xiang, D.K.; Li, S.N.; Yang, L.H.; Gao, L.F.; Feng, C. MicroRNA-495 Ameliorates Cardiac Microvascular Endothelial Cell Injury and Inflammatory Reaction by Suppressing the NLRP3 Inflammasome Signaling Pathway. Cell. Physiol. Biochem. 2018, 49, 798–815. [Google Scholar] [CrossRef]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.F.; Yu, T.; Chu, X.M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yuan, M.; Zhang, L.; Wu, B.; Sun, X. Adiponectin alleviates NLRP3-inflammasome-mediated pyroptosis of aortic endothelial cells by inhibiting FoxO4 in arteriosclerosis. Biochem. Biophys. Res. Commun. 2019, 514, 266–272. [Google Scholar] [CrossRef]
- Ge, X.; Meng, Q.; Wei, L.; Liu, J.; Li, M.; Liang, X.; Lin, F.; Zhang, Y.; Li, Y.; Liu, Z.; et al. Myocardial ischemia-reperfusion induced cardiac extracellular vesicles harbour proinflammatory features and aggravate heart injury. J. Extracell. Vesicles 2021, 10, e12072. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wang, S.; Chang, S.; Ren, D.; Shali, S.; Li, C.; Yang, H.; Huang, Z.; Ge, J. M2 macrophage-derived exosomes carry microRNA-148a to alleviate myocardial ischemia/reperfusion injury via inhibiting TXNIP and the TLR4/NF-κB/NLRP3 inflammasome signaling pathway. J. Mol. Cell. Cardiol. 2020, 142, 65–79. [Google Scholar] [CrossRef]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibáñez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef]
- Yue, R.; Lu, S.; Luo, Y.; Zeng, J.; Liang, H.; Qin, D.; Wang, X.; Wang, T.; Pu, J.; Hu, H. Mesenchymal stem cell-derived exosomal microRNA-182-5p alleviates myocardial ischemia/reperfusion injury by targeting GSDMD in mice. Cell Death Discov. 2022, 8, 202. [Google Scholar] [CrossRef]
- Wang, K.; Li, Y.; Qiang, T.; Chen, J.; Wang, X. Role of epigenetic regulation in myocardial ischemia/reperfusion injury. Pharmacol. Res. 2021, 170, 105743. [Google Scholar] [CrossRef]
- Archer, K.; Broskova, Z.; Bayoumi, A.S.; Teoh, J.P.; Davila, A.; Tang, Y.; Su, H.; Kim, I.M. Long Non-Coding RNAs as Master Regulators in Cardiovascular Diseases. Int. J. Mol. Sci. 2015, 16, 23651–23667. [Google Scholar] [CrossRef]
- Ding, S.; Liu, D.; Wang, L.; Wang, G.; Zhu, Y. Inhibiting MicroRNA-29a Protects Myocardial Ischemia-Reperfusion Injury by Targeting SIRT1 and Suppressing Oxidative Stress and NLRP3-Mediated Pyroptosis Pathway. J. Pharmacol. Exp. Ther. 2020, 372, 128–135. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, K.S.; Liu, L.; Li, S.L. MicroRNA-132 promotes oxidative stress-induced pyroptosis by targeting sirtuin 1 in myocardial ischaemia-reperfusion injury. Int. J. Mol. Med. 2020, 45, 1942–1950. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.J.; Ji, Y.X.; Zhang, P.; Deng, K.Q.; Gong, J.; Ren, S.; Wang, X.; Chen, I.; Wang, H.; et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat. Med. 2016, 22, 1131–1139. [Google Scholar] [CrossRef]
- Ritter, N.; Ali, T.; Kopitchinski, N.; Schuster, P.; Beisaw, A.; Hendrix, D.A.; Schulz, M.H.; Müller-McNicoll, M.; Dimmeler, S.; Grote, P. The lncRNA Locus Handsdown Regulates Cardiac Gene Programs and Is Essential for Early Mouse Development. Dev. Cell 2019, 50, 644–657. [Google Scholar] [CrossRef]
- Kang, H.; Yu, H.; Zeng, L.; Ma, H.; Cao, G. LncRNA Rian reduces cardiomyocyte pyroptosis and alleviates myocardial ischemia-reperfusion injury by regulating by the miR-17-5p/CCND1 axis. Hypertens. Res. 2022, 45, 976–989. [Google Scholar] [CrossRef]
- Sun, J.; Zhu, Y.M.; Liu, Q.; Hu, Y.H.; Li, C.; Jie, H.H.; Xu, G.H.; Xiao, R.J.; Xing, X.L.; Yu, S.C.; et al. LncRNA ROR modulates myocardial ischemia-reperfusion injury mediated by the miR-185-5p/CDK6 axis. Lab. Investig. 2022, 102, 505–514. [Google Scholar] [CrossRef]
- Li, C.; Song, H.; Chen, C.; Chen, S.; Zhang, Q.; Liu, D.; Li, J.; Dong, H.; Wu, Y.; Liu, Y. LncRNA PVT1 Knockdown Ameliorates Myocardial Ischemia Reperfusion Damage via Suppressing Gasdermin D-Mediated Pyroptosis in Cardiomyocytes. Front. Cardiovasc. Med. 2021, 8, 747802. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Zong, L.; Wang, W. CircANXA2 Promotes Myocardial Apoptosis in Myocardial Ischemia-Reperfusion Injury via Inhibiting miRNA-133 Expression. BioMed Res. Int. 2020, 2020, 8590861. [Google Scholar] [CrossRef]
- Bai, M.; Pan, C.L.; Jiang, G.X.; Zhang, Y.M.; Zhang, Z. CircHIPK3 aggravates myocardial ischemia-reperfusion injury by binding to miRNA-124-3p. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 10107–10114. [Google Scholar] [CrossRef]
- Chang, H.; Li, Z.B.; Wu, J.Y.; Zhang, L. Circ-100338 induces angiogenesis after myocardial ischemia-reperfusion injury by sponging miR-200a-3p. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6323–6332. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Hang, Y.; Lu, Y.; Li, D.; Shen, F.; Guan, P.; Dong, J.; Shi, L.; Hu, W. CircRNA circ-NNT mediates myocardial ischemia/reperfusion injury through activating pyroptosis by sponging miR-33a-5p and regulating USP46 expression. Cell Death Discov. 2021, 7, 370. [Google Scholar] [CrossRef]
- Zhong, Y.; Li, Y.P.; Yin, Y.Q.; Hu, B.L.; Gao, H. Dexmedetomidine inhibits pyroptosis by down-regulating miR-29b in myocardial ischemia reperfusion injury in rats. Int. Immunopharmacol. 2020, 86, 106768. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Hu, S.; Liu, J.J.; Huang, L.; Zhong, P.; Fan, Z.X.; Ye, P.; Chen, M.H. Piperine protects against pyroptosis in myocardial ischaemia/reperfusion injury by regulating the miR-383/RP105/AKT signalling pathway. J. Cell. Mol. Med. 2021, 25, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Chen, Y.; Sun, Y.; Gao, Q.; Li, H.; Yang, Z.; Wang, Y.; Jiang, X.; Yu, B. Target of MCC950 in Inhibition of NLRP3 Inflammasome Activation: A Literature Review. Inflammation 2020, 43, 17–23. [Google Scholar] [CrossRef]
- van Hout, G.P.; Bosch, L.; Ellenbroek, G.H.; de Haan, J.J.; van Solinge, W.W.; Cooper, M.A.; Arslan, F.; de Jager, S.C.; Robertson, A.A.; Pasterkamp, G.; et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur. Heart J. 2017, 38, 828–836. [Google Scholar] [CrossRef] [Green Version]
- Mastrocola, R.; Penna, C.; Tullio, F.; Femminò, S.; Nigro, D.; Chiazza, F.; Serpe, L.; Collotta, D.; Alloatti, G.; Cocco, M.; et al. Pharmacological Inhibition of NLRP3 Inflammasome Attenuates Myocardial Ischemia/Reperfusion Injury by Activation of RISK and Mitochondrial Pathways. Oxidative Med. Cell. Longev. 2016, 2016, 5271251. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Lv, Q.; Zheng, M.; Sun, H.; Shi, F. NLRP3 inflammasome inhibitor INF39 attenuated NLRP3 assembly in macrophages. Int. Immunopharmacol. 2021, 92, 107358. [Google Scholar] [CrossRef]
- Audia, J.P.; Yang, X.M.; Crockett, E.S.; Housley, N.; Haq, E.U.; O’Donnell, K.; Cohen, M.V.; Downey, J.M.; Alvarez, D.F. Caspase-1 inhibition by VX-765 administered at reperfusion in P2Y(12) receptor antagonist-treated rats provides long-term reduction in myocardial infarct size and preservation of ventricular function. Basic Res. Cardiol. 2018, 113, 32. [Google Scholar] [CrossRef]
- Rout, A.; Tantry, U.S.; Novakovic, M.; Sukhi, A.; Gurbel, P.A. Targeted pharmacotherapy for ischemia reperfusion injury in acute myocardial infarction. Expert Opin. Pharmacother. 2020, 21, 1851–1865. [Google Scholar] [CrossRef] [PubMed]
- Do Carmo, H.; Arjun, S.; Petrucci, O.; Yellon, D.M.; Davidson, S.M. The Caspase 1 Inhibitor VX-765 Protects the Isolated Rat Heart via the RISK Pathway. Cardiovasc. Drugs Ther. 2018, 32, 165–168. [Google Scholar] [CrossRef] [Green Version]
- Rossello, X.; Yellon, D.M. The RISK pathway and beyond. Basic Res. Cardiol. 2018, 113, 2. [Google Scholar] [CrossRef] [Green Version]
- Deftereos, S.G.; Beerkens, F.J.; Shah, B.; Giannopoulos, G.; Vrachatis, D.A.; Giotaki, S.G.; Siasos, G.; Nicolas, J.; Arnott, C.; Patel, S.; et al. Colchicine in Cardiovascular Disease: In-Depth Review. Circulation 2022, 145, 61–78. [Google Scholar] [CrossRef]
- Samuel, M.; Tardif, J.C.; Khairy, P.; Roubille, F.; Waters, D.D.; Grégoire, J.C.; Pinto, F.J.; Maggioni, A.P.; Diaz, R.; Berry, C.; et al. Cost-effectiveness of low-dose colchicine after myocardial infarction in the Colchicine Cardiovascular Outcomes Trial (COLCOT). Eur. Heart J. Qual. Care Clin. Outcomes 2021, 7, 486–495. [Google Scholar] [CrossRef]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Bakhta, O.; Blanchard, S.; Guihot, A.L.; Tamareille, S.; Mirebeau-Prunier, D.; Jeannin, P.; Prunier, F. Cardioprotective Role of Colchicine Against Inflammatory Injury in a Rat Model of Acute Myocardial Infarction. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 446–455. [Google Scholar] [CrossRef]
- Tang, Y.; Shi, C.; Qin, Y.; Wang, S.; Pan, H.; Chen, M.; Yu, X.; Lou, Y.; Fan, G. Network Pharmacology-Based Investigation and Experimental Exploration of the Antiapoptotic Mechanism of Colchicine on Myocardial Ischemia Reperfusion Injury. Front. Pharmacol. 2021, 12, 804030. [Google Scholar] [CrossRef]
- Huet, F.; Fauconnier, J.; Legall, M.; Sicard, P.; Lozza, C.; Lacampagne, A.; Roubille, F. Low-dose colchicine prevents sympathetic denervation after myocardial ischemia-reperfusion: A new potential protective mechanism. Future Sci. OA 2020, 7, Fso656. [Google Scholar] [CrossRef]
- Akodad, M.; Fauconnier, J.; Sicard, P.; Huet, F.; Blandel, F.; Bourret, A.; de Santa Barbara, P.; Aguilhon, S.; LeGall, M.; Hugon, G.; et al. Interest of colchicine in the treatment of acute myocardial infarct responsible for heart failure in a mouse model. Int. J. Cardiol. 2017, 240, 347–353. [Google Scholar] [CrossRef]
- Dia, M.; Paccalet, A.; Pillot, B.; Leon, C.; Ovize, M.; Crola Da Silva, C.; Bochaton, T.; Paillard, M. Myocardial Ischemia-Reperfusion and Diabetes: Lessons Learned From Bedside to Bench. Front. Cardiovasc. Med. 2021, 8, 660698. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Cui, R.; Wang, C.; Feng, Y.; Li, Z.; Tong, Y.; Qu, K.; Liu, C.; Zhang, J. Metformin protects against intestinal ischemia-reperfusion injury and cell pyroptosis via TXNIP-NLRP3-GSDMD pathway. Redox Biol. 2020, 32, 101534. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zhu, L.; Liu, J.; Zhu, T.; Xie, Z.; Sun, X.; Zhang, H. Metformin Protects against Oxidative Stress Injury Induced by Ischemia/Reperfusion via Regulation of the lncRNA-H19/miR-148a-3p/Rock2 Axis. Oxidative Med. Cell. Longev. 2019, 2019, 8768327. [Google Scholar] [CrossRef]
- Huijink, T.M.; Venema, L.H.; Posma, R.A.; de Vries, N.J.; Westerkamp, A.C.; Ottens, P.J.; Touw, D.J.; Nijsten, M.W.; Leuvenink, H.G.D. Metformin Preconditioning and Postconditioning to Reduce Ischemia Reperfusion Injury in an Isolated Ex Vivo Rat and Porcine Kidney Normothermic Machine Perfusion Model. Clin. Transl. Sci. 2021, 14, 222–230. [Google Scholar] [CrossRef]
- Park, J.M.; Shin, J.H.; Yang, S.W.; Lee, J.Y.; Lee, C.L.; Lim, J.S.; Song, K.H.; Kim, G.H.; Na, Y.G. Metformin and Sildenafil Attenuate Inflammation and Suppress Apoptosis After Ischemia/Reperfusion Injuries in Rat Urinary Bladder. Int. Neurourol. J. 2021, 25, 285–295. [Google Scholar] [CrossRef]
- Asghari, A.; Akbari, G.; Meghdadi, A.; Mortazavi, P. Effects of melatonin and metformin co-administration on testicular ischemia/reperfusion injury in rats. J. Pediatric Urol. 2016, 12, e410.e1–e410.e7. [Google Scholar] [CrossRef]
- Demir, M.; Yilmaz, B.; Kalyoncu, S.; Tuncer, M.; Bozdag, Z.; Ince, O.; Bozdayi, M.A.; Ulusal, H.; Taysi, S. Metformin reduces ovarian ischemia reperfusion injury in rats by improving oxidative/nitrosative stress. Taiwan. J. Obstet. Gynecol. 2021, 60, 45–50. [Google Scholar] [CrossRef]
- Zhao, D.; Yang, J.; Yang, L. Insights for Oxidative Stress and mTOR Signaling in Myocardial Ischemia/Reperfusion Injury under Diabetes. Oxidative Med. Cell. Longev. 2017, 2017, 6437467. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, Z.; Zheng, G.; Yu, L.; Yin, Y.; Mu, N.; Ma, H. Metformin promotes autophagy in ischemia/reperfusion myocardium via cytoplasmic AMPKα1 and nuclear AMPKα2 pathways. Life Sci. 2019, 225, 64–71. [Google Scholar] [CrossRef]
- Saisho, Y. Metformin and Inflammation: Its Potential Beyond Glucose-lowering Effect. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 196–205. [Google Scholar] [CrossRef]
- Bharath, L.P.; Nikolajczyk, B.S. The intersection of metformin and inflammation. Am. J. Physiol. Cell Physiol. 2021, 320, C873–C879. [Google Scholar] [CrossRef]
- Chen, X.; Li, X.; Zhang, W.; He, J.; Xu, B.; Lei, B.; Wang, Z.; Cates, C.; Rousselle, T.; Li, J. Activation of AMPK inhibits inflammatory response during hypoxia and reoxygenation through modulating JNK-mediated NF-κB pathway. Metabolism 2018, 83, 256–270. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, L.; Shi, X.; Yang, L.; Hua, F.; Ma, J.; Zhu, W.; Liu, X.; Xuan, R.; Shen, Y.; et al. Metformin protects against myocardial ischemia-reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway. Aging 2020, 12, 24270–24287. [Google Scholar] [CrossRef] [PubMed]
- Yoshitomi, O.; Cho, S.; Hara, T.; Shibata, I.; Maekawa, T.; Ureshino, H.; Sumikawa, K. Direct protective effects of dexmedetomidine against myocardial ischemia-reperfusion injury in anesthetized pigs. Shock 2012, 38, 92–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, Y.; Hirata, N.; Kawaguchi, R.; Tokinaga, Y.; Yamakage, M. Dexmedetomidine Maintains Its Direct Cardioprotective Effect Against Ischemia/Reperfusion Injury in Hypertensive Hypertrophied Myocardium. Anesth. Analg. 2018, 126, 443–452. [Google Scholar] [CrossRef]
- Marzilli, M.; Vinereanu, D.; Lopaschuk, G.; Chen, Y.; Dalal, J.J.; Danchin, N.; Etriby, E.; Ferrari, R.; Gowdak, L.H.; Lopatin, Y.; et al. Trimetazidine in cardiovascular medicine. Int. J. Cardiol. 2019, 293, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Ford, I.; Fox, K.; Challeton, J.P.; Correges, A.; Tendera, M.; Widimský, P.; Danchin, N. Efficacy and safety of trimetazidine after percutaneous coronary intervention (ATPCI): A randomised, double-blind, placebo-controlled trial. Lancet 2020, 396, 830–838. [Google Scholar] [CrossRef]
- Chen, X.; Lin, S.; Dai, S.; Han, J.; Shan, P.; Wang, W.; Huang, Z.; Ye, B.; Huang, W. Trimetazidine affects pyroptosis by targeting GSDMD in myocardial ischemia/reperfusion injury. Inflamm. Res. 2022, 71, 227–241. [Google Scholar] [CrossRef]
- Ye, B.; Chen, X.; Dai, S.; Han, J.; Liang, X.; Lin, S.; Cai, X.; Huang, Z.; Huang, W. Emodin alleviates myocardial ischemia/reperfusion injury by inhibiting gasdermin D-mediated pyroptosis in cardiomyocytes. Drug Des. Dev. Ther. 2019, 13, 975–990. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Lei, Z.; Rao, Z.; Yang, R.; Zheng, L.; Fan, Y.; Luan, F.; Zeng, N. Cardioprotective activity of ethyl acetate extract of Cinnamomi Ramulus against myocardial ischemia/reperfusion injury in rats via inhibiting NLRP3 inflammasome activation and pyroptosis. Phytomedicine 2021, 93, 153798. [Google Scholar] [CrossRef]
- Reforgiato, M.R.; Milano, G.; Fabriàs, G.; Casas, J.; Gasco, P.; Paroni, R.; Samaja, M.; Ghidoni, R.; Caretti, A.; Signorelli, P. Inhibition of ceramide de novo synthesis as a postischemic strategy to reduce myocardial reperfusion injury. Basic Res. Cardiol. 2016, 111, 12. [Google Scholar] [CrossRef] [PubMed]
- Bonezzi, F.; Piccoli, M.; Dei Cas, M.; Paroni, R.; Mingione, A.; Monasky, M.M.; Caretti, A.; Riganti, C.; Ghidoni, R.; Pappone, C.; et al. Sphingolipid Synthesis Inhibition by Myriocin Administration Enhances Lipid Consumption and Ameliorates Lipid Response to Myocardial Ischemia Reperfusion Injury. Front. Physiol. 2019, 10, 986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Cai, W.; Du, R.; Li, H.; Wang, B.; Zhou, Y.; Shen, D.; Shen, H.; Lan, Y.; Chen, L.; et al. Corrigendum: Sevoflurane Alleviates Myocardial Ischemia Reperfusion Injury by Inhibiting P2X7-NLRP3 Mediated Pyroptosis. Front. Mol. Biosci. 2022, 9, 901322. [Google Scholar] [CrossRef]
- Shokoples, B.G.; Paradis, P.; Schiffrin, E.L. P2X7 Receptors: An Untapped Target for the Management of Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 186–199. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, Z.; Liu, X.; Yin, H.Y.; Tang, Y.; Cao, X. P2X7 Receptor-Mediated Inflammation in Cardiovascular Disease. Front. Pharmacol. 2021, 12, 654425. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Ito, M.; Ichihara, M.; Ito, M. Molecular hydrogen as an emerging therapeutic medical gas for neurodegenerative and other diseases. Oxidative Med. Cell. Longev. 2012, 2012, 353152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Yang, H.; Fan, Y.; Li, L.; Fang, J.; Yang, W. Hydrogen-Rich Saline Attenuates Cardiac and Hepatic Injury in Doxorubicin Rat Model by Inhibiting Inflammation and Apoptosis. Mediat. Inflamm. 2016, 2016, 1320365. [Google Scholar] [CrossRef] [Green Version]
- Chi, J.; Li, Z.; Hong, X.; Zhao, T.; Bie, Y.; Zhang, W.; Yang, J.; Feng, Z.; Yu, Z.; Xu, Q.; et al. Inhalation of Hydrogen Attenuates Progression of Chronic Heart Failure via Suppression of Oxidative Stress and P53 Related to Apoptosis Pathway in Rats. Front. Physiol. 2018, 9, 1026. [Google Scholar] [CrossRef]
- Ohsawa, I.; Nishimaki, K.; Yamagata, K.; Ishikawa, M.; Ohta, S. Consumption of hydrogen water prevents atherosclerosis in apolipoprotein E knockout mice. Biochem. Biophys. Res. Commun. 2008, 377, 1195–1198. [Google Scholar] [CrossRef]
- Kawamura, T.; Huang, C.S.; Tochigi, N.; Lee, S.; Shigemura, N.; Billiar, T.R.; Okumura, M.; Nakao, A.; Toyoda, Y. Inhaled hydrogen gas therapy for prevention of lung transplant-induced ischemia/reperfusion injury in rats. Transplantation 2010, 90, 1344–1351. [Google Scholar] [CrossRef]
- Lee, J.W.; Kim, J.I.; Lee, Y.A.; Lee, D.H.; Song, C.S.; Cho, Y.J.; Han, J.S. Inhaled hydrogen gas therapy for prevention of testicular ischemia/reperfusion injury in rats. J. Pediatric Surg. 2012, 47, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; Ding, X.; A, R.; Zheng, M.; Li, Z.; Pan, S.; Yang, W. Hydrogen gas inhalation alleviates myocardial ischemia-reperfusion injury by the inhibition of oxidative stress and NLRP3-mediated pyroptosis in rats. Life Sci. 2021, 272, 119248. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Non-Coding RNAs | Experimental Model | Targeted Gene | Expression with MIRI | Mechanism | First Author/Year |
|---|---|---|---|---|---|
| MiRNA-29a | Myocardial cells hypoxia/ reoxygenation model | SIRT1 | Increased | Activate oxidative stress and NLRP3-mediated pyroptosis pathway | Ding et al. 2020 [61] |
| MiRNA-29b | MIR rat and myocardial cells hypoxia/ reoxygenation model | FoxO3a/ARC | Increased | Activate pyroptosis and inflammatory reaction | Zhong et al. 2020 [73] |
| MiRNA-132 | MIR rat and myocardial cells hypoxia/ reoxygenation model | SIRT1 | Increased | Activate oxidative stress and pyroptosis | Zhou et al. 2020 [62] |
| MiRNA-383 | MIR rat model | RP105 | Increased | Activate cardiomyocyte pyroptosis | Guo et al. 2021 [74] |
| LncRNA Rian | MIR rat and myocardial cells oxygen-glucose deprivation/reoxygenation model | MiRNA-17-5p | Decreased | Reduce cardiomyocyte pyroptosis | Kang et al. 2022 [65] |
| LncRNA ROR | Myocardial cells hypoxia/ reoxygenation model | MiRNA-185-5p | Increased | Activate cardiomyocyte pyroptosis | Sun et al. 2022 [66] |
| LncRNA PVT1 | MIR rat and myocardial cells hypoxia/ reoxygenation model | GSDMD | Increased | Activate cardiomyocyte pyrotosis | Li et al. 2021 [67] |
| CircRNA-NNT | MIR rat and myocardial cells hypoxia/reoxygenation model | MiRNA-33a-5p | Increased | Activate cardiomyocyte pyroptosis | Ye et al. 2021 [72] |
| Drug | Experimental Model | Mechanism of Action | Effect | First Author/Year |
|---|---|---|---|---|
| MCC950 | In a vivo pig model of myocardial infarction | Selectively inhibits NLRP3-inflammasome formation and reduces pyroptosis, IL-18, and IL-1b signaling. | Reduces infarct size circulating markers of damage and inflammation and the influx of myocardial neutrophil and preserves cardiac function. | Hout et al. 2017 [77] |
| INF4E | In a vitro rat model of MIRI | Inhibits the NLRP3 inflammasome, activates the prosurvival RISK pathway, and improves mitochondrial function. | Reduces infarct size and lactate dehydrogenase release and improves the ventricular pressure in the postischemic left. | Mastrocola et al. 2016 [78] |
| VX-756 | In a vitro rat model of MIRI | Selectively inhibits prodrug caspase 1 and actives the PI3K/Akt pathway (the reperfusion injury salvage kinase (RISK) pathway). | Reduces infarct size. | Carmo et al. 2018 [82] |
| Colchicine | In a vivo rat model of MIRI | Increases the level of IL-10 and decreases the level of cardiac TGF-β. | Reduces infarct size and inhibits the increased expression of inflammatory cytokines. | Bakhta et al. 2018 [87] |
| Metformin | In a vitro rat model of MIRI and ventricle myocytes hypoxi/reoxygenation model | Enhances the AMPK pathway and suppresses the activation of NLRP3 inflammasome. | Alleviates myocardial infarct size, attenuates cell apoptosis, inhibits myocardial fibrosis, and decreases the level of pro-inflammatory cytokines, such as TNF-α, IL-6, and IL-1β, as well as decreases the activation of NLRP3 inflammasome. | Zhang et al. 2020 [103] |
| Dexmedetomidine | In a vivo rat model of MIRI and ventricle myocytes hypoxi/reoxygenation model | Downregulating miR-29b to activate FoxO3a/ARC axis to attenuate cell pyroptosis and ameliorate inflammatory response. | Reduces the size of myocardial infarction and the expression levels of cellular inflammation and pyroptosis-related proteins or markers. | Zhong et al. 2020 [73] |
| Trimetazidine | In a vivo rat model of MIRI and ventricle myocytes hypoxi/reoxygenation model | Alleviates pyroptosis through the TLR4/MyD88/ NF-κB/NLRP3 inflammasome pathway. | Increases the viability of cardiomyocytes, reduces the infarct size, and inhibits noncanonical inflammasome signaling. | Chen et al. 2022 [108] |
| Emodin | In a vivo rat model of MIRI and ventricle myocytes hypoxi/reoxygenation model | Alleviates pyroptosis through the TLR4/MyD88/ NF-κB/NLRP3 inflammasome pathway. | Increases the rate of cell survival in vitro and decreases the myocardial infarct size in vivo. | Ye et al. 2019 [109] |
| Cinnamyl ethyl acetate | In a vivo rat model of MIRI | Suppresses NLRP3 inflammasome and subsequent pyroptosis-related signaling pathways. | Decreases myocardial infarct size and improves cardiac function, mitigates myocardial damage, and represses inflammatory response. | Peng et al. 2021 [110] |
| Sevoflurane | Patients with a history of myocardial ischemia who underwent abdominal surgery with Sevoflurane general anesthesia and ventricle myocytes hypoxi/reoxygenation model. | Inhibits the expression of IL-1β, IL-18, and GSDMD by inhibiting the P2X7-NLRP3 signaling pathway to regulate inflammatory reaction and pyroptosis. | Reduces myocardial infarct size, the expression of inflammatory factors, and infiltration of inflammatory cells. | Wu et al. 2022 [113] |
| Hydrogen | In a vivo rat model of MIRI | Inhibits oxidative stress and NLRP3-mediated pyroptosis. | Improves myocardial infarct size, no-reflow area, cardiac function, microstructure, and mitochondrial morphology. | Nie et al. 2021 [122] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Zhang, J.; Zhang, D.; Yu, P.; Zhang, J.; Yu, S. Research Progress on the Role of Pyroptosis in Myocardial Ischemia-Reperfusion Injury. Cells 2022, 11, 3271. https://doi.org/10.3390/cells11203271
Liu Y, Zhang J, Zhang D, Yu P, Zhang J, Yu S. Research Progress on the Role of Pyroptosis in Myocardial Ischemia-Reperfusion Injury. Cells. 2022; 11(20):3271. https://doi.org/10.3390/cells11203271
Chicago/Turabian StyleLiu, Yang, Jing Zhang, Deju Zhang, Peng Yu, Jun Zhang, and Shuchun Yu. 2022. "Research Progress on the Role of Pyroptosis in Myocardial Ischemia-Reperfusion Injury" Cells 11, no. 20: 3271. https://doi.org/10.3390/cells11203271