
Tumor-Induced T Cell Polarization by Schwann Cells
 , , and
, , and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Lines and Cell Cultures
2.3. Chemotaxis Assay
2.4. T Cell Analysis
2.5. PGE2 Analysis
2.6. TGF-β1 Production
2.7. Western Blot Analysis
2.8. Quantitative PCR
2.9. Statistical Analysis
3. Results
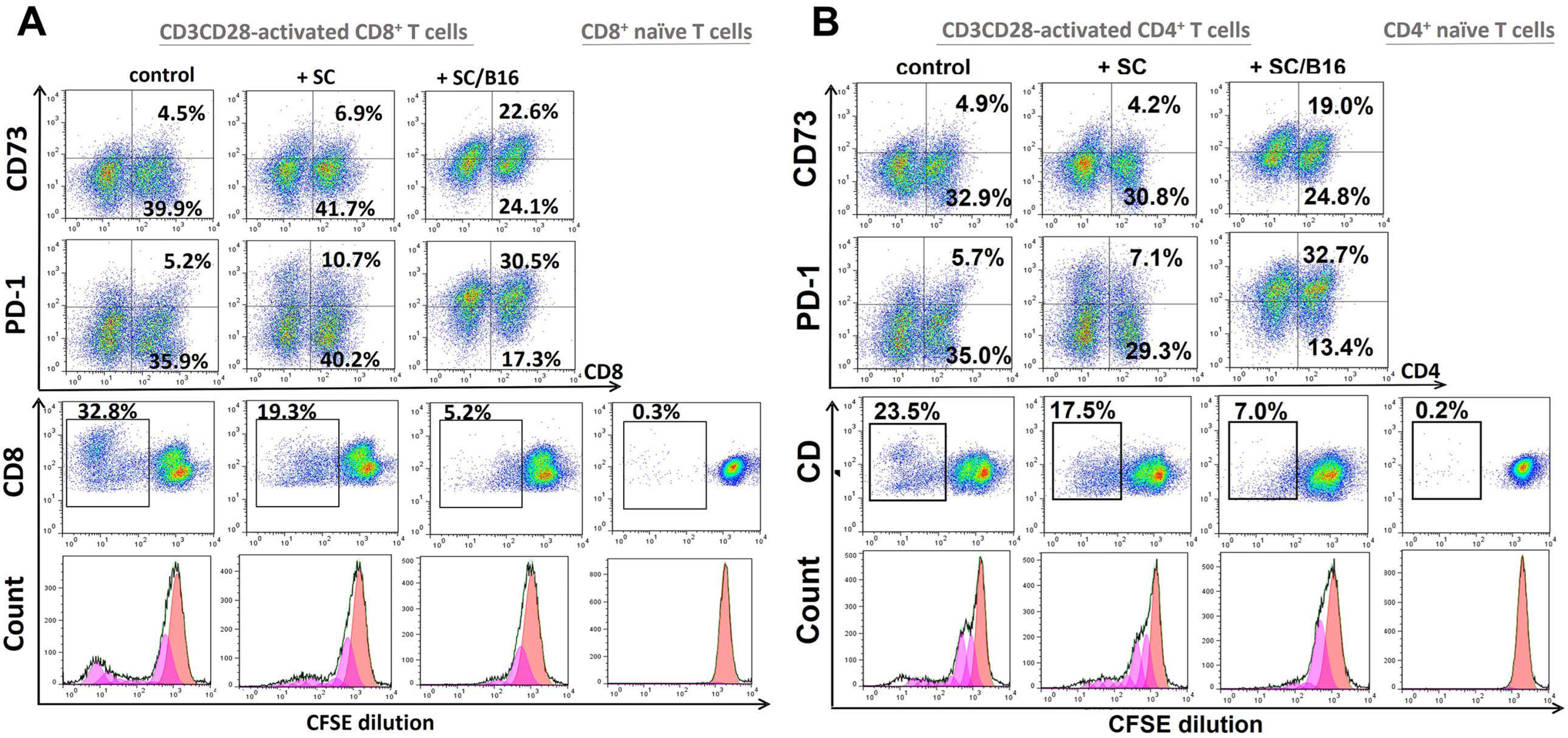
3.1. Polarization of T Cells by Tumor-Treated Schwann Cells
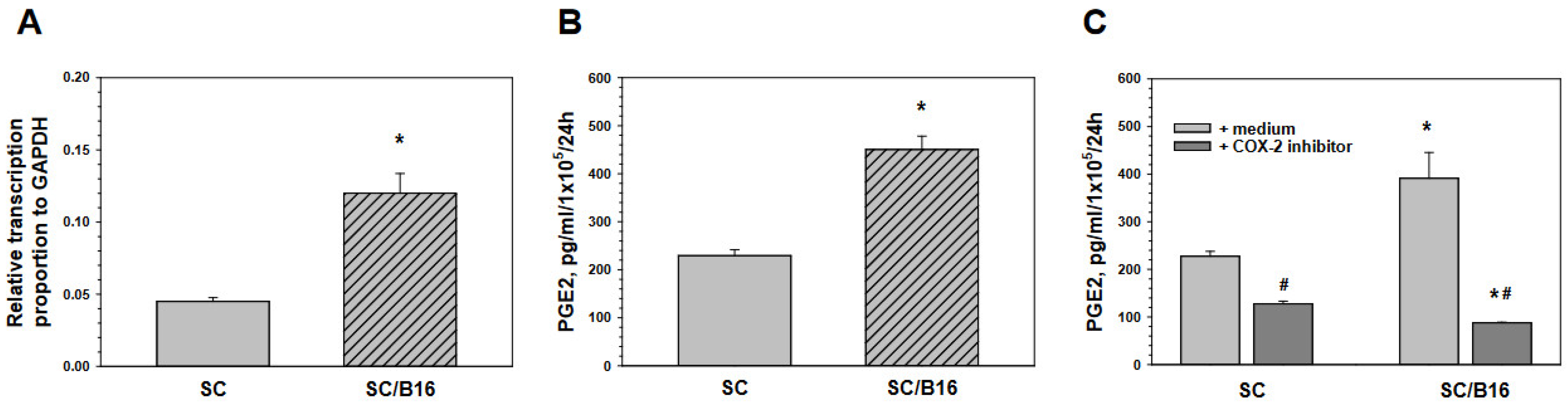
3.2. Schwann Cell-Derived Prostaglandin E2 Induces T Cell Polarization
3.3. Tumor-Derived TGF-β Activates Schwann Cells to Produce Prostaglandin E
3.4. Tumor-Induced Prostaglandin E Production by Schwann Cells Is Mediated by SMAD and ERK Signaling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, E.A.; Kessler, J.A. TGF-β Family Signaling in Neural and Neuronal Differentiation, Development, and Function. Cold Spring Harb. Perspect. Biol. 2017, 9, a022244. [Google Scholar] [CrossRef] [Green Version]
- Hiew, L.-F.; Poon, C.-H.; You, H.-Z.; Lim, L.-W. TGF-β/Smad Signalling in Neurogenesis: Implications for Neuropsychiatric Diseases. Cells 2021, 10, 1382. [Google Scholar] [CrossRef] [PubMed]
- Travis, M.A.; Sheppard, D. TGF-β activation and function in immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.Y.; Flavell, R.A. TGF-β and Regulatory T Cell in Immunity and Autoimmunity. J. Clin. Immunol. 2008, 28, 647–659. [Google Scholar] [CrossRef] [Green Version]
- Palomares, O.; Martín-Fontecha, M.; Lauener, R.; Traidl-Hoffmann, C.; Cavkaytar, O.; Akdis, M.; Akdis, C.A. Regulatory T cells and immune regulation of allergic diseases: Roles of IL-10 and TGF-β. Genes Immun. 2014, 15, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9, a022236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferdoushi, A.; Griffin, N.; Marsland, M.; Xu, X.; Faulkner, S.; Gao, F.; Liu, H.; King, S.J.; Denham, J.W.; van Helden, D.F.; et al. Tumor innervation and clinical outcome in pancreatic cancer. Sci. Rep. 2021, 11, 7390. [Google Scholar] [CrossRef] [PubMed]
- Albo, D.; Akay, C.L.; Marshall, C.L.; Wilks, J.A.; Verstovsek, G.; Liu, H.; Agarwal, N.; Berger, D.H.; Ayala, G.E. Neurogenesis in colorectal cancer is a marker of aggressive tumor behavior and poor outcomes. Cancer 2011, 117, 4834–4845. [Google Scholar] [CrossRef]
- Pundavela, J.; Demont, Y.; Jobling, P.; Lincz, L.F.; Roselli, S.; Thorne, R.F.; Bond, D.; Bradshaw, R.A.; Walker, M.M.; Hondermarck, H. ProNGF correlates with Gleason score and is a potential driver of nerve infiltration in prostate cancer. Am. J. Pathol. 2014, 184, 3156–3162. [Google Scholar] [CrossRef]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic Nerve Development Contributes to Prostate Cancer Progression. Science 2013, 341, 1236361. [Google Scholar] [CrossRef] [Green Version]
- Reavis, H.D.; Chen, H.I.; Drapkin, R. Tumor Innervation: Cancer Has Some Nerve. Trends Cancer 2020, 6, 1059–1067. [Google Scholar] [CrossRef]
- Silverman, D.A.; Martinez, V.K.; Dougherty, P.M.; Myers, J.N.; Calin, G.A.; Amit, M. Cancer-Associated Neurogenesis and Nerve-Cancer Cross-talk. Cancer Res. 2021, 81, 1431–1440. [Google Scholar] [CrossRef]
- Bunimovich, Y.L.; Keskinov, A.A.; Shurin, G.V.; Shurin, M.R. Schwann cells: A new player in the tumor microenvironment. Cancer Immunol. Immunother. 2017, 66, 959–968. [Google Scholar] [CrossRef] [Green Version]
- Martyn, G.V.; Shurin, G.V.; Keskinov, A.A.; Bunimovich, Y.L.; Shurin, M.R. Schwann cells shape the neuro-immune environs and control cancer progression. Cancer Immunol. Immunother. 2019, 68, 1819–1829. [Google Scholar] [CrossRef]
- Zhou, Y.; Shurin, G.V.; Zhong, H.; Bunimovich, Y.L.; Han, B.; Shurin, M.R. Schwann Cells Augment Cell Spreading and Metastasis of Lung Cancer. Cancer Res. 2018, 78, 5927–5939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.H.; Shurin, G.V.; Khosravi, H.; Kazi, R.; Kruglov, O.; Shurin, M.R.; Bunimovich, Y.L. Immunomodulation by Schwann cells in disease. Cancer Immunol. Immunother. 2020, 69, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.-W.; Li, Y.-C.; Ma, S.-R.; Mao, L.; Yu, G.-T.; Bu, L.-L.; Kulkarni, A.B.; Zhang, W.-F.; Sun, Z.-J. Specific blockade CD73 alters the “exhausted” phenotype of T cells in head and neck squamous cell carcinoma. Int. J. Cancer 2018, 143, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Kurachi, M. CD8(+) T cell exhaustion. Semin Immunopathol. 2019, 41, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Al-Taei, S.; Salimu, J.; Spary, L.K.; Clayton, A.; Lester, J.F.; Tabi, Z. Prostaglandin E(2)-mediated adenosinergic effects on CD14(+) cells: Self-amplifying immunosuppression in cancer. Oncoimmunology 2016, 6, e1268308. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Yao, J.; Niu, X. The Signaling Pathway of PGE and Its Regulatory Role in T Cell Differentiation. Mediat. Inflamm. 2021, 2021, 9087816. [Google Scholar] [CrossRef]
- Sulaiman, W.; Dreesen, T.D. Effect of local application of transforming growth factor-β at the nerve repair site following chronic axotomy and denervation on the expression of regeneration-associated genes. Laboratory investigation. J. Neurosurg. 2014, 121, 859–874. [Google Scholar] [CrossRef]
- Li, S.; Gu, X.; Yi, S. The Regulatory Effects of Transforming Growth Factor-β on Nerve Regeneration. Cell Transpl. 2017, 26, 381–394. [Google Scholar] [CrossRef] [Green Version]
- Harding, P.; Balasubramanian, L.; Swegan, J.; Stevens, A.; Glass, W.F. Transforming growth factor beta regulates cyclooxygenase-2 in glomerular mesangial cells. Kidney Int. 2006, 69, 1578–1585. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.-S.; Cheng, R.-H.; Chang, M.-C.; Lee, J.-J.; Chang, H.-H.; Huang, W.-L.; Yeung, S.-Y.; Chang, Y.-C.; Jeng, J.-H. TGF-β1 stimulates cyclooxygenase-2 expression and PGE2 production of human dental pulp cells: Role of ALK5/Smad2 and MEK/ERK signal transduction pathways. J. Formos. Med. Assoc. 2017, 116, 748–754. [Google Scholar] [CrossRef]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shurin, M.R.; Shurin, G.V.; Zlotnikov, S.B.; Bunimovich, Y.L. The Neuroimmune Axis in the Tumor Microenvironment. J. Immunol. 2020, 204, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Chen, S.; Chen, M. Schwann Cells in the Tumor Microenvironment: Need More Attention. J. Oncol. 2022, 2022, 1058667. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, J.; Han, B.; Zhong, R.; Zhong, H. Schwann cells promote lung cancer proliferation by promoting the M2 polarization of macrophages. Cell Immunol. 2020, 357, 104211. [Google Scholar] [CrossRef]
- Deborde, S.; Omelchenko, T.; Lyubchik, A.; Zhou, Y.; He, S.; McNamara, W.F.; Chernichenko, N.; Lee, S.Y.; Barajas, F.; Chen, C.H.; et al. Schwann cells induce cancer cell dispersion and invasion. J. Clin. Investig. 2016, 126, 1538–1554. [Google Scholar] [CrossRef] [Green Version]
- Yurteri, Ü.; Çifcibaşı, K.; Friess, H.; Ceyhan, G.O.; Istvanffy, R.; Demir, I.E. Schwann Cells in Peripheral Cancers: Bystanders or Promoters? Adv. Biol. 2022, 6, e2200033. [Google Scholar] [CrossRef]
- He, X.; Xu, C. PD-1: A Driver or Passenger of T Cell Exhaustion? Mol. Cell 2020, 77, 930–931. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Schillebeeckx, I.; Earls, J.; Flanagan, K.C.; Hiken, J.; Bode, A.; Armstrong, J.R.; Messina, D.N.; Adkins, D.; Ley, J.; Alborelli, I.; et al. T cell subtype profiling measures exhaustion and predicts anti-PD-1 response. Sci. Rep. 2022, 12, 1342. [Google Scholar] [CrossRef]
- Smith, C.J.; Snyder, C.M. Inhibitory Molecules PD-1, CD73 and CD39 Are Expressed by CD8(+) T Cells in a Tissue-Dependent Manner and Can Inhibit T Cell Responses to Stimulation. Front. Immunol. 2021, 12, 704862. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Fan, J.; Zhang, M.; Qin, L.; Dominguez, D.; Long, A.; Wang, G.; Ma, R.; Li, H.; Zhang, Y.; et al. CD73 expression on effector T cells sustained by TGF-β facilitates tumor resistance to anti-4-1BB/CD137 therapy. Nat. Commun. 2019, 10, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, F.; Cao, W.; Zhu, W.; Lam, N.; Li, L.; Gaddam, S.; Wang, Y.; Kim, C.; Lambert, S.; Zhang, H.; et al. The cell-surface 5′-nucleotidase CD73 defines a functional T memory cell subset that declines with age. Cell Rep. 2021, 37, 109981. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Briceño, P.; Rivas-Yañez, E.; Rosemblatt, M.V.; Parra-Tello, B.; Farías, P.; Vargas, L.; Simon, V.; Cárdenas, C.; Lladser, A.; Salazar-Onfray, F.; et al. CD73 Ectonucleotidase Restrains CD8+ T Cell Metabolic Fitness and Anti-tumoral Activity. Front. Cell Dev. Biol. 2021, 9, 638037. [Google Scholar] [CrossRef]
- Panigrahi, S.; Bazdar, D.A.; Albakri, M.; Ferrari, B.; Antonelli, C.J.; Freeman, M.L.; Dubyak, G.; Zender, C.; Sieg, S.F. CD8+CD73+ T cells in the tumor microenvironment of head and neck cancer patients are linked to diminished T cell infiltration and activation in tumor tissue. Eur. J. Immunol. 2020, 50, 2055–2066. [Google Scholar] [CrossRef]
- Shevchenko, I.; Mathes, A.; Groth, C.; Karakhanova, S.; Müller, V.; Utikal, J.; Werner, J.; Bazhin, A.V.; Umansky, V. Enhanced expression of CD39 and CD73 on T cells in the regulation of anti-tumor immune responses. Oncoimmunology 2020, 9, 1744946. [Google Scholar] [CrossRef] [Green Version]
- Haanen, J.B.A.G.; Peters, S. Minus Times Minus Equals Plus. J. Clin. Oncol. 2022, 40, 3453–3455. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Liu, K.; Liu, C.; Wang, X.; Ma, W.; Xu, W.; Wu, J.; Sun, C. Tumor accomplice: T cell exhaustion induced by chronic inflammation. Front. Immunol. 2022, 13, 979116. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; McLennan, I.S.; Koishi, K.; Hendry, I.A. Transforming growth factor-beta 2 is anterogradely and retrogradely transported in motoneurons and up-regulated after nerve injury. Neuroscience 2000, 97, 735–742. [Google Scholar] [CrossRef]
- Einheber, S.; Hannocks, M.J.; Metz, C.N.; Rifkin, D.B.; Salzer, J.L. Transforming growth factor-beta 1 regulates axon/Schwann cell interactions. J. Cell Biol. 1995, 129, 443–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLennan, I.S.; Koishi, K. The transforming growth factor-betas: Multifaceted regulators of the development and maintenance of skeletal muscles, motoneurons and Schwann cells. Int. J. Dev. Biol. 2002, 46, 559–567. [Google Scholar] [PubMed]
- Fischer, S.M. Prostaglandins and cancer. Front Biosci. 1997, 2, d482–d500. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Dubois, R.N. Prostaglandins and cancer. Gut 2006, 55, 115–122. [Google Scholar] [CrossRef]
- Coceani, F. Prostaglandins and the Central Nervous System. Arch. Intern. Med. 1974, 133, 119–129. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Andreasson, K.I.; Isakson, P.C.; Worley, P.F. Cyclooxygenases and the central nervous system. Prostaglandins 1997, 54, 601–624. [Google Scholar] [CrossRef]
- Yagami, T.; Koma, H.; Yamamoto, Y. Pathophysiological Roles of Cyclooxygenases and Prostaglandins in the Central Nervous System. Mol. Neurobiol. 2016, 53, 4754–4771. [Google Scholar] [CrossRef]
- Zhang, J.; Rivest, S. Anti-inflammatory effects of prostaglandin E2 in the central nervous system in response to brain injury and circulating lipopolysaccharide. J. Neurochem. 2001, 76, 855–864. [Google Scholar] [CrossRef]
- Satoh, T.; Furuta, K.; Suzuki, M.; Watanabe, Y. Prostaglandin J2 and its metabolites promote neurite outgrowth induced by nerve growth factor in PC12 cells. Biochem. Biophys. Res. Commun. 1999, 258, 50–53. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D. Prostaglandins, COX-2, and sensory perception. Gut 2000, 47, iv66. [Google Scholar] [CrossRef] [Green Version]
- Johansson, T.; Narumiya, S.; Zeilhofer, H.U. Contribution of peripheral versus central EP1 prostaglandin receptors to inflammatory pain. Neurosci. Lett. 2011, 495, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Güllner, H.-G. Prostaglandin actions on the adrenergic nervous system. Klin. Wochenschr. 1983, 61, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, S.F.; Hsiao, H.Y.; Mak, O.T. Prostaglandins and cyclooxygenases in glial cells during brain inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Deadwyler, G.D.; Dang, I.; Nelson, J.; Srikanth, M.; De Vries, G.H. Prostaglandin E(2) metabolism is activated in Schwann cell lines derived from human NF1 malignant peripheral nerve sheath tumors. Neuron Glia Biol. 2004, 1, 149–155. [Google Scholar] [CrossRef]
- Takahashi, M.; Kawaguchi, M.; Shimada, K.; Konishi, N.; Furuya, H.; Nakashima, T. Cyclooxygenase-2 expression in Schwann cells and macrophages in the sciatic nerve after single spinal nerve injury in rats. Neurosci. Lett. 2004, 363, 203–206. [Google Scholar] [CrossRef]
- Sreeramkumar, V.; Fresno, M.; Cuesta, N. Prostaglandin E2 and T cells: Friends or foes? Immunol. Cell Biol. 2012, 90, 579–586. [Google Scholar] [CrossRef] [Green Version]
- Maseda, D.; Ricciotti, E.; Crofford, L.J. Prostaglandin regulation of T cell biology. Pharmacol. Res. 2019, 149, 104456. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shurin, G.V.; Vats, K.; Kruglov, O.; Bunimovich, Y.L.; Shurin, M.R. Tumor-Induced T Cell Polarization by Schwann Cells. Cells 2022, 11, 3541. https://doi.org/10.3390/cells11223541
Shurin GV, Vats K, Kruglov O, Bunimovich YL, Shurin MR. Tumor-Induced T Cell Polarization by Schwann Cells. Cells. 2022; 11(22):3541. https://doi.org/10.3390/cells11223541
Chicago/Turabian StyleShurin, Galina V., Kavita Vats, Oleg Kruglov, Yuri L. Bunimovich, and Michael R. Shurin. 2022. "Tumor-Induced T Cell Polarization by Schwann Cells" Cells 11, no. 22: 3541. https://doi.org/10.3390/cells11223541