
Pan-Cancer Analysis Identifies MNX1 and Associated Antisense Transcripts as Biomarkers for Cancer


Abstract
:1. Introduction
2. Methods
2.1. Data Availability, Sample Selection and Expression Analysis
2.2. Prediction of Targets
2.3. Differential Expression Analysis
2.4. Gene Ontology Analysis
2.5. Statistical Analysis
2.6. Clustering Analysis
2.7. Survival Analysis
3. Results
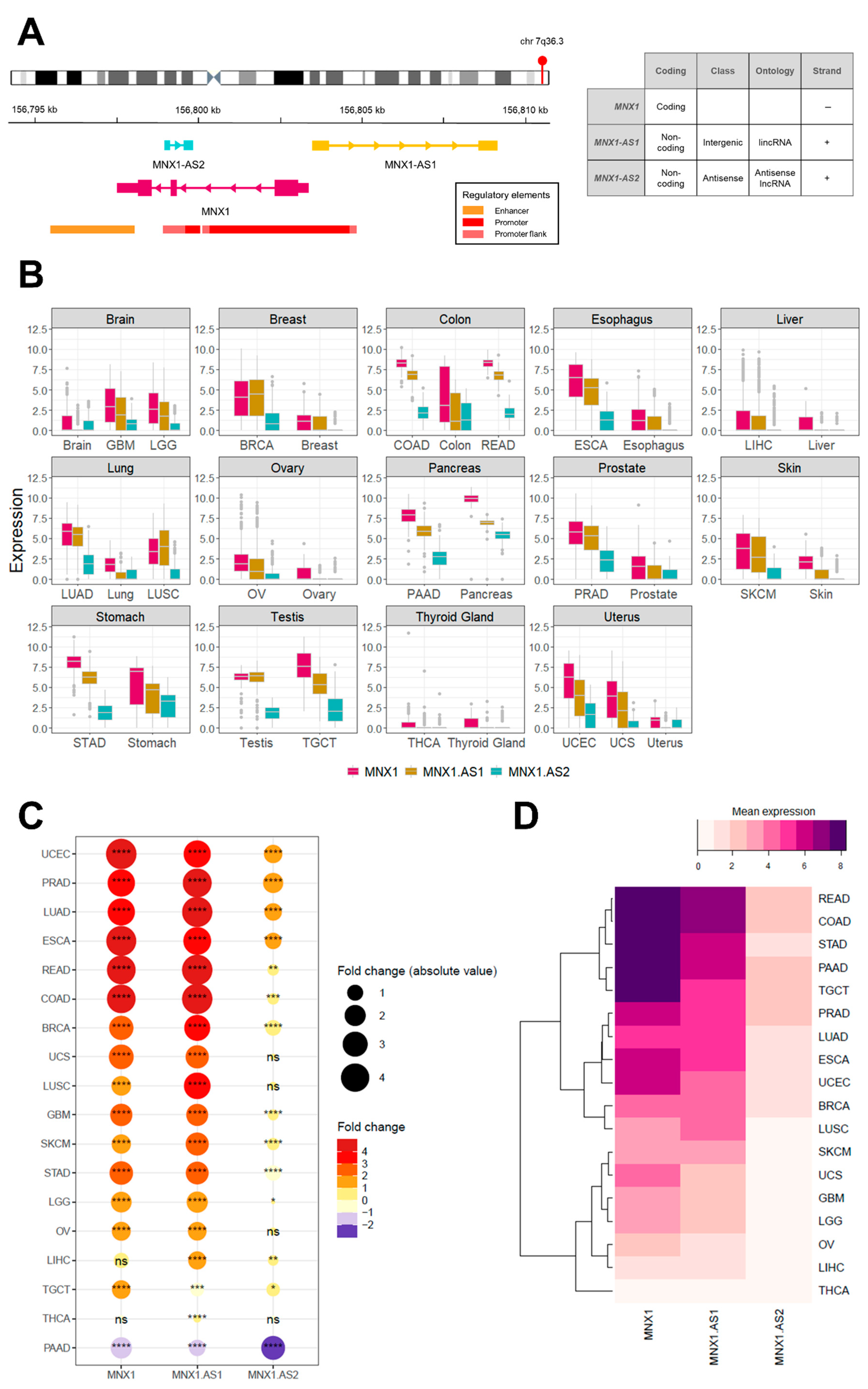
3.1. MNX1, MNX1-AS1, and MNX1-AS2 Are Dysregulated in Most Cancers
3.2. Diagnostic Biomarker Potential for MNX1, MNX1-AS1, and MNX1-AS2
3.3. MNX1, MNX1-AS1, and MNX1-AS2 Correlate with Oncologic Clinicopathological Features
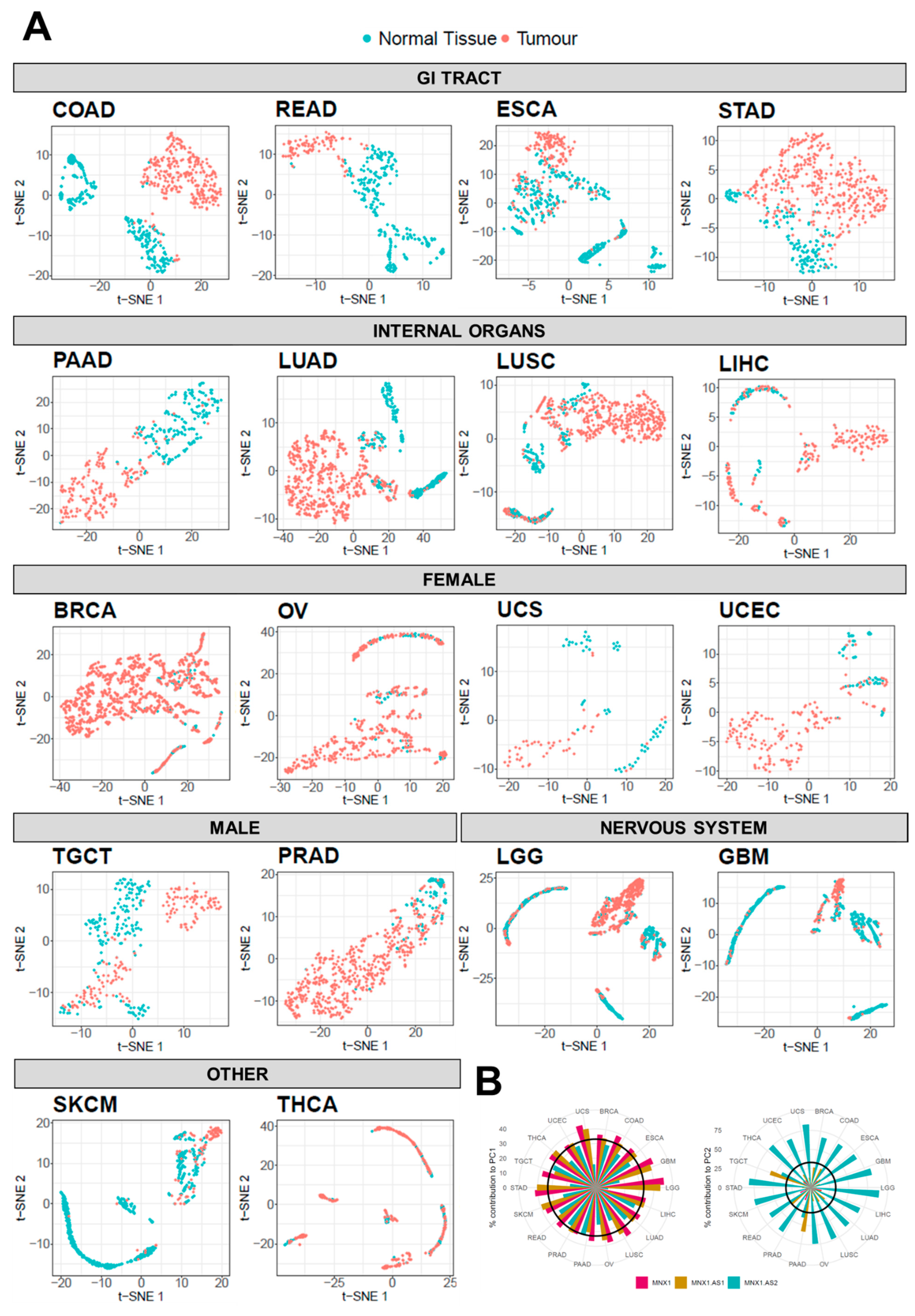
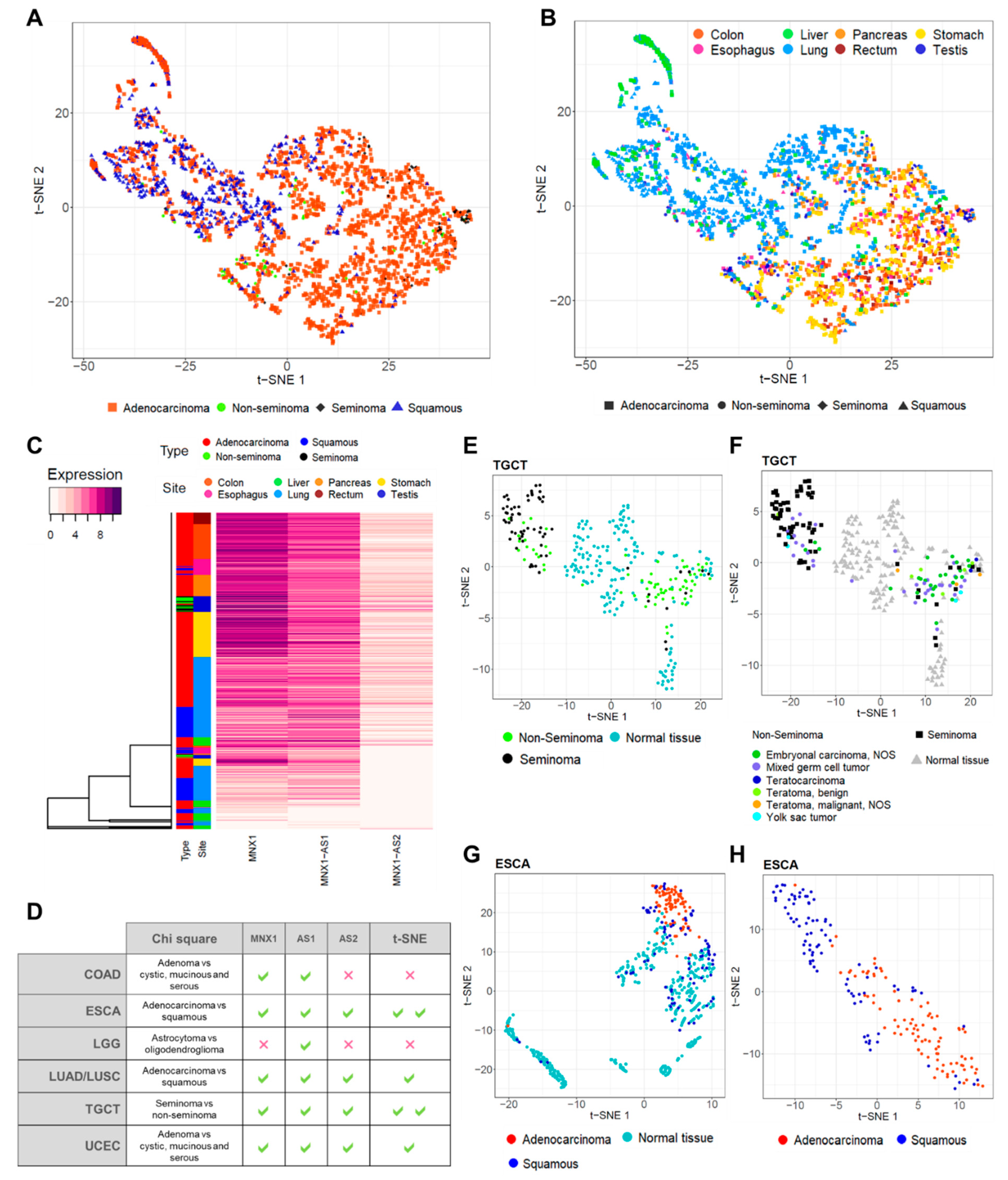
3.4. MNX1, MNX1-AS1, and MNX1-AS2 Distinguish Tumour Subtypes
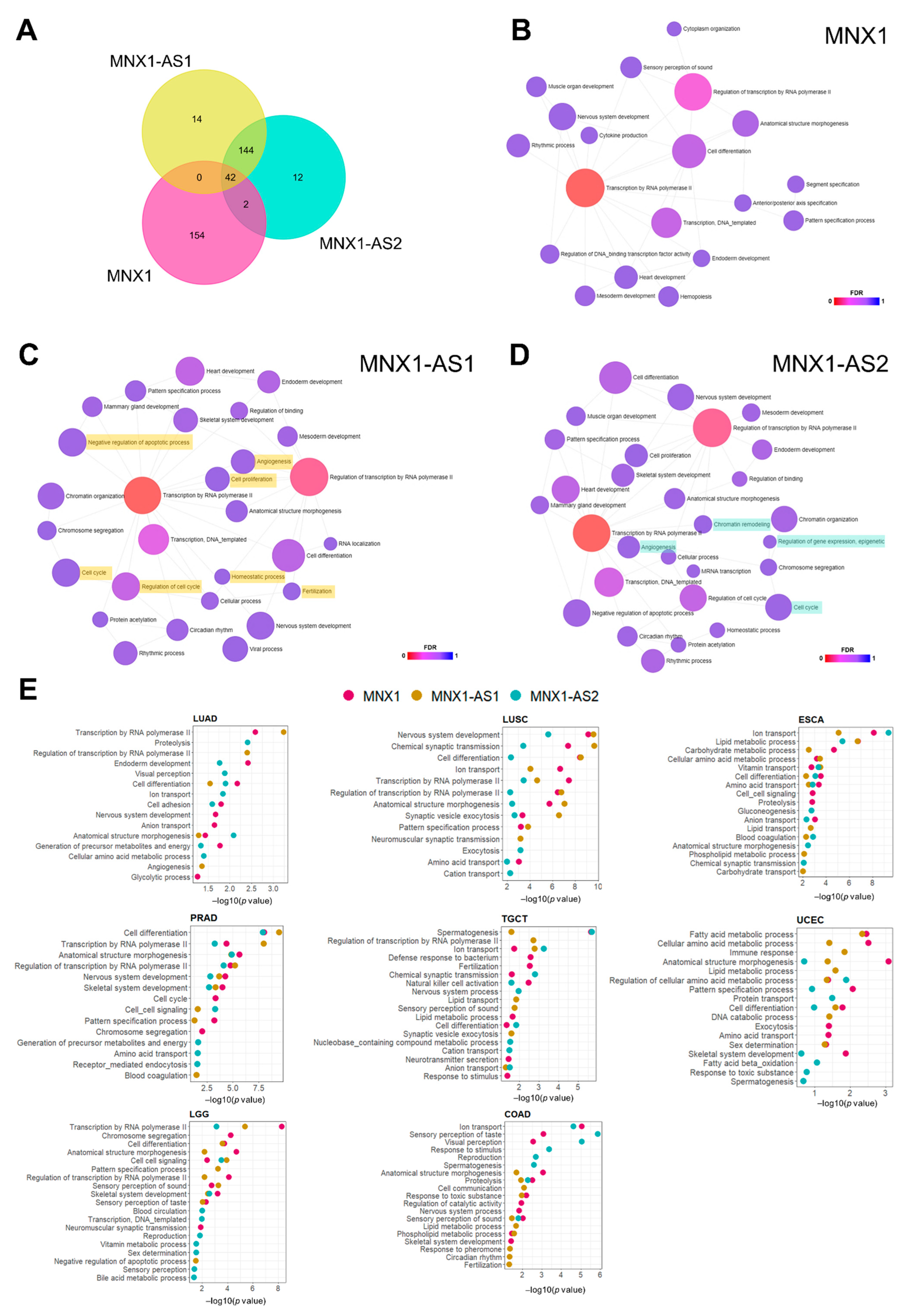
3.5. Prediction of Biological Functions of MNX1, MNX1-AS1, and MNX1-AS2 in Cancer
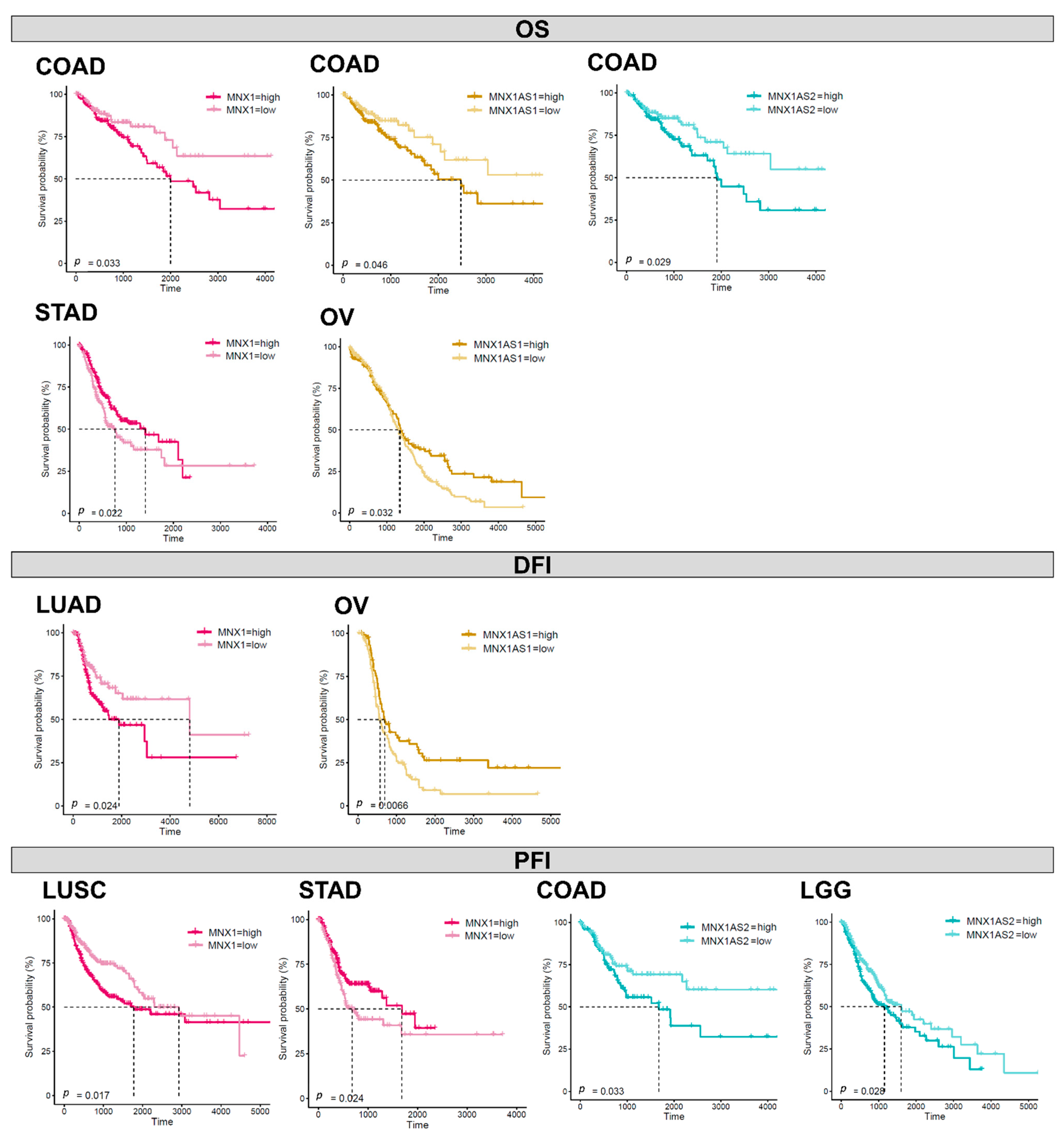
3.6. Prognostic Biomarker Potential for MNX1, MNX1-AS1, and MNX1-AS2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ahmad, A.S.; Ormiston-Smith, N.; Sasieni, P.D. Trends in the Lifetime Risk of Developing Cancer in Great Britain: Comparison of Risk for those Born from 1930 to 1960. Br. J. Cancer 2015, 112, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.; Creighton, C.J.; Davis, C.; Donehower, L.; Drummond, J.; Wheeler, D.; Ally, A.; Balasundaram, M.; Birol, I.; Butterfield, Y.S. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N. The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Han, B.; Mendelsohn, M.; Smith, M.; Jessell, T.M.; Sockanathan, S. Requirement for the Homeobox Gene Hb9 in the Consolidation of Motor Neuron Identity. Neuron 1999, 23, 659–674. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Arber, S.; Jessell, T.M.; Edlund, H. Selective Agenesis of the Dorsal Pancreas in Mice Lacking Homeobox Gene Hlxb9. Nat. Genet. 1999, 23, 67–70. [Google Scholar] [CrossRef]
- Thaler, J.; Harrison, K.; Sharma, K.; Lettieri, K.; Kehrl, J.; Pfaff, S.L. Active Suppression of Interneuron Programs within Developing Motor Neurons Revealed by Analysis of Homeodomain Factor HB9. Neuron 1999, 23, 675–687. [Google Scholar] [CrossRef] [Green Version]
- Harrison, K.A.; Thaler, J.; Pfaff, S.L.; Gu, H.; Kehrl, J.H. Pancreas Dorsal Lobe Agenesis and Abnormal Islets of Langerhans in Hlxb9-Deficient Mice. Nat. Genet. 1999, 23, 71–75. [Google Scholar] [CrossRef]
- Tosi, S.; Kamel, Y.M.; Owoka, T.; Federico, C.; Truong, T.H.; Saccone, S. Paediatric Acute Myeloid Leukaemia with the T (7; 12)(q36; p13) Rearrangement: A Review of the Biological and Clinical Management Aspects. Biomark. Res. 2015, 3, 21. [Google Scholar] [CrossRef] [Green Version]
- Nagel, S.; Scherr, M.; Quentmeier, H.; Kaufmann, M.; Zaborski, M.; Drexler, H.G.; MacLeod, R. HLXB9 Activates IL6 in Hodgkin Lymphoma Cell Lines and is Regulated by PI3K Signalling Involving E2F3. Leukemia 2005, 19, 841–846. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Wang, M.; Zhu, Y.; Zhu, W.; Yang, T.; Li, H.; Lin, S.; Dai, C.; Deng, Y.; Song, D. Expression, Clinical Significance, and Functional Prediction of MNX1 in Breast Cancer. Mol. Ther.-Nucleic Acids 2018, 13, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Neufing, P.J.; Kalionis, B.; Horsfall, D.J.; Ricciardelli, C.; Stahl, J.; Vivekanandan, S.; Raymond, W.; Tilley, W.D. Expression and Localization of Homeodomain Proteins DLX4/HB9 in Normal and Malignant Human Breast Tissues. Anticancer. Res. 2003, 23, 1479–1488. [Google Scholar] [PubMed]
- Zhang, L.; Wang, J.; Wang, Y.; Zhang, Y.; Castro, P.; Shao, L.; Sreekumar, A.; Putluri, N.; Guha, N.; Deepak, S. MNX1 is Oncogenically Upregulated in African-American Prostate Cancer. Cancer Res. 2016, 76, 6290–6298. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Wu, R.; Li, G.; Liu, C.; Tan, L.; Xiao, K.; Ye, Y.; Qin, Z. Motor Neuron and Pancreas Homeobox 1/HLXB9 Promotes Sustained Proliferation in Bladder Cancer by Upregulating CCNE1/2. J. Exp. Clin. Cancer Res. 2018, 37, 154. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Pan, Q.; Lu, Y.; Jiang, X.; Zhang, S.; Wu, J. MNX1 Promotes Cell Proliferation and Activates Wnt/Β-catenin Signaling in Colorectal Cancer. Cell Biol. Int. 2019, 43, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Leotta, C.G.; Federico, C.; Brundo, M.V.; Tosi, S.; Saccone, S. HLXB9 Gene Expression, and Nuclear Location during in Vitro Neuronal Differentiation in the SK-N-BE Neuroblastoma Cell Line. PLoS ONE 2014, 9, e105481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Chen, S.; Zhao, D.; Yan, J.; Chen, J.; Yang, C.; Zheng, G. MNX1 Reduces Sensitivity to Anoikis by Activating TrkB in Human Glioma Cells. Mol. Med. Rep. 2018, 18, 3271–3279. [Google Scholar] [CrossRef]
- Wilkens, L.; Jaggi, R.; Hammer, C.; Inderbitzin, D.; Giger, O.; von Neuhoff, N. The Homeobox Gene HLXB9 is Upregulated in a Morphological Subset of Poorly Differentiated Hepatocellular Carcinoma. Virchows Arch. 2011, 458, 697–708. [Google Scholar] [CrossRef]
- Desai, S.S.; Modali, S.D.; Parekh, V.I.; Kebebew, E.; Agarwal, S.K. GSK-3β Protein Phosphorylates and Stabilizes HLXB9 Protein in Insulinoma Cells to Form a Targetable Mechanism of Controlling Insulinoma Cell Proliferation. J. Biol. Chem. 2014, 289, 5386–5398. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Zhou, H. Clinical Effects and Molecular Mechanisms of lncRNA MNX1-AS1 in Malignant Tumors. Am. J. Transl. Res. 2020, 12, 7593. [Google Scholar]
- Li, J.; Jin, W.; Zhang, Z.; Chu, J.; Yang, H.; Li, C.; Dong, R.; Zhao, C. The prognostic utility and clinical outcomes of MNX1-AS1 expression in cancers: A systematic review and meta-analysis. Res. Sq. 2020. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N. Visualizing and Interpreting Cancer Genomics Data Via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Vivian, J.; Rao, A.A.; Nothaft, F.A.; Ketchum, C.; Armstrong, J.; Novak, A.; Pfeil, J.; Narkizian, J.; Deran, A.D.; Musselman-Brown, A. Toil Enables Reproducible, Open Source, Big Biomedical Data Analyses. Nat. Biotechnol. 2017, 35, 314–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.; Tang, Q.; He, J.; Li, L.; Yang, N.; Yu, S.; Wang, M.; Zhang, Y.; Lin, J.; Cui, T. RNAInter v4.0: RNA Interactome Repository with Redefined Confidence Scoring System and Improved Accessibility. Nucleic Acids Res. 2022, 50, D326–D332. [Google Scholar]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; Van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D. JASPAR 2020: Update of the Open-Access Database of Transcription Factor Binding Profiles. Nucleic Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef]
- Lin, G.; Chai, J.; Yuan, S.; Mai, C.; Cai, L.; Murphy, R.W.; Zhou, W.; Luo, J. VennPainter: A Tool for the Comparison and Identification of Candidate Genes Based on Venn Diagrams. PLoS ONE 2016, 11, e0154315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Maaten, L.; Hinton, G. Visualizing Data using T-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Li, J.; Li, Q.; Li, D.; Shen, Z.; Zhang, K.; Bi, Z.; Li, Y. Long Non-Coding RNA MNX1-AS1 Promotes Progression of Triple Negative Breast Cancer by Enhancing Phosphorylation of Stat3. Front. Oncol. 2020, 10, 1108. [Google Scholar] [CrossRef]
- Wu, Q.; Luo, X.; Liu, J.; Lu, Y.; Wang, Y.; Qi, J.; Liu, Z.; Huang, Q.; Liu, Z.; Lu, J. MYC-Activated lncRNA MNX1-AS1 Promotes the Progression of Colorectal Cancer by Stabilizing YB1. Cancer Res. 2021, 81, 2636–2650. [Google Scholar] [CrossRef]
- Chu, J.; Li, H.; Xing, Y.; Jia, J.; Sheng, J.; Yang, L.; Sun, K.; Qu, Y.; Zhang, Y.; Yin, H. LncRNA MNX1-AS1 Promotes Progression of Esophageal Squamous Cell Carcinoma by Regulating miR-34a/SIRT1 Axis. Biomed. Pharmacother. 2019, 116, 109029. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, L.; Lu, X.; Wang, K.; Ning, X.; Liu, Z. Upregulated Expression of MNX1-AS1 Long Noncoding RNA Predicts Poor Prognosis in Gastric Cancer. Bosn. J. Basic Med. Sci. 2019, 19, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Xu, Y.; Wang, J.; Yang, X.; Wen, L.; Feng, J. lncRNA MNX1-AS1 Promotes Glioblastoma Progression through Inhibition of miR-4443. Oncol. Res. 2019, 27, 341. [Google Scholar] [CrossRef] [PubMed]
- Ji, D.; Wang, Y.; Sun, B.; Yang, J.; Luo, X. Long Non-Coding RNA MNX1-AS1 Promotes Hepatocellular Carcinoma Proliferation and Invasion through Targeting miR-218-5p/COMMD8 Axis. Biochem. Biophys. Res. Commun. 2019, 513, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Han, L.; Liu, Z.; Gao, N. Long Noncoding RNA MNX1-AS1 Contributes to Lung Cancer Progression through the miR-527/BRF2 Pathway. J. Cell. Physiol. 2019, 234, 13843–13850. [Google Scholar] [CrossRef]
- Shen, Y.; Lv, M.; Fang, Y.; Lu, J.; Wu, Y. LncRNA MNX1-AS1 Promotes Ovarian Cancer Process Via Targeting the miR-744-5p/SOX12 Axis. J. Ovarian Res. 2021, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Li, H.; Li, F.; Liu, P.; Zhao, X. Long Noncoding RNA MNX1-AS1 Knockdown Inhibits Cell Proliferation and Migration in Ovarian Cancer. Cancer Biother. Radiopharm. 2017, 32, 91–99. [Google Scholar] [CrossRef]
- Li, Z.; Wang, F.; Zhang, S. Knockdown of Lnc RNA MNX1-AS1 Suppresses Cell Proliferation, Migration, and Invasion in Prostate Cancer. FEBS Open Bio. 2019, 9, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Long, W.; Rui, C.; Song, X.; Dai, X.; Xue, X.; Lu, Y.; Shen, R.; Li, J.; Li, J.; Ding, H. Distinct Expression Profiles of lncRNAs between Early-Onset Preeclampsia and Preterm Controls. Clin. Chim. Acta. 2016, 463, 193–199. [Google Scholar] [CrossRef]
- Cao, H.; Wahlestedt, C.; Kapranov, P. Strategies to Annotate and Characterize Long Noncoding RNAs: Advantages and Pitfalls. Trends Genet. 2018, 34, 704–721. [Google Scholar] [CrossRef]
- Carlevaro-Fita, J.; Lanzós, A.; Feuerbach, L.; Hong, C.; Mas-Ponte, D.; Pedersen, J.S.; Johnson, R. Cancer LncRNA Census Reveals Evidence for Deep Functional Conservation of Long Noncoding RNAs in Tumorigenesis. Commun. Biol. 2020, 3, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G. The GENCODE V7 Catalog of Human Long Noncoding RNAs: Analysis of their Gene Structure, Evolution, and Expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattioli, K.; Volders, P.; Gerhardinger, C.; Lee, J.C.; Maass, P.G.; Melé, M.; Rinn, J.L. High-Throughput Functional Analysis of lncRNA Core Promoters Elucidates Rules Governing Tissue Specificity. Genome Res. 2019, 29, 344–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, Q.; Li, Y.; Wang, Z.; Zhou, S.; Sun, H.; Yu, G. The Evolution and Expression Pattern of Human Overlapping lncRNA and Protein-Coding Gene Pairs. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Cao, G.; Jing, L.; Lin, S.; Wang, X.; Zhang, J.; Wang, M.; Liu, W.; Lv, C. Analysing the Relationship between Lnc RNA and Protein-coding Gene and the Role of Lnc RNA as Ce RNA in Pulmonary Fibrosis. J. Cell. Mol. Med. 2014, 18, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.W.; Karakasheva, T.A.; Lee, D.; Lee, J.; Long, Q.; Bass, A.J.; Wong, K.K.; Rustgi, A.K. Comparative Transcriptomes of Adenocarcinomas and Squamous Cell Carcinomas Reveal Molecular Similarities that Span Classical Anatomic Boundaries. PLoS Genet. 2017, 13, e1006938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, E.; Wendik, B.; Srivastava, M.; Pacho, F.; Töchterle, S.; Salvenmoser, W.; Meyer, D. Pronephric Tubule Morphogenesis in Zebrafish Depends on Mnx Mediated Repression of Irx1b within the Intermediate Mesoderm. Dev. Biol. 2016, 411, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.E. Transcriptional Promiscuity in Testes. Curr. Biol. 1996, 6, 768–769. [Google Scholar] [CrossRef] [Green Version]
- Xia, B.; Yan, Y.; Baron, M.; Wagner, F.; Barkley, D.; Chiodin, M.; Kim, S.Y.; Keefe, D.L.; Alukal, J.P.; Boeke, J.D. Widespread Transcriptional Scanning in the Testis Modulates Gene Evolution Rates. Cell 2020, 180, 248–262. [Google Scholar] [CrossRef]
- Som, A.; Wen, S.; Tu, S. Stem Cell Origin of Testicular Seminoma. Clin. Genitourin. Cancer 2013, 11, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Lei, Z.; Li, A.; Jiang, Y.; Ji, J. Differentiation of Testicular Seminoma and Nonseminomatous Germ Cell Tumor on Magnetic Resonance Imaging. Medicine 2019, 98, e17937. [Google Scholar] [CrossRef]
- Conway, J.R.; Warner, J.L.; Rubinstein, W.S.; Miller, R.S. Next-Generation Sequencing and the Clinical Oncology Workflow: Data Challenges, Proposed Solutions, and a Call to Action. JCO Precis. Oncol. 2019, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Xu, L.; Shou, T.; Chen, Q. Systematic Analysis Identifies Three-lncRNA Signature as a Potentially Prognostic Biomarker for Lung Squamous Cell Carcinoma using Bioinformatics Strategy. Transl. Lung Cancer Res. 2019, 8, 614. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wu, F.; Han, Z.; Hu, M.; Lin, W.; Li, Y.; Cao, M. Biomarkers (mRNAs and Non-Coding RNAs) for the Diagnosis and Prognosis of Colorectal Cancer–From the Body Fluid to Tissue Level. Front. Oncol. 2021, 11, 1478. [Google Scholar] [CrossRef]
- Wang, L.; Yu, Z.; Sun, S.; Peng, J.; Xiao, R.; Chen, S.; Zuo, X.; Cheng, Q.; Xia, Y. Long Non-Coding RNAs: Potential Molecular Biomarkers for Gliomas Diagnosis and Prognosis. Rev. Neurosci. 2017, 28, 375–380. [Google Scholar] [CrossRef]
- Yu, K.; Berry, G.J.; Rubin, D.L.; Re, C.; Altman, R.B.; Snyder, M. Association of Omics Features with Histopathology Patterns in Lung Adenocarcinoma. Cell Syst. 2017, 5, 620–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrara, S.; Soldà, G.; Di Leo, M.; Rahal, D.; Peano, C.; Giunta, M.; Lamonaca, L.; Auriemma, F.; Anderloni, A.; Fugazza, A. Side-by-Side Comparison of Next-Generation Sequencing, Cytology, and Histology in Diagnosing Locally Advanced Pancreatic Adenocarcinoma. Gastrointest Endosc. 2021, 93, 597–604. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organ | Breast | Colon | Oesophagus | Brain | Liver | Lung | Ovary | Pancreas | Prostate | Skin | Stomach | Testis | Thyroid | Uterus | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TCGA Cancer and Abbreviation | Breast Cancer BRCA | Colon Adenocarcinoma COAD | Rectum Adenocarcinoma READ | Oesophageal Carcinoma ESCA | Glioblastoma Multiforme GBM | Brain Lower Grade Glioma LGG | Liver Hepatocellular Carcinoma LIHC | Lung Adenocarcinoma LUAD | Lung Squamous Cell Carcinoma LUSC | Ovarian Serous Cystadenocarcinoma OV | Pancreatic Adenocarcinoma PAAD | Prostate Adenocarcinoma PRAD | Skin Cutaneous Melanoma SKCM | Stomach Adenocarcinoma STAD | Testicular Germ Cell Tumours TGCT | Thyroid Carcinoma THCA | Uterine Corpus Endometrial Carcinoma UCEC | Uterine Carcinosarcoma UCS |
| Samples | ||||||||||||||||||
| Normal tissue (GTEx) | 179 | 307 | 307 | 650 | 1141 | 1141 | 109 | 288 | 288 | 86 | 167 | 100 | 788 | 173 | 164 | 279 | 77 | 77 |
| Primary tumour (TCGA) | 1212 | 331 | 103 | 195 | 171 | 523 | 421 | 574 | 548 | 427 | 183 | 548 | 102 | 450 | 154 | 571 | 204 | 57 |
| Clinical stage | ||||||||||||||||||
| Stage I | 182 (15.6%) | 44 (14.3%) | 12 (11.7%) | 18 (9.2%) | N/A | N/A | 169 (40.1%) | 306 (53.3%) | 269 (48.7%) | 1 (0.2%) | 21 (11.5%) | N/A | 3 (2.5%) | 58 (12.9%) | 104 (67.5%) | 283 (49.6%) | N/A | 22 (38.6%) |
| Stage II | 617 (53.0%) | 110 (35.8%) | 24 (23.3%) | 76 (39.0%) | N/A | N/A | 86 (20.4%) | 135 (23.5%) | 178 (32.2%) | 24 (5.6%) | 147 (80.3%) | N/A | 66 (55.0%) | 121 (26.9%) | 12 (7.8%) | 51 (8.9%) | N/A | 5 (8.8%) |
| Stage III | 248 (21.3%) | 82 (26.7%) | 33 (32.0%) | 55 (28.2%) | N/A | N/A | 77 (18.3%) | 96 (16.7%) | 89 (16.1%) | 332 (77.2%) | 3 (1.6%) | N/A | 2 (1.7%) | 169 (37.6%) | 14 (9.1%) | 112 (19.6%) | N/A | 20 (35.1%) |
| Stage IV | 20 (1.7%) | 40 (13.0%) | 13 (12.6%) | 9 (4.6%) | N/A | N/A | 5 (1.2%) | 28 (4.9%) | 8 (1.4%) | 63 (14.7%) | 4 (2.2%) | N/A | 27 (22.5%) | 41 (9.1%) | 0 (0.0%) | 55 (9.6%) | N/A | 10 (17.5%) |
| N/A | 97 (8.3%) | 31 (10.1%) | 21 (20.4%) | 37 (19.0%) | N/A | N/A | 84 (20.0%) | 9 (1.6%) | 8 (1.4%) | 10 (2.3%) | 8 (4.4%) | N/A | 22 (18.3%) | 61 (13.6%) | 24 (15.6%) | 70 (12.3%) | N/A | 0 (0.0%) |
| Age GTEx (years) | ||||||||||||||||||
| Age < 50 | 64 (35.7%) | 108 (35.1%) | 252 (38.7%) | 178 (15.6%) | 343 (68.9%) | 29 (26.6%) | 92 (32.1%) | 38 (44.2%) | 67 (40.1%) | 44 (44.0%) | 266 (33.8%) | 72 (41.6%) | 62 (37.9%) | 86 (30.9%) | 38 (49.3%) | |||
| Age > 50 | 115 (64.3%) | 199 (64.9%) | 398 (61.3%) | 963 (84.4%) | 155 (31.1%) | 80 (73.3%) | 195 (67.9%) | 48 (55.8%) | 100 (59.9%) | 56 (56.0%) | 522 (66.2%) | 101 (58.4%) | 102 (62.1%) | 193 (69.1%) | 39 (50.6%) | |||
| Age TCGA (years) | ||||||||||||||||||
| Age < 50 | 328 (30.1%) | 44 (15.4%) | 17 (18.7%) | 27 (14.9%) | 36 (23.7%) | 343 (68.9%) | 71 (19.6%) | 34 (8.3%) | 20 (3.7%) | 101 (23.9%) | 24 (13.5%) | 34 (6.9%) | 15 (14.7%) | 34 (8.3%) | 122 (96.1%) | 272 (55.9%) | 11 (6.2%) | 0 (0.0%) |
| Age > 50 | 763 (69.9%) | 242 (84.6%) | 74 (81.3%) | 154 (85.1%) | 116 (76.3%) | 155 (31.1%) | 291 (80.4%) | 375 (91.7%) | 519 (96.3%) | 322 (76.1%) | 154 (86.5%) | 461 (93.1%) | 87 (85.3%) | 375 (91.7%) | 5 (3.9%) | 215 (44.1%) | 166 (93.8%) | 57 (100.0%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ragusa, D.; Tosi, S.; Sisu, C. Pan-Cancer Analysis Identifies MNX1 and Associated Antisense Transcripts as Biomarkers for Cancer. Cells 2022, 11, 3577. https://doi.org/10.3390/cells11223577
Ragusa D, Tosi S, Sisu C. Pan-Cancer Analysis Identifies MNX1 and Associated Antisense Transcripts as Biomarkers for Cancer. Cells. 2022; 11(22):3577. https://doi.org/10.3390/cells11223577
Chicago/Turabian StyleRagusa, Denise, Sabrina Tosi, and Cristina Sisu. 2022. "Pan-Cancer Analysis Identifies MNX1 and Associated Antisense Transcripts as Biomarkers for Cancer" Cells 11, no. 22: 3577. https://doi.org/10.3390/cells11223577