

Preclinical Study of Plasmodium Immunotherapy Combined with Radiotherapy for Solid Tumors

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cell Culture and Parasites
2.3. Tumor Models and Animal Grouping
2.4. Treatment Regimens
2.5. Luciferase In Vivo Imaging
2.6. Flank Tumor Rechallenge Experiments
2.7. Western Blotting
2.8. Single Cell Sample Preparation and Isolation
2.9. Flow Cytometry Analysis
2.10. Immunohistochemical Analysis
2.11. Statistical Analysis
3. Results
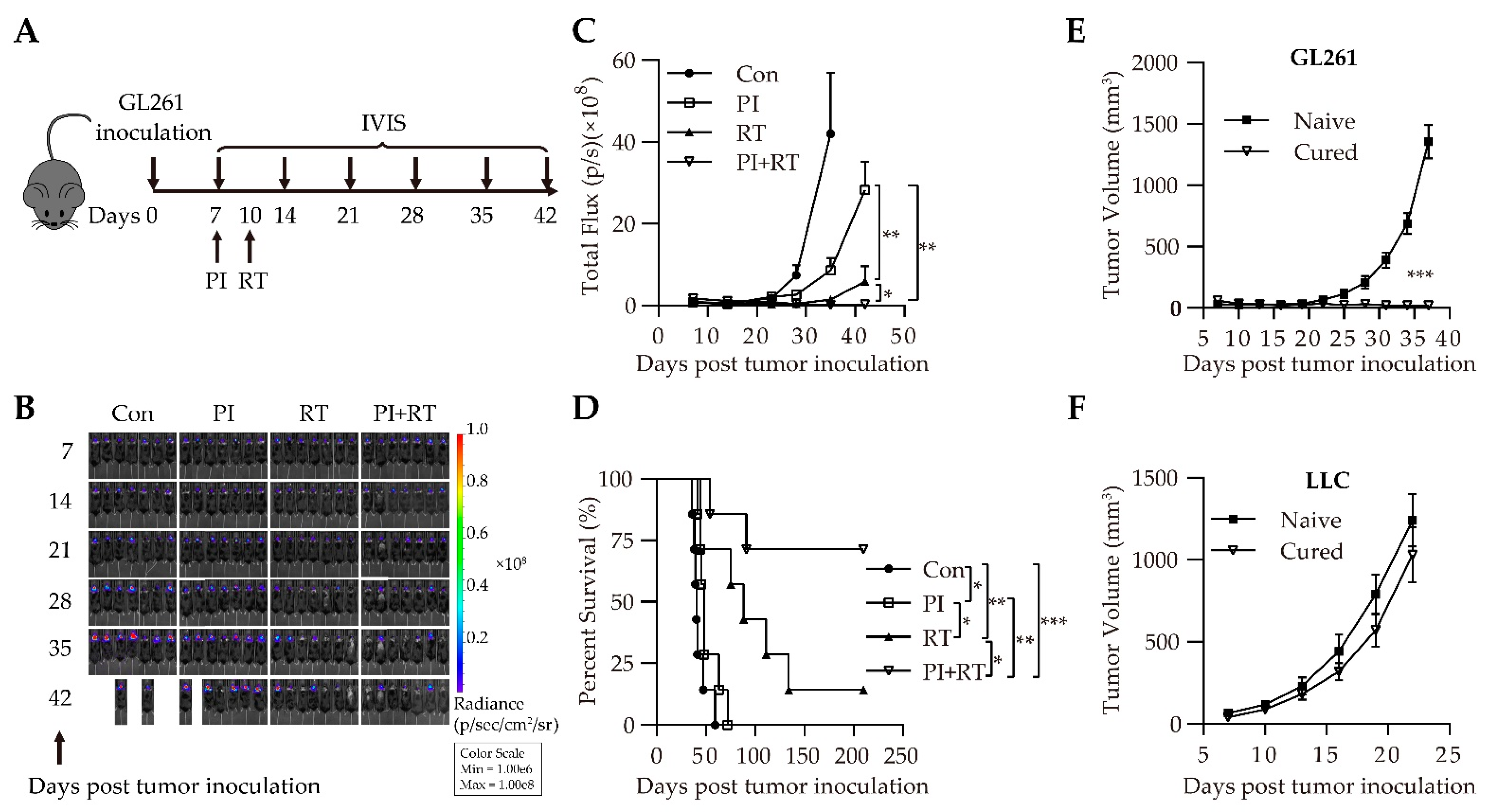
3.1. Antitumor Effect of the Combination of Plasmodium Immunotherapy (PI) and Radiotherapy (RT) in Orthotopic Glioma Model
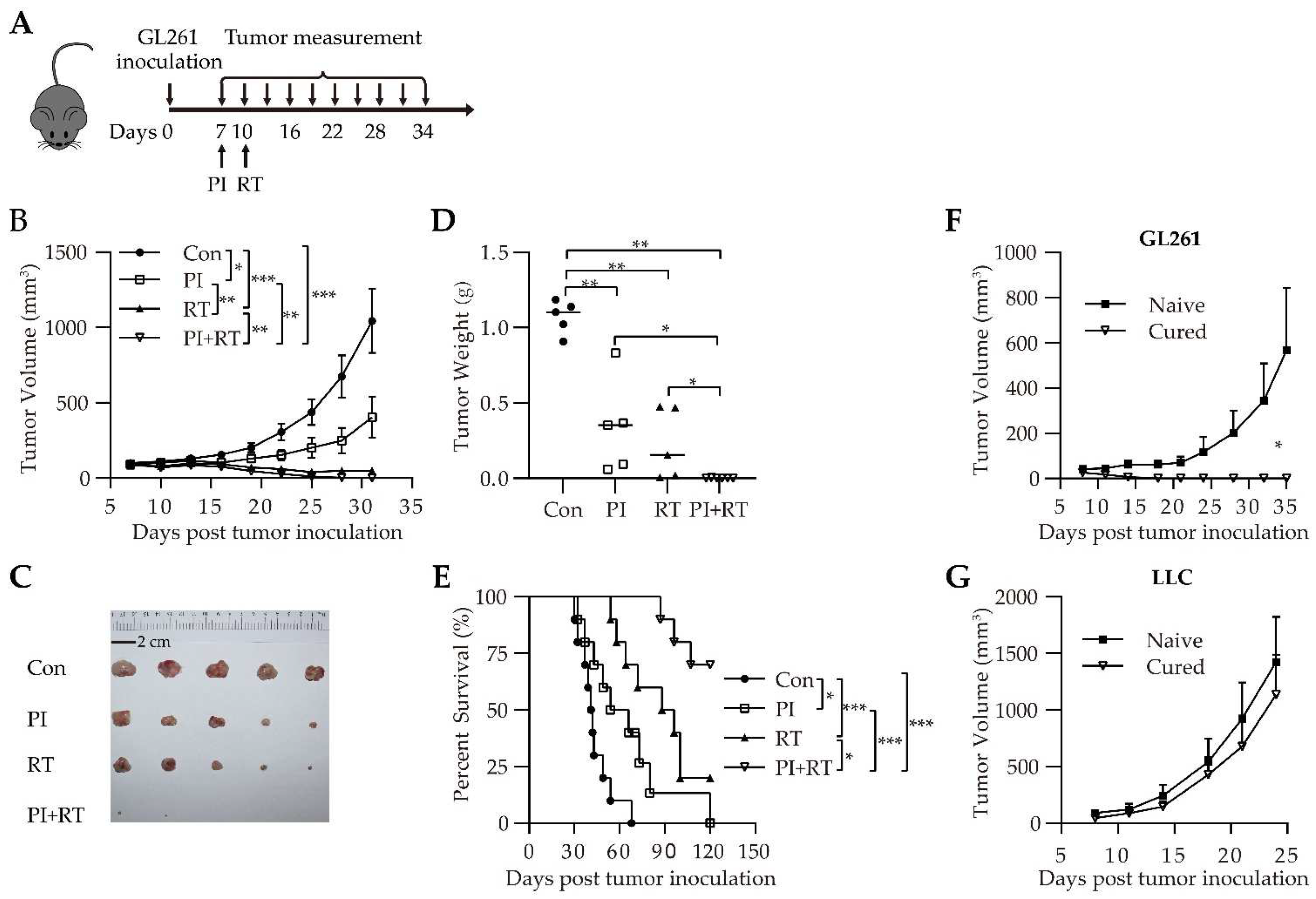
3.2. Antitumor Effect of the Combination of PI and RT in Subcutaneous Glioma Model
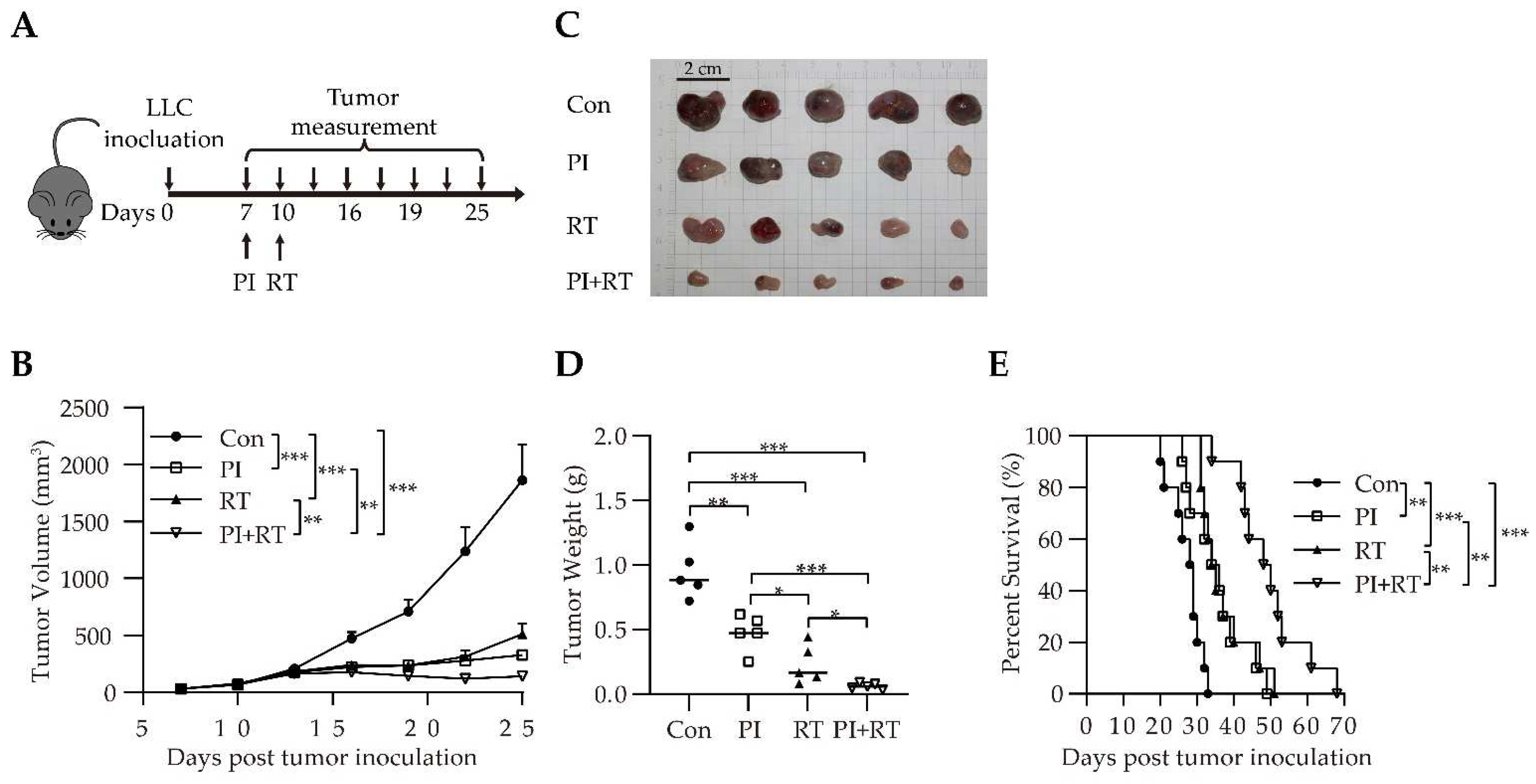
3.3. Antitumor Effect of the Combination of PI and RT in Lung Cancer Model
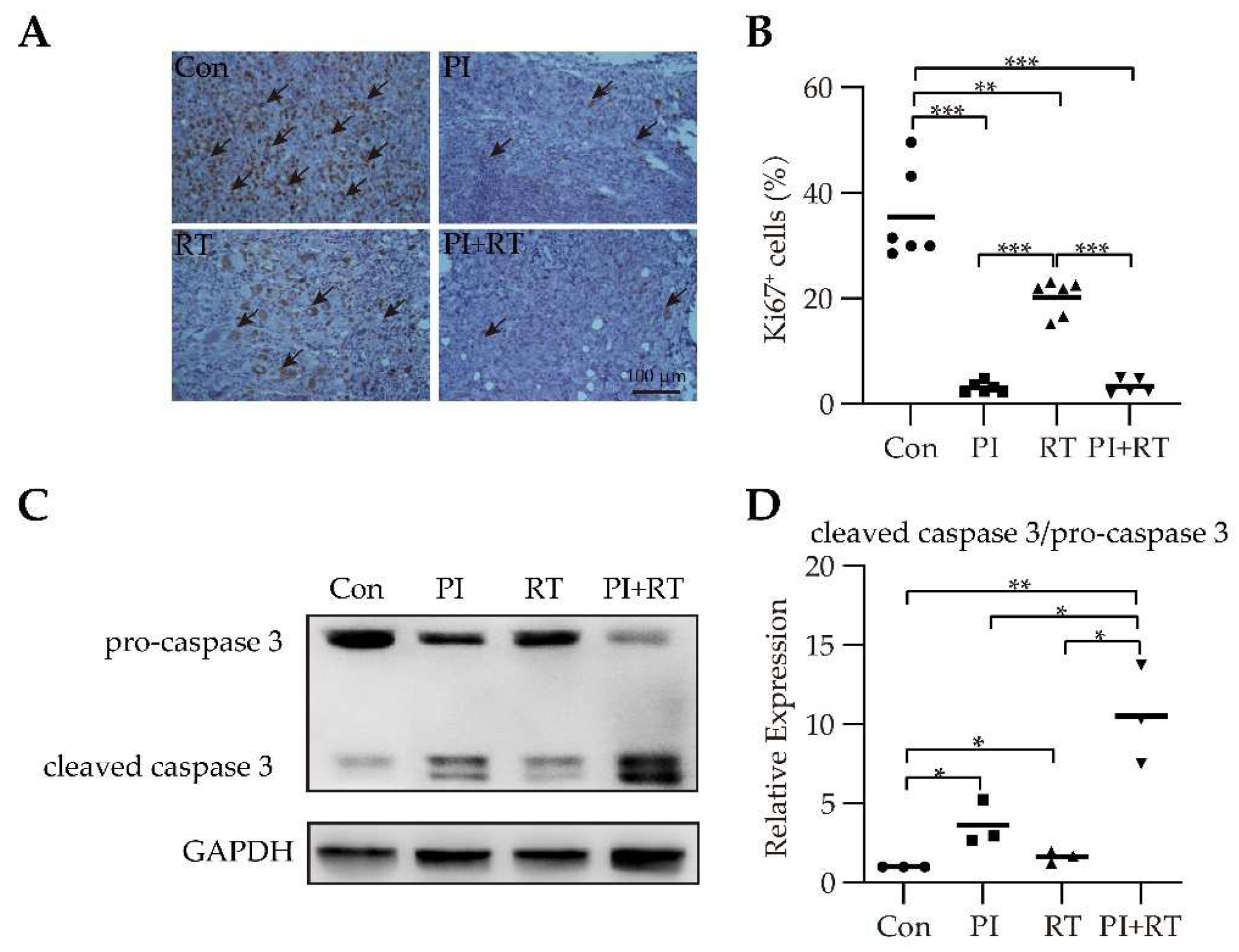
3.4. The Effect of PI and RT Combination on Lung Cancer Cell Proliferation and Apoptosis
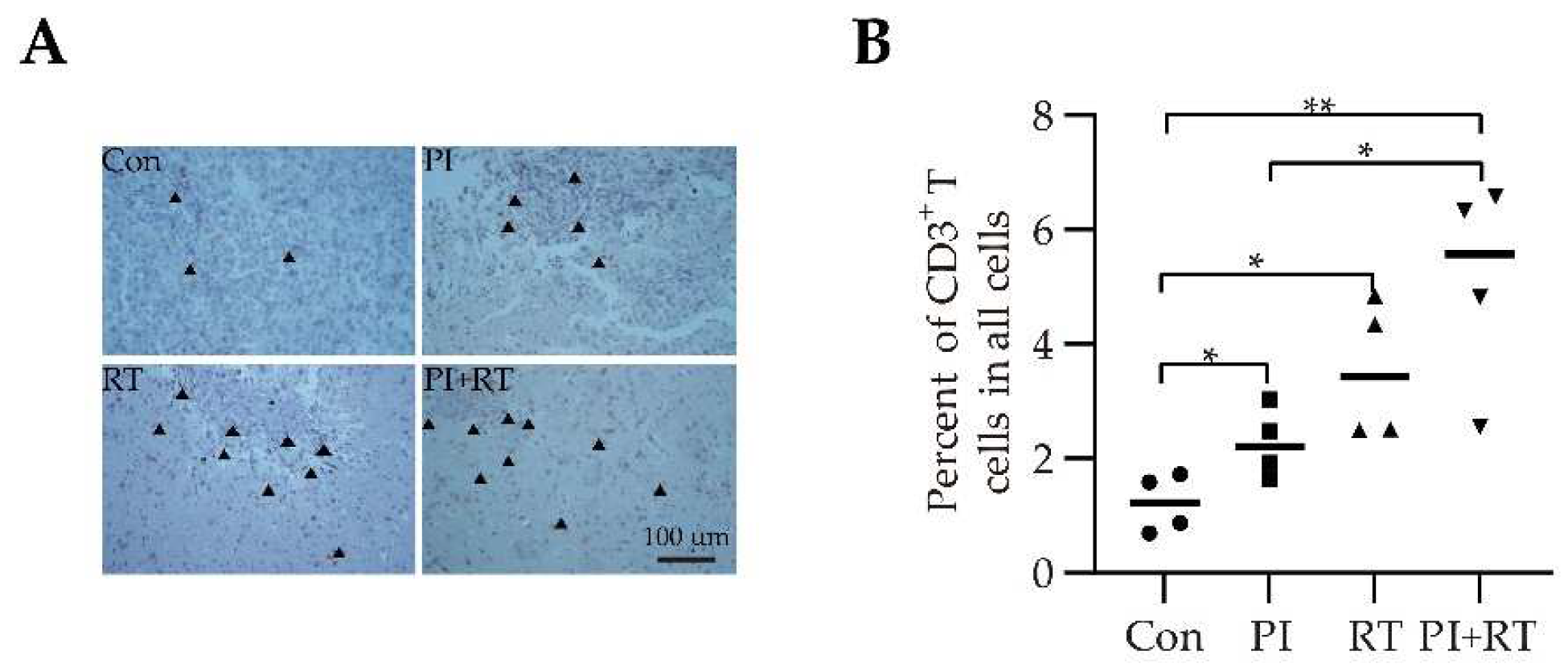
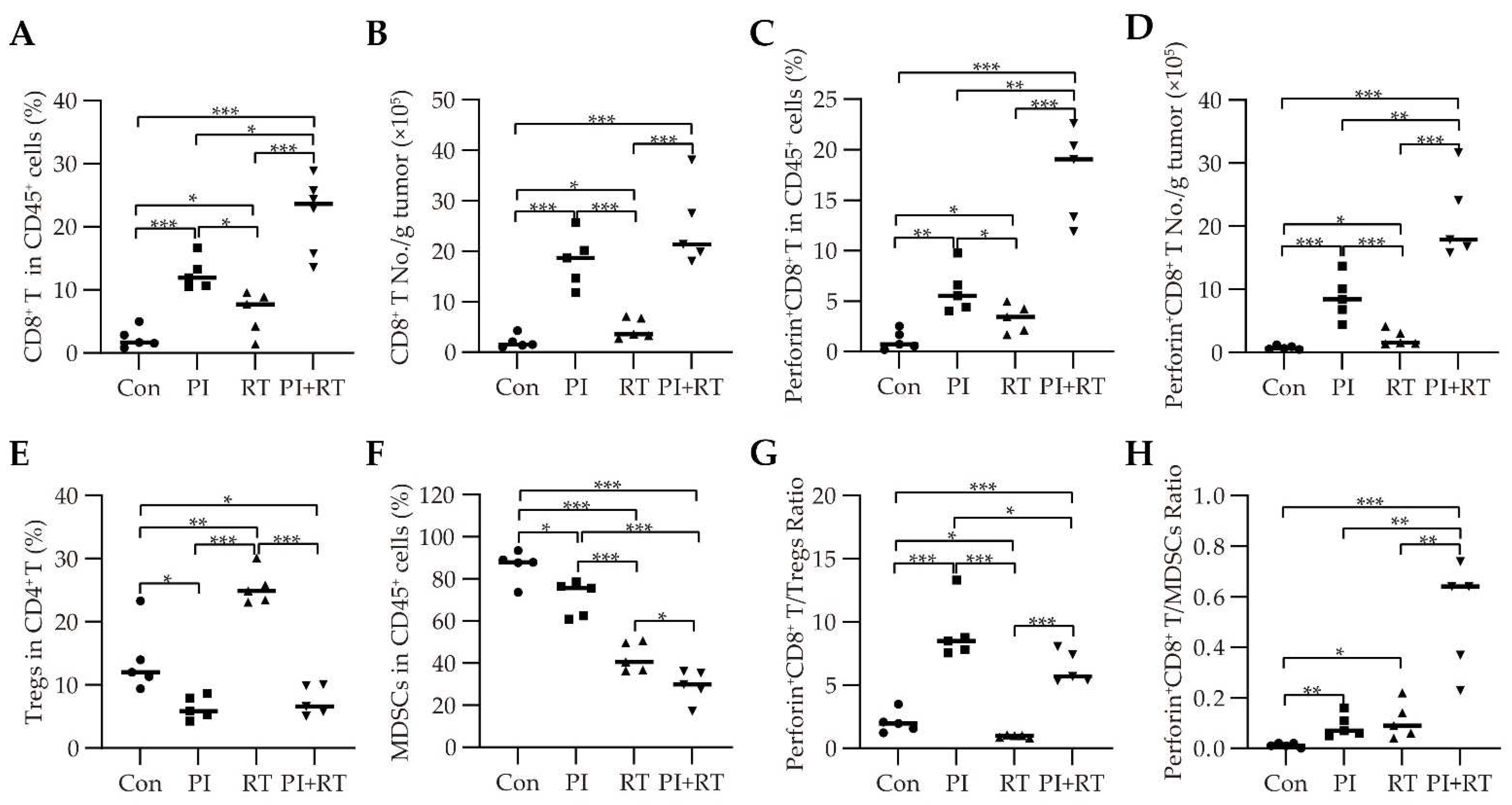
3.5. The Effect of PI and RT Combination on the Immune Profiles in Tumor Microenvironment
3.6. The Effect of PI and RT Combination on the Immune Profiles in the Spleens
3.7. The Effect of PI and RT Combination on the Peripheral Blood
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). GLOBOCAN 2020: Cancer Today. The Global Cancer Observatory 2020 December. 2020. Available online: https://gco.iarc.fr/today/fact-sheets-cancers (accessed on 26 July 2022).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27 (Suppl. S2), S87–S97. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224. [Google Scholar] [CrossRef]
- Wang, M.; Wang, S.; Desai, J.; Trapani, J.A.; Neeson, P.J. Therapeutic strategies to remodel immunologically cold tumors. Clin. Transl. Immunol. 2020, 9, e1226. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, D.; Saw, P.E.; Song, E. Turning cold tumors hot: From molecular mechanisms to clinical applications. Trends Immunol. 2022, 43, 523–545. [Google Scholar] [CrossRef]
- Chen, X.; Qin, L.; Hu, W.; Adah, D. The mechanisms of action of Plasmodium infection against cancer. Cell Commun. Signal. 2021, 19, 74. [Google Scholar] [CrossRef]
- Stevenson, M.M.; Riley, E.M. Innate immunity to malaria. Nat. Rev. Immunol. 2004, 4, 169–180. [Google Scholar] [CrossRef]
- Chen, L.; He, Z.; Qin, L.; Li, Q.; Shi, X.; Zhao, S.; Chen, L.; Zhong, N.; Chen, X. Antitumor effect of malaria parasite infection in a murine Lewis lung cancer model through induction of innate and adaptive immunity. PLoS ONE 2011, 6, e24407. [Google Scholar] [CrossRef] [PubMed]
- Adah, D.; Yang, Y.; Liu, Q.; Gadidasu, K.; Tao, Z.; Yu, S.; Dai, L.; Li, X.; Zhao, S.; Qin, L.; et al. Plasmodium infection inhibits the expansion and activation of MDSCs and Tregs in the tumor microenvironment in a murine Lewis lung cancer model. Cell Commun. Signal. 2019, 17, 32. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Ma, M.; Qin, L.; Kang, Z.; Adah, D.; Tao, Z.; Li, X.; Dai, L.; Zhao, S.; Chen, X.; et al. Plasmodium infection inhibits triple negative 4T1 breast cancer potentially through induction of CD8(+) T cell-mediated antitumor responses in mice. Biomed. Pharmacother. 2021, 138, 111406. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, Q.; Wang, J.; Zhao, S.; Nashun, B.; Qin, L.; Chen, X. Plasmodium infection inhibits tumor angiogenesis through effects on tumor-associated macrophages in a murine implanted hepatoma model. Cell Commun. Signal. 2020, 18, 157. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Q.; Lu, J.; Adah, D.; Yu, S.; Zhao, S.; Yao, Y.; Qin, L.; Qin, L.; Chen, X. Exosomes from Plasmodium-infected hosts inhibit tumor angiogenesis in a murine Lewis lung cancer model. Oncogenesis 2017, 6, e351. [Google Scholar] [CrossRef]
- Qin, L.; Zhong, M.; Adah, D.; Qin, L.; Chen, X.; Ma, C.; Fu, Q.; Zhu, X.; Li, Z.; Wang, N.; et al. A novel tumour suppressor lncRNA F630028O10Rik inhibits lung cancer angiogenesis by regulating miR-223-3p. J. Cell. Mol. Med. 2020, 24, 3549–3559. [Google Scholar] [CrossRef]
- Liang, Y.; Chen, X.; Tao, Z.; Ma, M.; Adah, D.; Li, X.; Dai, L.; Ding, W.; Fanuel, S.; Zhao, S.; et al. Plasmodium infection prevents recurrence and metastasis of hepatocellular carcinoma possibly via inhibition of the epithelialmesenchymal transition. Mol. Med. Rep. 2021, 23, 418. [Google Scholar] [CrossRef]
- Atun, R.; Jaffray, D.A.; Barton, M.B.; Bray, F.; Baumann, M.; Vikram, B.; Hanna, T.P.; Knaul, F.M.; Lievens, Y.; Lui, T.Y.; et al. Expanding global access to radiotherapy. Lancet Oncol. 2015, 16, 1153–1186. [Google Scholar] [CrossRef]
- Citrin, D.E. Recent Developments in Radiotherapy. N. Engl. J. Med. 2017, 377, 1065–1075. [Google Scholar] [CrossRef]
- Cytlak, U.M.; Dyer, D.P.; Honeychurch, J.; Williams, K.J.; Travis, M.A.; Illidge, T.M. Immunomodulation by radiotherapy in tumour control and normal tissue toxicity. Nat. Rev. Immunol. 2022, 22, 124–138. [Google Scholar] [CrossRef]
- Mortezaee, K.; Parwaie, W.; Motevaseli, E.; Mirtavoos-Mahyari, H.; Musa, A.E.; Shabeeb, D.; Esmaely, F.; Najafi, M.; Farhood, B. Targets for improving tumor response to radiotherapy. Int. Immunopharmacol. 2019, 76, 105847. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef]
- Pilones, K.A.; Vanpouille-Box, C.; Demaria, S. Combination of radiotherapy and immune checkpoint inhibitors. Semin. Radiat. Oncol. 2015, 25, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef]
- Moore, C.; Hsu, C.C.; Chen, W.M.; Chen, B.P.C.; Han, C.; Story, M.; Aguilera, T.; Pop, L.M.; Hannan, R.; Fu, Y.X.; et al. Personalized Ultrafractionated Stereotactic Adaptive Radiotherapy (PULSAR) in Preclinical Models Enhances Single-Agent Immune Checkpoint Blockade. Int. J. Radiat. Oncol. Biol. Phys. 2021, 110, 1306–1316. [Google Scholar] [CrossRef]
- Yokouchi, H.; Yamazaki, K.; Chamoto, K.; Kikuchi, E.; Shinagawa, N.; Oizumi, S.; Hommura, F.; Nishimura, T.; Nishimura, M. Anti-OX40 monoclonal antibody therapy in combination with radiotherapy results in therapeutic antitumor immunity to murine lung cancer. Cancer Sci. 2008, 99, 361–367. [Google Scholar] [CrossRef]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K. Lung Cancer: Understanding Its Molecular Pathology and the 2015 WHO Classification. Front. Oncol. 2017, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Maleki Vareki, S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 157. [Google Scholar] [CrossRef] [PubMed]
- Tandon, N.; Thakkar, K.N.; LaGory, E.L.; Liu, Y.; Giaccia, A.J. Generation of stable expression mammalian cell lines using lentivirus. Bio-Protocol 2018, 8, e3073. [Google Scholar] [CrossRef]
- Lal, S.; Lacroix, M.; Tofilon, P.; Fuller, G.N.; Sawaya, R.; Lang, F.F. An implantable guide-screw system for brain tumor studies in small animals. J. Neurosurg. 2000, 92, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Yam, C.S.; Hajjar, A.M. Whole Spleen Flow Cytometry Assay. Bio-Protocol 2013, 3, e834. [Google Scholar] [CrossRef]
- Li, X.; Fang, F.; Gao, Y.; Tang, G.; Xu, W.; Wang, Y.; Kong, R.; Tuyihong, A.; Wang, Z. ROS Induced by KillerRed Targeting Mitochondria (mtKR) Enhances Apoptosis Caused by Radiation via Cyt c/Caspase-3 Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 4528616. [Google Scholar] [CrossRef]
- Eriksson, D.; Stigbrand, T. Radiation-induced cell death mechanisms. Tumour Biol. 2010, 31, 363–372. [Google Scholar] [CrossRef]
- St Paul, M.; Ohashi, P.S. The Roles of CD8(+) T Cell Subsets in Antitumor Immunity. Trends Cell Biol. 2020, 30, 695–704. [Google Scholar] [CrossRef]
- Kagi, D.; Vignaux, F.; Ledermann, B.; Burki, K.; Depraetere, V.; Nagata, S.; Hengartner, H.; Golstein, P. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science 1994, 265, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Mu, S.; Wang, Y.; Wang, H.; Cai, L.; Li, W.; Hu, Y. Prognostic role of myeloid-derived suppressor cells in cancers: A systematic review and meta-analysis. BMC Cancer 2018, 18, 1220. [Google Scholar] [CrossRef] [PubMed]
- Damuzzo, V.; Pinton, L.; Desantis, G.; Solito, S.; Marigo, I.; Bronte, V.; Mandruzzato, S. Complexity and challenges in defining myeloid-derived suppressor cells. Cytom. Part B Clin. Cytom. 2015, 88, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Baras, A.S.; Drake, C.; Liu, J.J.; Gandhi, N.; Kates, M.; Hoque, M.O.; Meeker, A.; Hahn, N.; Taube, J.M.; Schoenberg, M.P.; et al. The ratio of CD8 to Treg tumor-infiltrating lymphocytes is associated with response to cisplatin-based neoadjuvant chemotherapy in patients with muscle invasive urothelial carcinoma of the bladder. Oncoimmunology 2016, 5, e1134412. [Google Scholar] [CrossRef]
- Shah, W.; Yan, X.; Jing, L.; Zhou, Y.; Chen, H.; Wang, Y. A reversed CD4/CD8 ratio of tumor-infiltrating lymphocytes and a high percentage of CD4(+)FOXP3(+) regulatory T cells are significantly associated with clinical outcome in squamous cell carcinoma of the cervix. Cell. Mol. Immunol. 2011, 8, 59–66. [Google Scholar] [CrossRef]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef]
- Liu, F.; Lang, R.; Zhao, J.; Zhang, X.; Pringle, G.A.; Fan, Y.; Yin, D.; Gu, F.; Yao, Z.; Fu, L. CD8(+) cytotoxic T cell and FOXP3(+) regulatory T cell infiltration in relation to breast cancer survival and molecular subtypes. Breast Cancer Res. Treat. 2011, 130, 645–655. [Google Scholar] [CrossRef]
- Engwerda, C.R.; Beattie, L.; Amante, F.H. The importance of the spleen in malaria. Trends Parasitol. 2005, 21, 75–80. [Google Scholar] [CrossRef]
- Zhai, D.; An, D.; Wan, C.; Yang, K. Radiotherapy: Brightness and darkness in the era of immunotherapy. Transl. Oncol. 2022, 19, 101366. [Google Scholar] [CrossRef]
- Tritz, Z.P.; Ayasoufi, K.; Johnson, A.J. Anti-PD-1 checkpoint blockade monotherapy in the orthotopic GL261 glioma model: The devil is in the detail. Neuro-Oncol. Adv. 2021, 3, vdab066. [Google Scholar] [CrossRef] [PubMed]
- Trachet, E.E.; Urs, S.; Wong, A.; Wise, S.; Franklin, M.R. Radiation response in preclinical orthotopic models of brain cancer. Cancer Res. 2019, 79, 1928. [Google Scholar] [CrossRef]
- Scheithauer, H.; Belka, C.; Lauber, K.; Gaipl, U.S. Immunological aspects of radiotherapy. Radiat. Oncol. 2014, 9, 185. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keam, S.; Gill, S.; Ebert, M.A.; Nowak, A.K.; Cook, A.M. Enhancing the efficacy of immunotherapy using radiotherapy. Clin. Transl. Immunol. 2020, 9, e1169. [Google Scholar] [CrossRef]
- Yap, T.A.; Parkes, E.E.; Peng, W.; Moyers, J.T.; Curran, M.A.; Tawbi, H.A. Development of Immunotherapy Combination Strategies in Cancer. Cancer Discov. 2021, 11, 1368–1397. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Larkin, J.; Tabernero, J.; Bonini, C. Enhancing anti-tumour efficacy with immunotherapy combinations. Lancet 2021, 397, 1010–1022. [Google Scholar] [CrossRef]
- Sindoni, A.; Minutoli, F.; Ascenti, G.; Pergolizzi, S. Combination of immune checkpoint inhibitors and radiotherapy: Review of the literature. Crit. Rev. Oncol. Hematol. 2017, 113, 63–70. [Google Scholar] [CrossRef]
- Ko, E.C.; Formenti, S.C. Radiotherapy and checkpoint inhibitors: A winning new combination? Ther. Adv. Med. Oncol. 2018, 10, 1758835918768240. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Abiko, K.; Matsumura, N.; Hamanishi, J.; Horikawa, N.; Murakami, R.; Yamaguchi, K.; Yoshioka, Y.; Baba, T.; Konishi, I.; Mandai, M. IFN-gamma from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br. J. Cancer 2015, 112, 1501–1509. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, J.; Cai, Y.; Fu, S.; Zhang, N.; Fu, X.; Li, L. IFN-gamma-mediated inhibition of lung cancer correlates with PD-L1 expression and is regulated by PI3K-AKT signaling. Int. J. Cancer 2018, 143, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Wang, C.; Wang, B.; Yang, J.; Wang, Y.; Luo, F.; Xu, J.; Zhao, C.; Liu, R.; Chu, Y. The IFN-gamma/PD-L1 axis between T cells and tumor microenvironment: Hints for glioma anti-PD-1/PD-L1 therapy. J. Neuroinflamm. 2018, 15, 290. [Google Scholar] [CrossRef] [PubMed]
- Mojic, M.; Takeda, K.; Hayakawa, Y. The Dark Side of IFN-gamma: Its Role in Promoting Cancer Immunoevasion. Int. J. Mol. Sci. 2017, 19, 89. [Google Scholar] [CrossRef]
- Bai, J.; Gao, Z.; Li, X.; Dong, L.; Han, W.; Nie, J. Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PD-L1 blockade. Oncotarget 2017, 8, 110693–110707. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Tsoi, J.; Onyshchenko, M.; Abril-Rodriguez, G.; Ross-Macdonald, P.; Wind-Rotolo, M.; Champhekar, A.; Medina, E.; Torrejon, D.Y.; Shin, D.S.; et al. Conserved Interferon-gamma Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell 2021, 39, 122. [Google Scholar] [CrossRef]
- Mandai, M.; Hamanishi, J.; Abiko, K.; Matsumura, N.; Baba, T.; Konishi, I. Dual Faces of IFNgamma in Cancer Progression: A Role of PD-L1 Induction in the Determination of Pro- and Antitumor Immunity. Clin. Cancer Res. 2016, 22, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, K.R.; Crabtree, J.N.; Dent, A.E. Innate immunity to malaria—The role of monocytes. Immunol. Rev. 2020, 293, 8–24. [Google Scholar] [CrossRef]
- Kurup, S.P.; Butler, N.S.; Harty, J.T. T cell-mediated immunity to malaria. Nat. Rev. Immunol. 2019, 19, 457–471. [Google Scholar] [CrossRef]
- Urban, B.C.; Todryk, S. Malaria pigment paralyzes dendritic cells. J. Biol. 2006, 5, 4. [Google Scholar] [CrossRef][Green Version]
- Elliott, S.R.; Spurck, T.P.; Dodin, J.M.; Maier, A.G.; Voss, T.S.; Yosaatmadja, F.; Payne, P.D.; McFadden, G.I.; Cowman, A.F.; Rogerson, S.J.; et al. Inhibition of dendritic cell maturation by malaria is dose dependent and does not require Plasmodium falciparum erythrocyte membrane protein 1. Infect. Immun. 2007, 75, 3621–3632. [Google Scholar] [CrossRef] [PubMed]
- Scorza, T.; Magez, S.; Brys, L.; De Baetselier, P. Hemozoin is a key factor in the induction of malaria-associated immunosuppression. Parasite Immunol. 1999, 21, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Millington, O.R.; Di Lorenzo, C.; Phillips, R.S.; Garside, P.; Brewer, J.M. Suppression of adaptive immunity to heterologous antigens during Plasmodium infection through hemozoin-induced failure of dendritic cell function. J. Biol. 2006, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.S.; Schofield, L. Natural regulatory T cells in malaria: Host or parasite allies? PLoS Pathog. 2010, 6, e1000771. [Google Scholar] [CrossRef]
- Mukherjee, S.; Ghosh, S.; Sengupta, A.; Sarkar, S.; Keswani, T.; Chatterjee, R.; Bhattacharyya, A. IL-6 dependent expansion of inflammatory MDSCs (CD11b+ Gr-1+) promote Th-17 mediated immune response during experimental cerebral malaria. Cytokine 2022, 155, 155910. [Google Scholar] [CrossRef]
- Mackroth, M.S.; Abel, A.; Steeg, C.; Schulze Zur Wiesch, J.; Jacobs, T. Acute Malaria Induces PD1+CTLA4+ Effector T Cells with Cell-Extrinsic Suppressor Function. PLoS Pathog. 2016, 12, e1005909. [Google Scholar] [CrossRef]
- Eberl, G. Immunity by equilibrium. Nat. Rev. Immunol. 2016, 16, 524–532. [Google Scholar] [CrossRef]
- Liu, G.; Qin, L.; Li, Y.; Zhao, S.; Shugay, M.; Yan, Y.; Ye, Y.; Chen, Y.; Huang, C.; Bayaer, N.; et al. Subsequent malaria enhances virus-specific T cell immunity in SIV-infected Chinese rhesus macaques. Cell Commun. Signal. 2022, 20, 101. [Google Scholar] [CrossRef]
- Swearingen, K.E.; Lindner, S.E. Plasmodium Parasites Viewed through Proteomics. Trends Parasitol. 2018, 34, 945–960. [Google Scholar] [CrossRef]
- Cao, Y.; Feng, Y.H.; Gao, L.W.; Li, X.Y.; Jin, Q.X.; Wang, Y.Y.; Xu, Y.Y.; Jin, F.; Lu, S.L.; Wei, M.J. Artemisinin enhances the anti-tumor immune response in 4T1 breast cancer cells in vitro and in vivo. Int. Immunopharmacol. 2019, 70, 110–116. [Google Scholar] [CrossRef]
- Barradell, L.B.; Fitton, A. Artesunate. A review of its pharmacology and therapeutic efficacy in the treatment of malaria. Drugs 1995, 50, 714–741. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Woon, C.Y.; Liu, C.G.; Cheng, J.T.; You, M.; Sethi, G.; Wong, A.L.; Ho, P.C.; Zhang, D.; Ong, P.; et al. Repurposing Artemisinin and its Derivatives as Anticancer Drugs: A Chance or Challenge? Front. Pharmacol. 2021, 12, 828856. [Google Scholar] [CrossRef] [PubMed]
- Koks, C.A.; Garg, A.D.; Ehrhardt, M.; Riva, M.; Vandenberk, L.; Boon, L.; De Vleeschouwer, S.; Agostinis, P.; Graf, N.; Van Gool, S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer 2015, 136, E313–E325. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, Z.; Ding, W.; Cheng, Z.; Feng, Y.; Kang, Z.; Qiu, R.; Zhao, S.; Hu, W.; Zhou, F.; Wu, D.; et al. Preclinical Study of Plasmodium Immunotherapy Combined with Radiotherapy for Solid Tumors. Cells 2022, 11, 3600. https://doi.org/10.3390/cells11223600
Tao Z, Ding W, Cheng Z, Feng Y, Kang Z, Qiu R, Zhao S, Hu W, Zhou F, Wu D, et al. Preclinical Study of Plasmodium Immunotherapy Combined with Radiotherapy for Solid Tumors. Cells. 2022; 11(22):3600. https://doi.org/10.3390/cells11223600
Chicago/Turabian StyleTao, Zhu, Wenting Ding, Zhipeng Cheng, Yinfang Feng, Zhongkui Kang, Runmin Qiu, Siting Zhao, Wen Hu, Fang Zhou, Donghai Wu, and et al. 2022. "Preclinical Study of Plasmodium Immunotherapy Combined with Radiotherapy for Solid Tumors" Cells 11, no. 22: 3600. https://doi.org/10.3390/cells11223600
APA StyleTao, Z., Ding, W., Cheng, Z., Feng, Y., Kang, Z., Qiu, R., Zhao, S., Hu, W., Zhou, F., Wu, D., Duan, Z., Qin, L., & Chen, X. (2022). Preclinical Study of Plasmodium Immunotherapy Combined with Radiotherapy for Solid Tumors. Cells, 11(22), 3600. https://doi.org/10.3390/cells11223600