

Empagliflozin and Dapagliflozin Increase Na+ and Inward Rectifier K+ Current Densities in Human Cardiomyocytes Derived from Induced Pluripotent Stem Cells (hiPSC-CMs)
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. hiPSC-CM Culture and Infection
2.2. Chinese Hamster Ovary (CHO) Cell Culture and Transfection
2.3. Ca2+/Calmodulin-Dependent Kinase II (CaMKII) Silencing in CHO Cells
2.4. Patch-Clamp Recordings
2.4.1. Solutions
2.4.2. Pulse Protocols and Analysis
2.5. Statistical Analysis
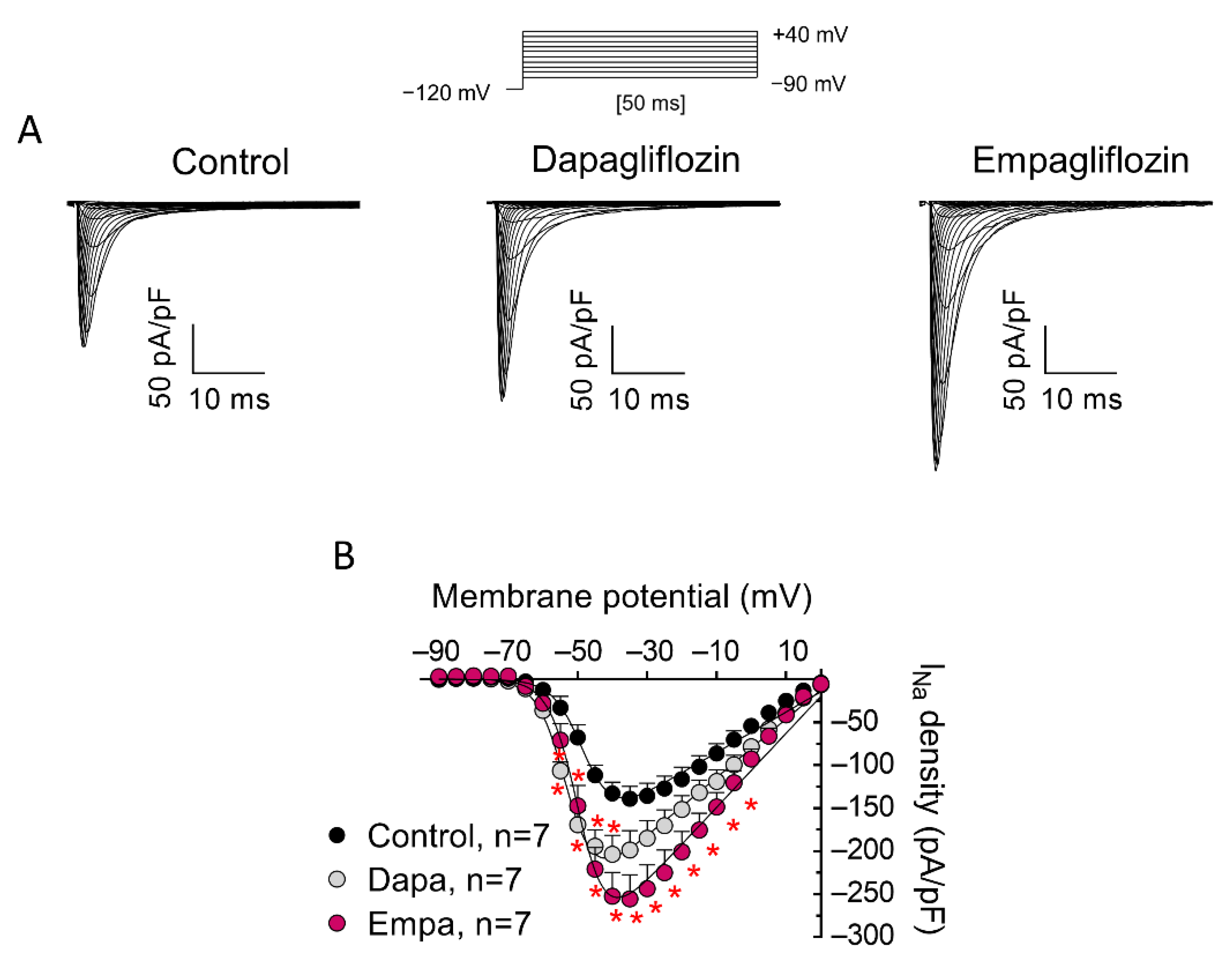
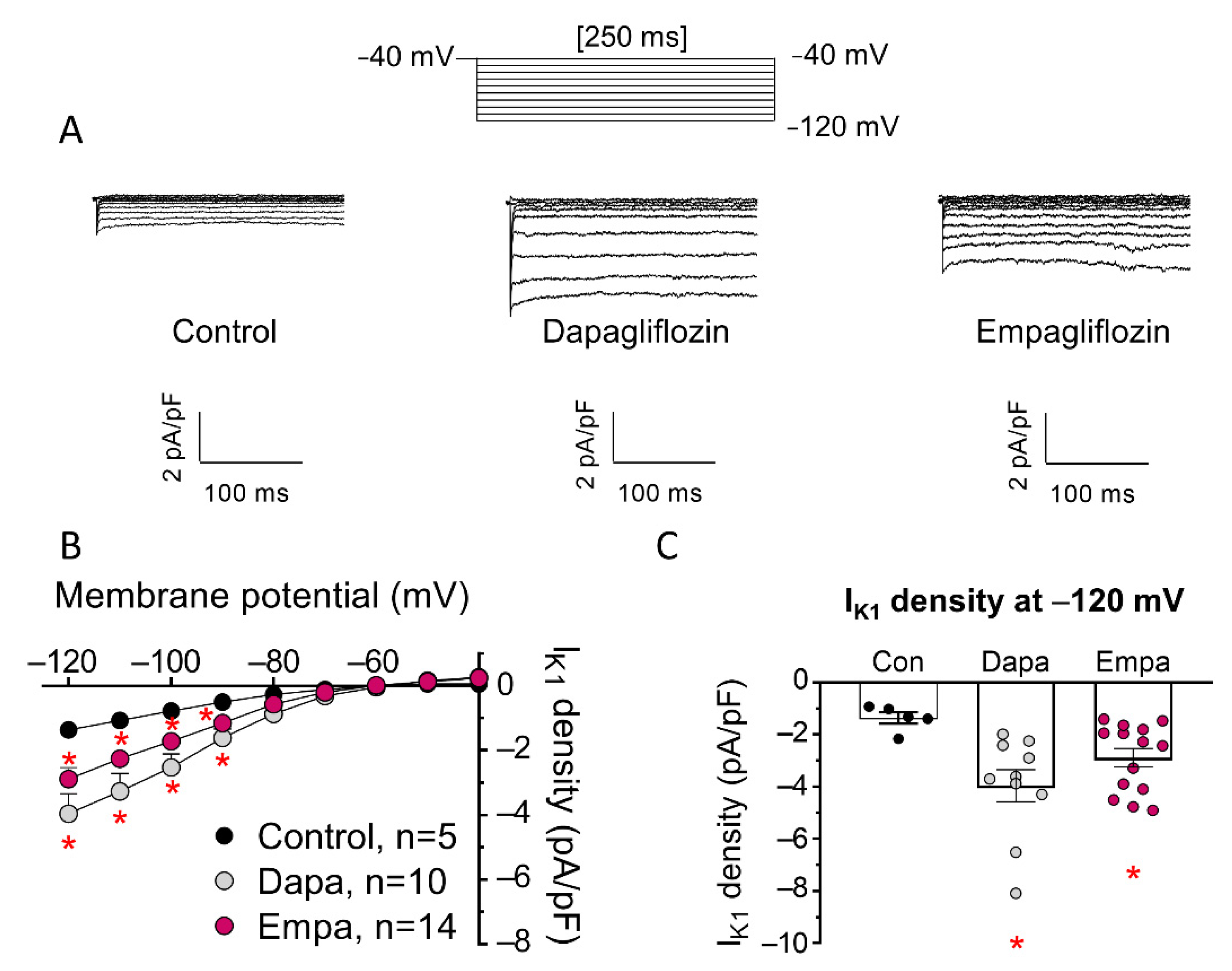
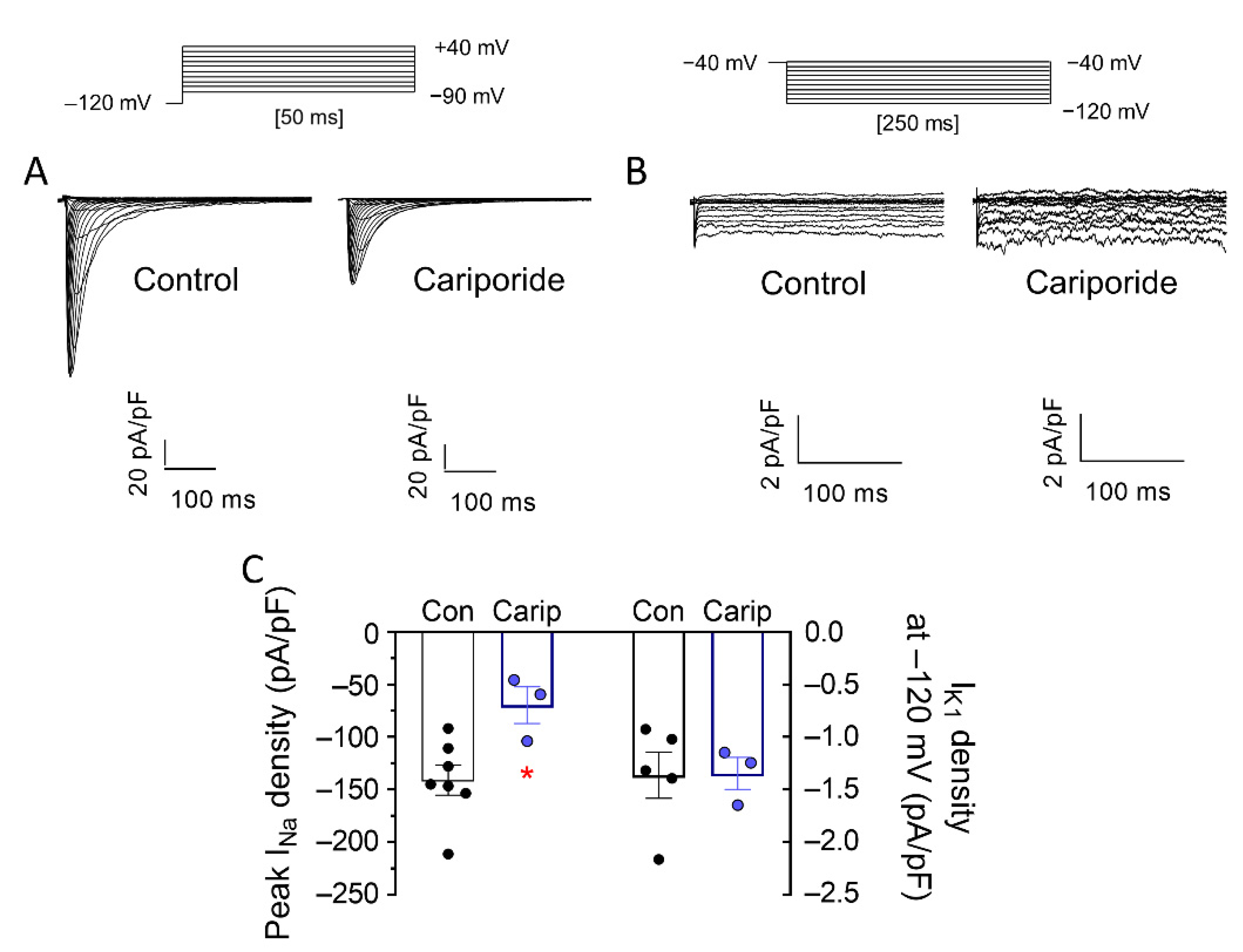
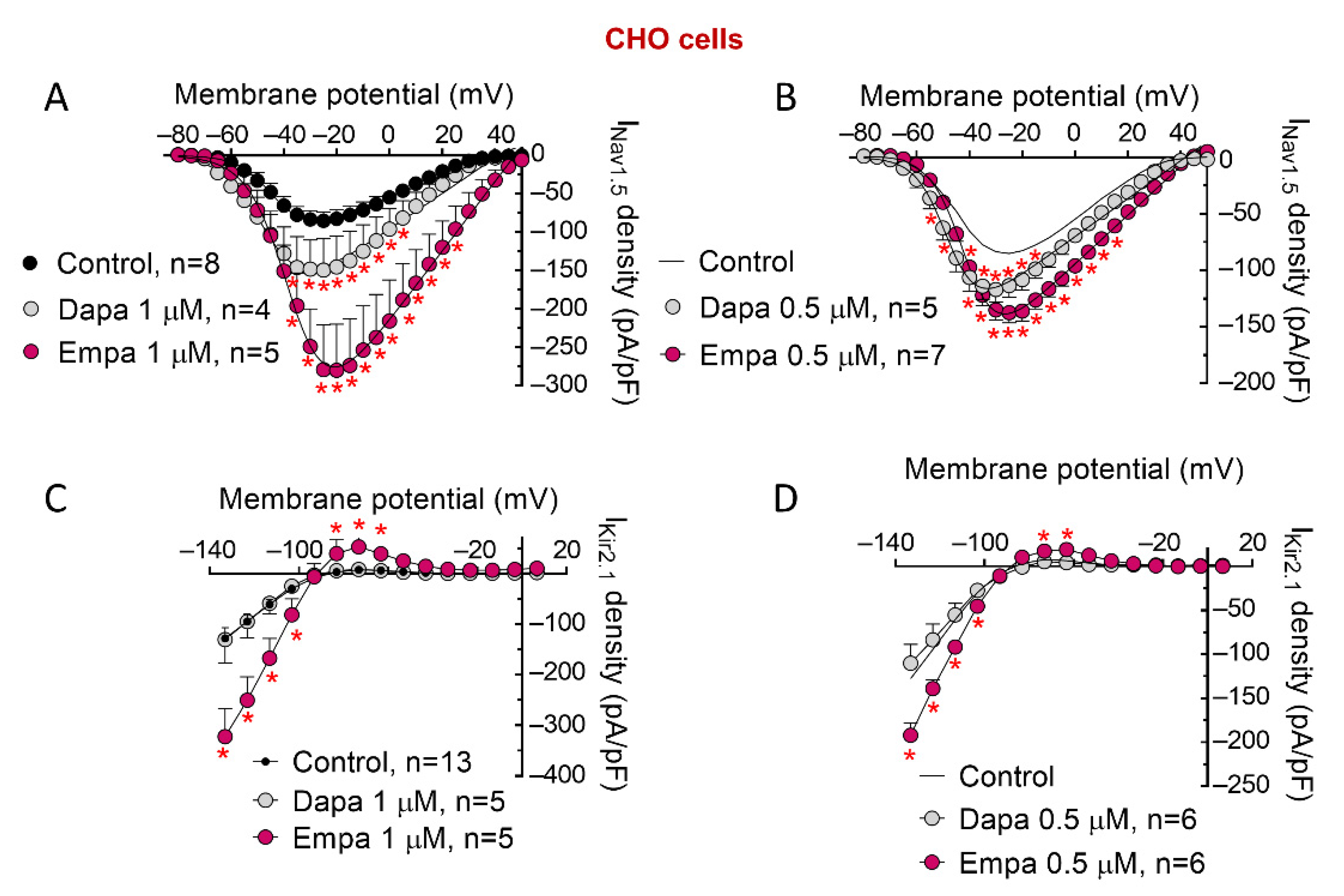
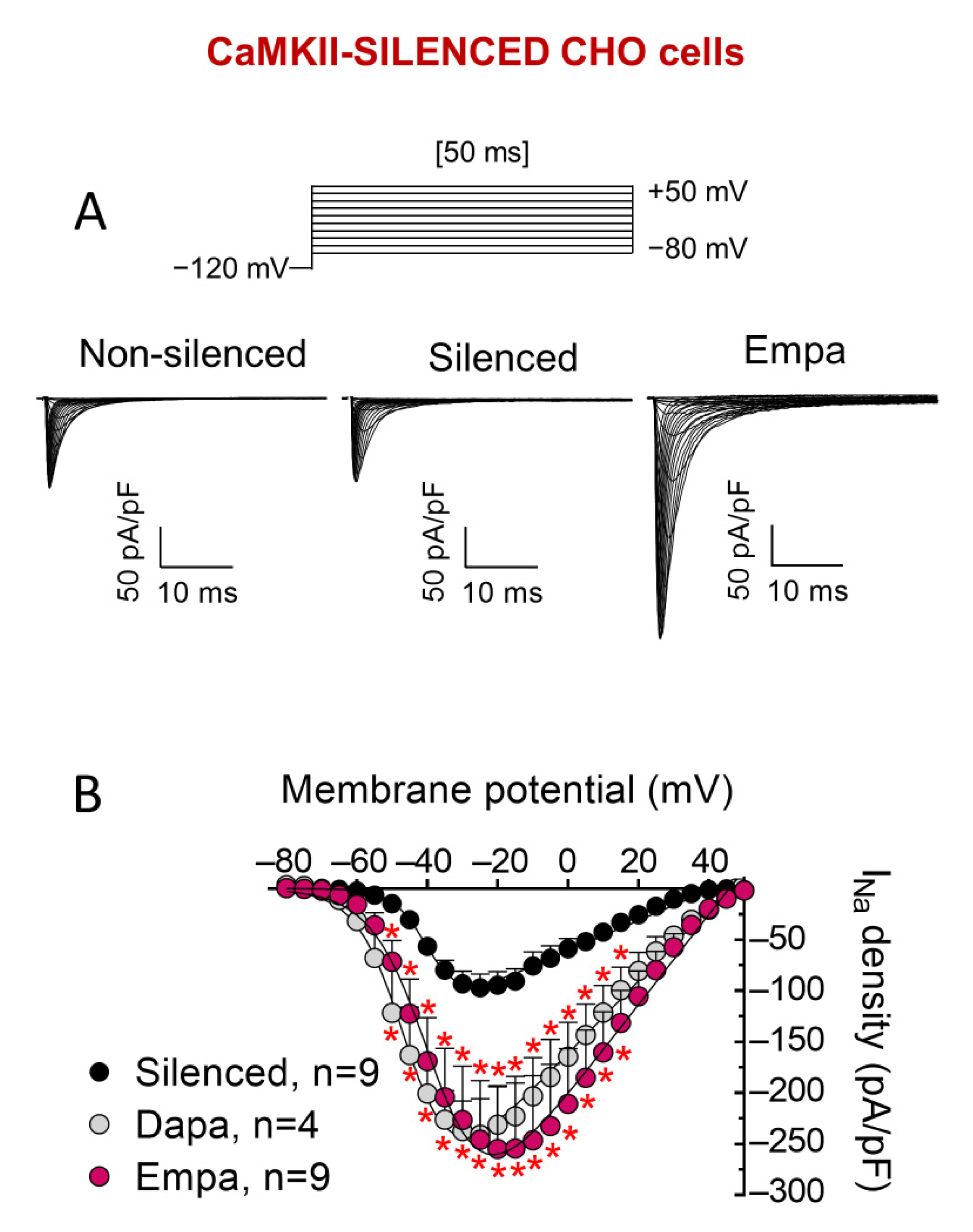
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Braunwald, E. Gliflozins in the Management of Cardiovascular Disease. N. Engl. J. Med. 2022, 386, 2024–2034. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Curtain, J.P.; Docherty, K.F.; Jhund, P.S.; Petrie, M.C.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; et al. Effect of Dapagliflozin on Ventricular Arrhythmias, Resuscitated Cardiac Arrest, or Sudden Death in DAPA-HF. Eur. Heart J. 2021, 42, 3727–3738. [Google Scholar] [CrossRef]
- Fernandes, G.C.; Fernandes, A.; Cardoso, R.; Penalver, J.; Knijnik, L.; Mitrani, R.D.; Myerburg, R.J.; Goldberger, J.J. Association of SGLT2 Inhibitors with Arrhythmias and Sudden Cardiac Death in Patients with Type 2 Diabetes or Heart Failure: A Meta-Analysis of 34 Randomized Controlled Trials. Heart Rhythm 2021, 18, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Mustroph, J.; Baier, M.J.; Pabel, S.; Stehle, T.; Trum, M.; Provaznik, Z.; Mohler, P.J.; Musa, H.; Hund, T.J.; Sossalla, S.; et al. Empagliflozin Inhibits Cardiac Late Sodium Current by Ca/Calmodulin-Dependent Kinase II. Circulation 2022, 146, 1259–1261. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, B.; Mira Hernández, J.; Shen, E.Y.; Habibi, N.R.; Bossuyt, J.; Bers, D.M. Empagliflozin Reverses Late Na+ Current Enhancement and Cardiomyocyte Proarrhythmia in a Translational Murine Model of Heart Failure with Preserved Ejection Fraction. Circulation 2022, 145, 1029–1031. [Google Scholar] [CrossRef]
- Philippaert, K.; Kalyaanamoorthy, S.; Fatehi, M.; Long, W.; Soni, S.; Byrne, N.J.; Barr, A.; Singh, J.; Wong, J.; Palechuk, T.; et al. Cardiac Late Sodium Channel Current Is a Molecular Target for the Sodium/Glucose Cotransporter 2 Inhibitor Empagliflozin. Circulation 2021, 143, 2188–2204. [Google Scholar] [CrossRef] [PubMed]
- Durak, A.; Olgar, Y.; Degirmenci, S.; Akkus, E.; Tuncay, E.; Turan, B. A SGLT2 Inhibitor Dapagliflozin Suppresses Prolonged Ventricular-Repolarization through Augmentation of Mitochondrial Function in Insulin-Resistant Metabolic Syndrome Rats. Cardiovasc. Diabetol. 2018, 17, 144. [Google Scholar] [CrossRef]
- Milstein, M.L.; Musa, H.; Balbuena, D.P.; Anumonwo, J.M.B.; Auerbach, D.S.; Furspan, P.B.; Hou, L.; Hu, B.; Schumacher, S.M.; Vaidyanathan, R.; et al. Dynamic Reciprocity of Sodium and Potassium Channel Expression in a Macromolecular Complex Controls Cardiac Excitability and Arrhythmia. Proc. Natl. Acad. Sci. USA 2012, 109, E2134–E2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nattel, S.; Maguy, A.; Le Bouter, S.; Yeh, Y.-H. Arrhythmogenic Ion-Channel Remodeling in the Heart: Heart Failure, Myocardial Infarction, and Atrial Fibrillation. Physiol. Rev. 2007, 87, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Valdivia, C.R.; Chu, W.W.; Pu, J.; Foell, J.D.; Haworth, R.A.; Wolff, M.R.; Kamp, T.J.; Makielski, J.C. Increased Late Sodium Current in Myocytes from a Canine Heart Failure Model and from Failing Human Heart. J. Mol. Cell. Cardiol. 2005, 38, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-R.; Lau, C.-P.; Leung, T.-K.; Nattel, S. Ionic Current Abnormalities Associated with Prolonged Action Potentials in Cardiomyocytes from Diseased Human Right Ventricles. Heart Rhythm 2004, 1, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.; Armoundas, A.A.; Tian, Y.; DiSilvestre, D.; Burysek, M.; Halperin, V.; O’Rourke, B.; Kass, D.A.; Marbán, E.; Tomaselli, G.F. Molecular Correlates of Altered Expression of Potassium Currents in Failing Rabbit Myocardium. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2077–H2087. [Google Scholar] [CrossRef]
- Pogwizd, S.M.; Schlotthauer, K.; Li, L.; Yuan, W.; Bers, D.M. Arrhythmogenesis and Contractile Dysfunction in Heart Failure: Roles of Sodium-Calcium Exchange, Inward Rectifier Potassium Current, and Residual Beta-Adrenergic Responsiveness. Circ. Res. 2001, 88, 1159–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husti, Z.; Varró, A.; Baczkó, I. Arrhythmogenic Remodeling in the Failing Heart. Cells 2021, 10, 3203. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Marín, P.; Tinaquero, D.; Utrilla, R.G.; Cebrián, J.; González-Guerra, A.; Crespo-García, T.; Cámara-Checa, A.; Rubio-Alarcón, M.; Dago, M.; Alfayate, S.; et al. Tbx5 Variants Disrupt Nav1.5 Function Differently in Patients Diagnosed with Brugada or Long QT Syndrome. Cardiovasc. Res. 2022, 118, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Hernández, M.; Matamoros, M.; Alfayate, S.; Nieto-Marín, P.; Utrilla, R.G.; Tinaquero, D.; de Andrés, R.; Crespo, T.; Ponce-Balbuena, D.; Willis, B.C.; et al. Brugada Syndrome Trafficking-Defective Nav1.5 Channels Can Trap Cardiac Kir2.1/2.2 Channels. JCI Insight 2018, 3, e96291. [Google Scholar] [CrossRef] [PubMed]
- Caballero, R.; Dolz-Gaitón, P.; Gómez, R.; Amorós, I.; Barana, A.; González de la Fuente, M.; Osuna, L.; Duarte, J.; López-Izquierdo, A.; Moraleda, I.; et al. Flecainide Increases Kir2.1 Currents by Interacting with Cysteine 311, Decreasing the Polyamine-Induced Rectification. Proc. Natl. Acad. Sci. USA 2010, 107, 15631–15636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matamoros, M.; Pérez-Hernández, M.; Guerrero-Serna, G.; Amorós, I.; Barana, A.; Núñez, M.; Ponce-Balbuena, D.; Sacristán, S.; Gómez, R.; Tamargo, J.; et al. Nav1.5 N-Terminal Domain Binding to α1-Syntrophin Increases Membrane Density of Human Kir2.1, Kir2.2 and Nav1.5 Channels. Cardiovasc. Res. 2016, 110, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuurbier, C.J.; Baartscheer, A.; Schumacher, C.A.; Fiolet, J.W.T.; Coronel, R. Sodium-Glucose Co-Transporter 2 Inhibitor Empagliflozin Inhibits the Cardiac Na+/H+ Exchanger 1: Persistent Inhibition under Various Experimental Conditions. Cardiovasc. Res. 2021, 117, 2699–2701. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.J.; Park, K.C.; Tokar, S.; Eykyn, T.R.; Fuller, W.; Pavlovic, D.; Swietach, P.; Shattock, M.J. Off-Target Effects of Sodium-Glucose Co-Transporter 2 Blockers: Empagliflozin Does Not Inhibit Na+/H+ Exchanger-1 or Lower [Na+]i in the Heart. Cardiovasc. Res. 2021, 117, 2794–2806. [Google Scholar] [CrossRef] [PubMed]
- Ponce-Balbuena, D.; Guerrero-Serna, G.; Valdivia, C.R.; Caballero, R.; Diez-Guerra, F.J.; Jiménez-Vázquez, E.N.; Ramírez, R.J.; Monteiro da Rocha, A.; Herron, T.J.; Campbell, K.F.; et al. Cardiac Kir2.1 and NaV1.5 Channels Traffic Together to the Sarcolemma to Control Excitability. Circ. Res. 2018, 122, 1501–1516. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Dybkova, N.; Rasenack, E.C.L.; Jacobshagen, C.; Fabritz, L.; Kirchhof, P.; Maier, S.K.G.; Zhang, T.; Hasenfuss, G.; Brown, J.H.; et al. Ca2+/Calmodulin-Dependent Protein Kinase II Regulates Cardiac Na+ Channels. J. Clin. Invest. 2006, 116, 3127–3138. [Google Scholar] [CrossRef]
- Ashpole, N.M.; Herren, A.W.; Ginsburg, K.S.; Brogan, J.D.; Johnson, D.E.; Cummins, T.R.; Bers, D.M.; Hudmon, A. Ca2+/Calmodulin-Dependent Protein Kinase II (CaMKII) Regulates Cardiac Sodium Channel NaV1.5 Gating by Multiple Phosphorylation Sites. J. Biol. Chem. 2012, 287, 19856–19869. [Google Scholar] [CrossRef] [Green Version]
- Mustroph, J.; Wagemann, O.; Lücht, C.M.; Trum, M.; Hammer, K.P.; Sag, C.M.; Lebek, S.; Tarnowski, D.; Reinders, J.; Perbellini, F.; et al. Empagliflozin Reduces Ca/Calmodulin-Dependent Kinase II Activity in Isolated Ventricular Cardiomyocytes. ESC Heart Fail. 2018, 5, 642–648. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Zou, R.; Shi, W.; Zhou, N.; Chen, S.; Zhou, H.; Chen, X.; Wu, Y. SGLT2 Inhibitor Dapagliflozin Reduces Endothelial Dysfunction and Microvascular Damage during Cardiac Ischemia/Reperfusion Injury through Normalizing the XO-SERCA2-CaMKII-Coffilin Pathways. Theranostics 2022, 12, 5034–5050. [Google Scholar] [CrossRef]
- Chadda, K.R.; Jeevaratnam, K.; Lei, M.; Huang, C.L.-H. Sodium Channel Biophysics, Late Sodium Current and Genetic Arrhythmic Syndromes. Pflug. Arch. 2017, 469, 629–641. [Google Scholar] [CrossRef]
- Utrilla, R.G.; Nieto-Marín, P.; Alfayate, S.; Tinaquero, D.; Matamoros, M.; Pérez-Hernández, M.; Sacristán, S.; Ondo, L.; de Andrés, R.; Díez-Guerra, F.J.; et al. Kir2.1-Nav1.5 Channel Complexes Are Differently Regulated than Kir2.1 and Nav1.5 Channels Alone. Front. Physiol. 2017, 8, 903. [Google Scholar] [CrossRef] [PubMed]
- Trum, M.; Riechel, J.; Lebek, S.; Pabel, S.; Sossalla, S.T.; Hirt, S.; Arzt, M.; Maier, L.S.; Wagner, S. Empagliflozin Inhibits Na+/H+ Exchanger Activity in Human Atrial Cardiomyocytes. ESC Heart Fail. 2020, 7, 4429–4437. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class Effects of SGLT2 Inhibitors in Mouse Cardiomyocytes and Hearts: Inhibition of Na+/H+ Exchanger, Lowering of Cytosolic Na+ and Vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Burgess, D.E.; Schroder, E.A.; Elayi, C.S.; Anderson, C.L.; January, C.T.; Sun, B.; Immadisetty, K.; Kekenes-Huskey, P.M.; Delisle, B.P. Long QT Syndrome Type 2: Emerging Strategies for Correcting Class 2 KCNH2 (HERG) Mutations and Identifying New Patients. Biomolecules 2020, 10, 1144. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Takanari, H.; Qile, M.; Nalos, L.; Houtman, M.J.C.; Romunde, F.L.; Heukers, R.; van Bergen En Henegouwen, P.M.P.; Vos, M.A.; van der Heyden, M.A.G. Class III Antiarrhythmic Drugs Amiodarone and Dronedarone Impair KIR 2.1 Backward Trafficking. J. Cell Mol. Med. 2017, 21, 2514–2523. [Google Scholar] [CrossRef]
- de Git, K.C.G.; de Boer, T.P.; Vos, M.A.; van der Heyden, M.A.G. Cardiac Ion Channel Trafficking Defects and Drugs. Pharmacol. Ther. 2013, 139, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Komoroski, B.; Vachharajani, N.; Feng, Y.; Li, L.; Kornhauser, D.; Pfister, M. Dapagliflozin, a Novel, Selective SGLT2 Inhibitor, Improved Glycemic Control over 2 Weeks in Patients with Type 2 Diabetes Mellitus. Clin. Pharmacol. Ther. 2009, 85, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Kasichayanula, S.; Liu, X.; Lacreta, F.; Griffen, S.C.; Boulton, D.W. Clinical Pharmacokinetics and Pharmacodynamics of Dapagliflozin, a Selective Inhibitor of Sodium-Glucose Co-Transporter Type 2. Clin. Pharmacokinet. 2014, 53, 17–27. [Google Scholar] [CrossRef]
- Scheen, A.J. Pharmacokinetic and Pharmacodynamic Profile of Empagliflozin, a Sodium Glucose Co-Transporter 2 Inhibitor. Clin. Pharmacokinet. 2014, 53, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Laffel, L.M.B.; Tamborlane, W.V.; Yver, A.; Simons, G.; Wu, J.; Nock, V.; Hobson, D.; Hughan, K.S.; Kaspers, S.; Marquard, J. Pharmacokinetic and Pharmacodynamic Profile of the Sodium-Glucose Co-Transporter-2 Inhibitor Empagliflozin in Young People with Type 2 Diabetes: A Randomized Trial. Diabet. Med. 2018, 35, 1096–1104. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hiPSC-CMs | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Activation | Inactivation | ||||||||
| τact (ms) | Vh (mV) | k | τfinact (ms) | Afinact (%) | τsinact (ms) | Asinact (%) | Vh (mV) | k | |
| Control | 0.27 ± 0.02 | −47.2 ± 1.6 | 4.7 ± 0.4 | 1.5 ± 0.1 | 83.1 ± 3.7 | 6.1 ± 0.9 | 16.9 ± 3.7 | −97.3 ± 4.5 | 5.6 ± 0.2 |
| Dapa | 0.29 ± 0.01 | −55.5 ± 2.8 * | 4.6 ± 0.6 | 1.7 ± 0.1 | 85.3 ± 2.9 | 8.2 ± 1.3 | 14.7 ± 2.9 | −96.2 ± 4.3 | 5.7 ± 0.4 |
| Empa | 0.27 ± 0.03 | −48.5 ± 0.8 | 4.5 ± 0.3 | 1.5 ± 0.1 | 83.2 ± 1.7 | 6.0 ± 0.5 | 16.8 ± 1.7 | −108.6 ± 4.4 * | 5.4 ± 0.3 |
| CHO cells | |||||||||
| Activation | Inactivation | ||||||||
| τact (ms) | Vh (mV) | k | τfinact (ms) | Afinact (%) | τsinact (ms) | Asinact (%) | Vh (mV) | k | |
| Control | 0.22 ± 0.02 | −37.6 ± 2.5 | 6.3 ± 0.4 | 1.3 ± 0.1 | 86.5 ± 1.3 | 8.8 ± 1.1 | 13.5 ± 1.3 | −81.5 ± 2.2 | 5.7 ± 0.3 |
| Dapa | 0.22 ± 0.01 | −45.7 ± 3.1 * | 6.2 ± 0.4 | 1.5 ± 0.1 | 88.7 ± 1.1 | 9.2 ± 1.2 | 11.3 ± 1.1 | −83.7 ± 4.4 | 5.8 ± 0.4 |
| Empa | 0.21 ± 0.01 | −40.4 ± 4.8 | 6.3 ± 0.8 | 1.3 ± 0.05 | 85.5 ± 2.7 | 5.7 ± 0.7 | 14.5 ± 2.7 | −94.5 ± 0.8 * | 6.1 ± 0.4 |
| CaMKII-silenced CHO cells | |||||||||
| Activation | Inactivation | ||||||||
| τact (ms) | Vh (mV) | k | τfinact (ms) | Afinact (%) | τsinact (ms) | Asinact (%) | Vh (mV) | k | |
| Control | 0.25 ± 0.03 | −40.6 ± 1.8 | 6.3 ± 0.7 | 1.0 ± 0.08 # | 82.7 ± 2.8 | 6.9 ± 0.6 # | 17.3 ± 2.8 | −76.2 ± 1.5 # | 5.4 ± 0.6 |
| Dapa | 0.23 ± 0.03 | −48.8 ± 3.5 * | 6.1 ± 0.7 | 1.2 ± 0.1 # | 83.7 ± 2.9 | 5.4 ± 0.8 # | 16.3 ± 2.9 | −76.9 ± 4.1 # | 5.7 ± 0.1 |
| Empa | 0.22 ± 0.02 | −38.6 ± 1.0 | 6.5 ± 0.5 | 1.1 ± 0.1 # | 85.0 ± 2.4 | 6.5 ± 0.5 | 15.0 ± 2.4 | −91.4 ± 2.2 *# | 5.5 ± 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dago, M.; Crespo-García, T.; Cámara-Checa, A.; Rapún, J.; Rubio-Alarcón, M.; Marín, M.; Tamargo, J.; Caballero, R.; Delpón, E. Empagliflozin and Dapagliflozin Increase Na+ and Inward Rectifier K+ Current Densities in Human Cardiomyocytes Derived from Induced Pluripotent Stem Cells (hiPSC-CMs). Cells 2022, 11, 3707. https://doi.org/10.3390/cells11233707
Dago M, Crespo-García T, Cámara-Checa A, Rapún J, Rubio-Alarcón M, Marín M, Tamargo J, Caballero R, Delpón E. Empagliflozin and Dapagliflozin Increase Na+ and Inward Rectifier K+ Current Densities in Human Cardiomyocytes Derived from Induced Pluripotent Stem Cells (hiPSC-CMs). Cells. 2022; 11(23):3707. https://doi.org/10.3390/cells11233707
Chicago/Turabian StyleDago, María, Teresa Crespo-García, Anabel Cámara-Checa, Josu Rapún, Marcos Rubio-Alarcón, María Marín, Juan Tamargo, Ricardo Caballero, and Eva Delpón. 2022. "Empagliflozin and Dapagliflozin Increase Na+ and Inward Rectifier K+ Current Densities in Human Cardiomyocytes Derived from Induced Pluripotent Stem Cells (hiPSC-CMs)" Cells 11, no. 23: 3707. https://doi.org/10.3390/cells11233707