


UBA52 Is Crucial in HSP90 Ubiquitylation and Neurodegenerative Signaling during Early Phase of Parkinson’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Cell Culture, Differentiation and Treatments
2.3. Plasmids, Cloning, Mutagenesis and Transfection
2.4. siRNA Mediated Knockdown
2.5. Animals and Stereotactic Neurosurgery
2.6. Animal Behavior Assessment
2.7. Cell Viability
2.8. mRNA Expression by RT-PCR & qPCR
2.9. Protein Extraction and Western Blot
2.10. Immunocytochemistry/Immunohistochemistry and Counterstaining
2.11. Thioflavin-S (Th-S) Assay
2.12. Confocal Microscopy
2.13. Co-Immunoprecipitation (Co-IP)
2.14. Mass Spectrometry
2.15. In Vitro Ubiquitylation Assay
2.16. Proteasome Activity
2.17. Statistical Analysis
3. Results
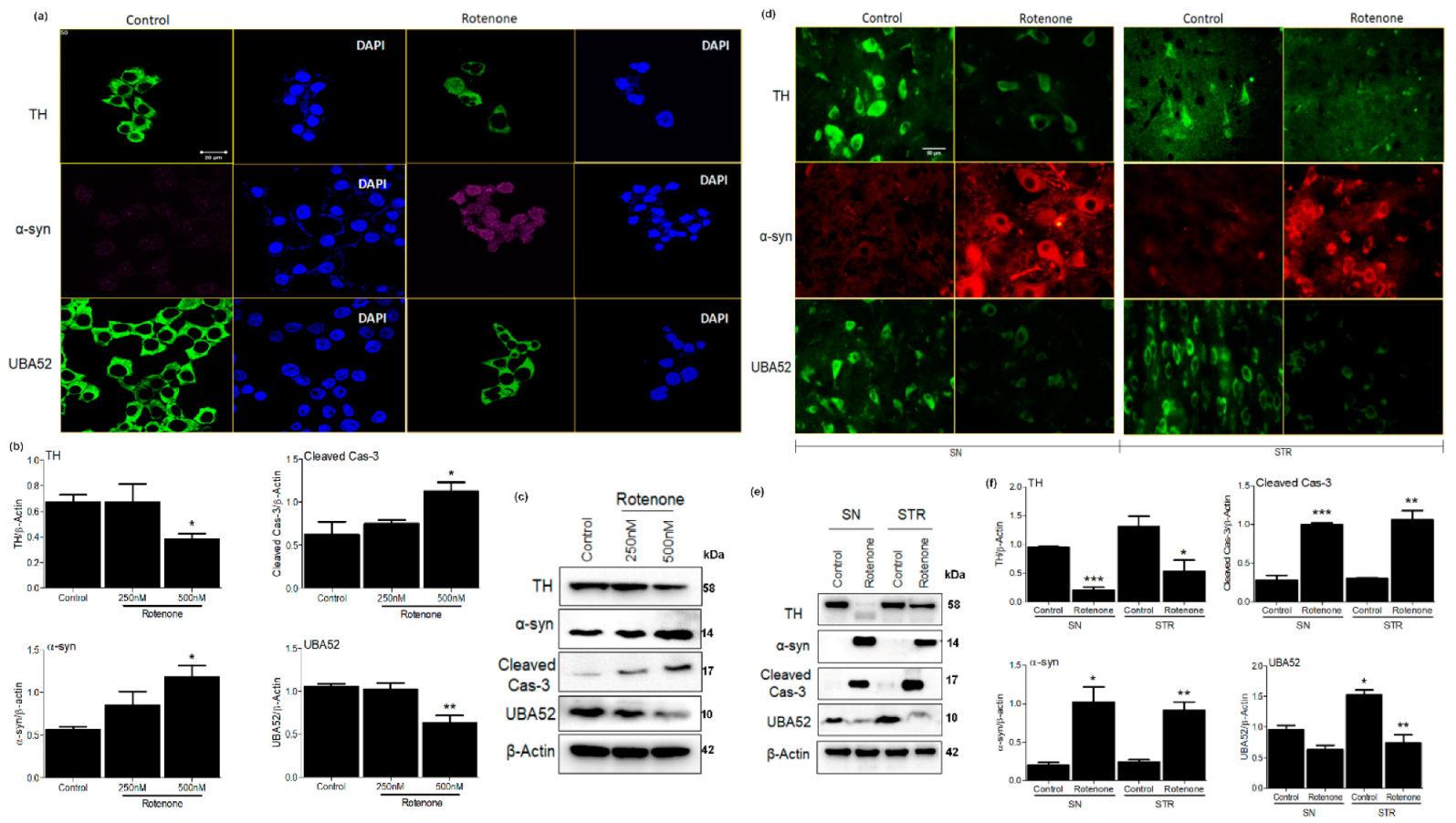
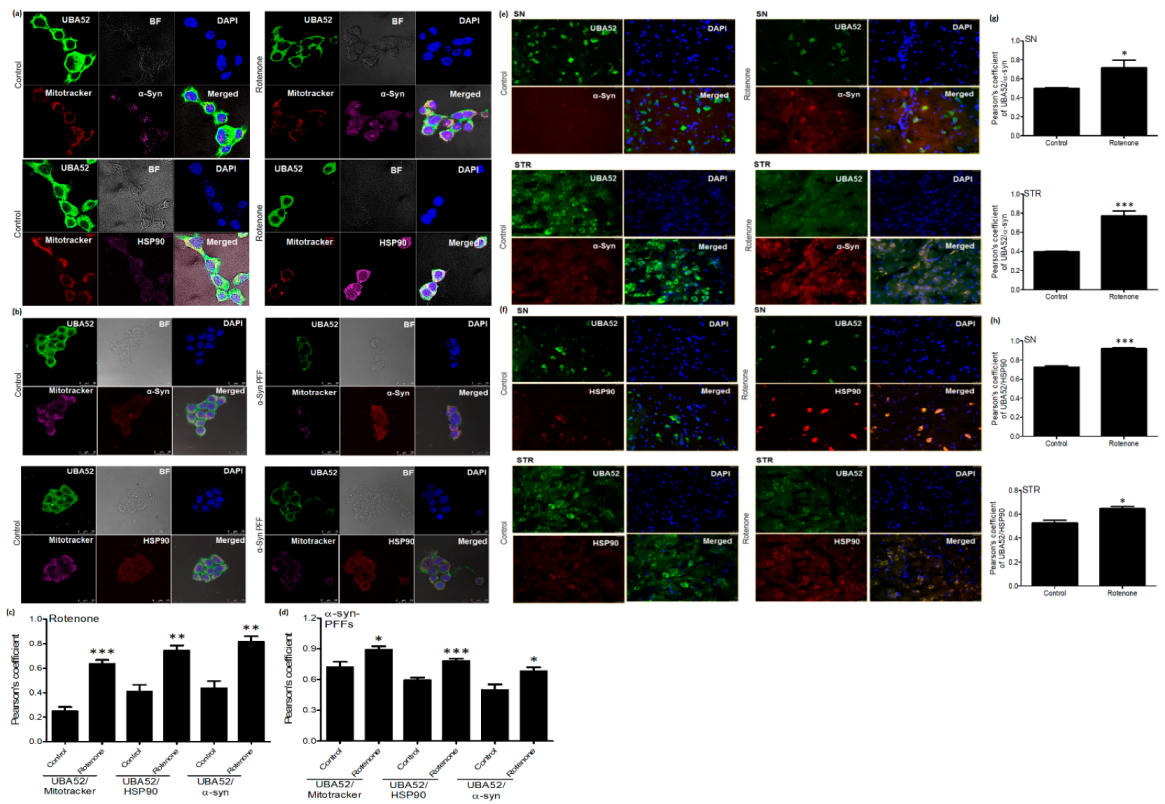
3.1. Parkinson’s Disease-Specific Pathological Markers and UBA52
3.2. UBA52 Surplus Attenuates the PD-Specific Pathological Markers and Neuronal Death
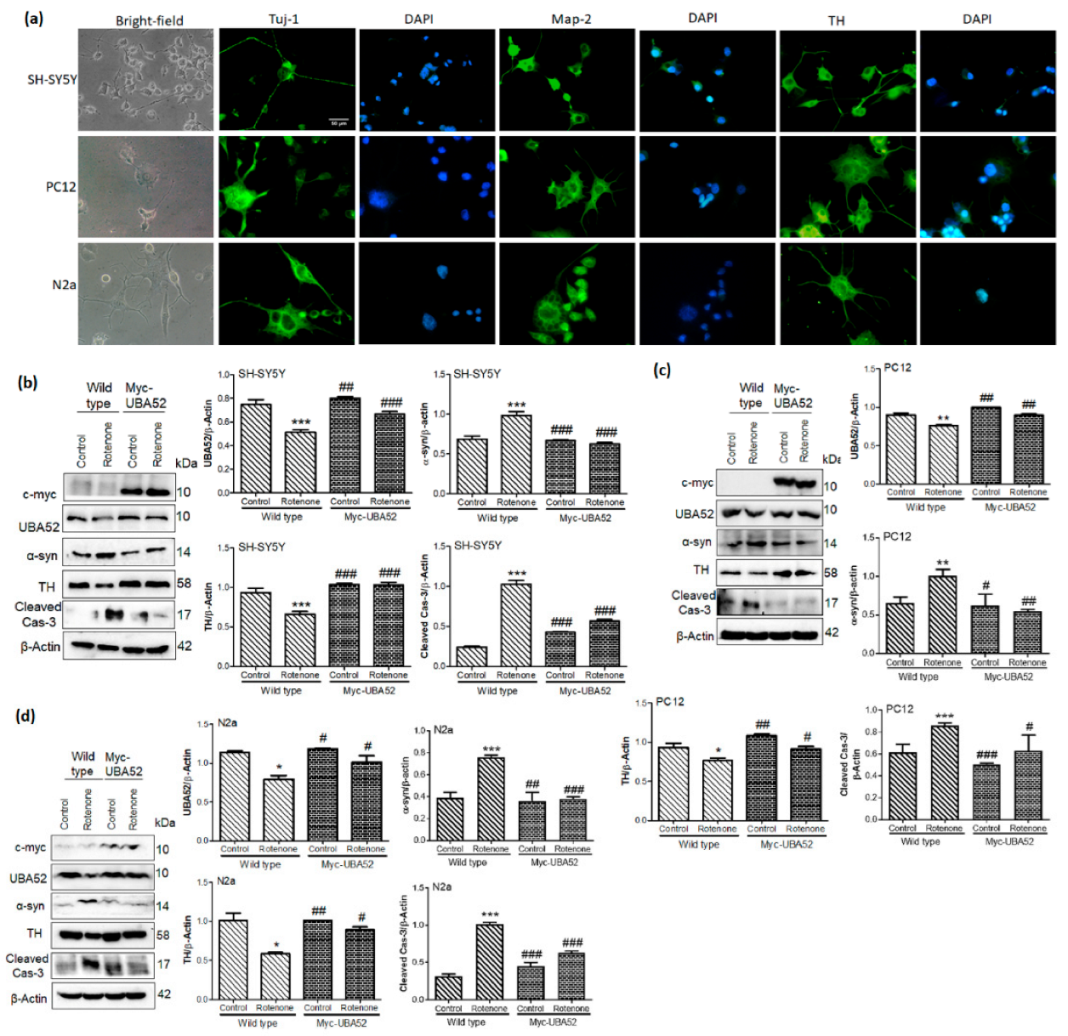
3.3. UBA52 Revokes the PD-Specific Pathophysiological Markers in Differentiated Neuronal Cells
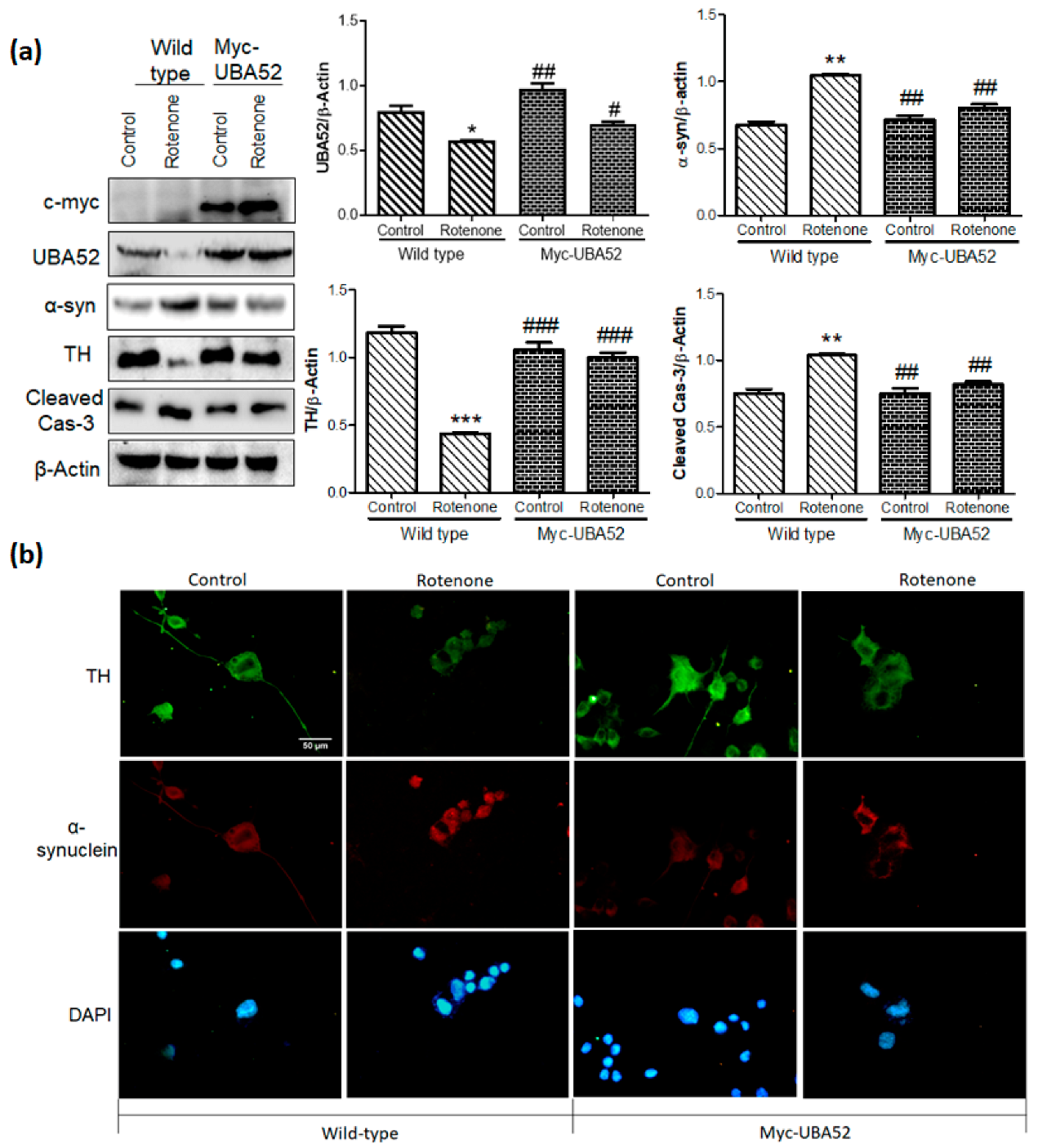
3.4. Effect of UBA52 on PD-Specific Pathological Markers in Experimental In Vitro Chronic Model
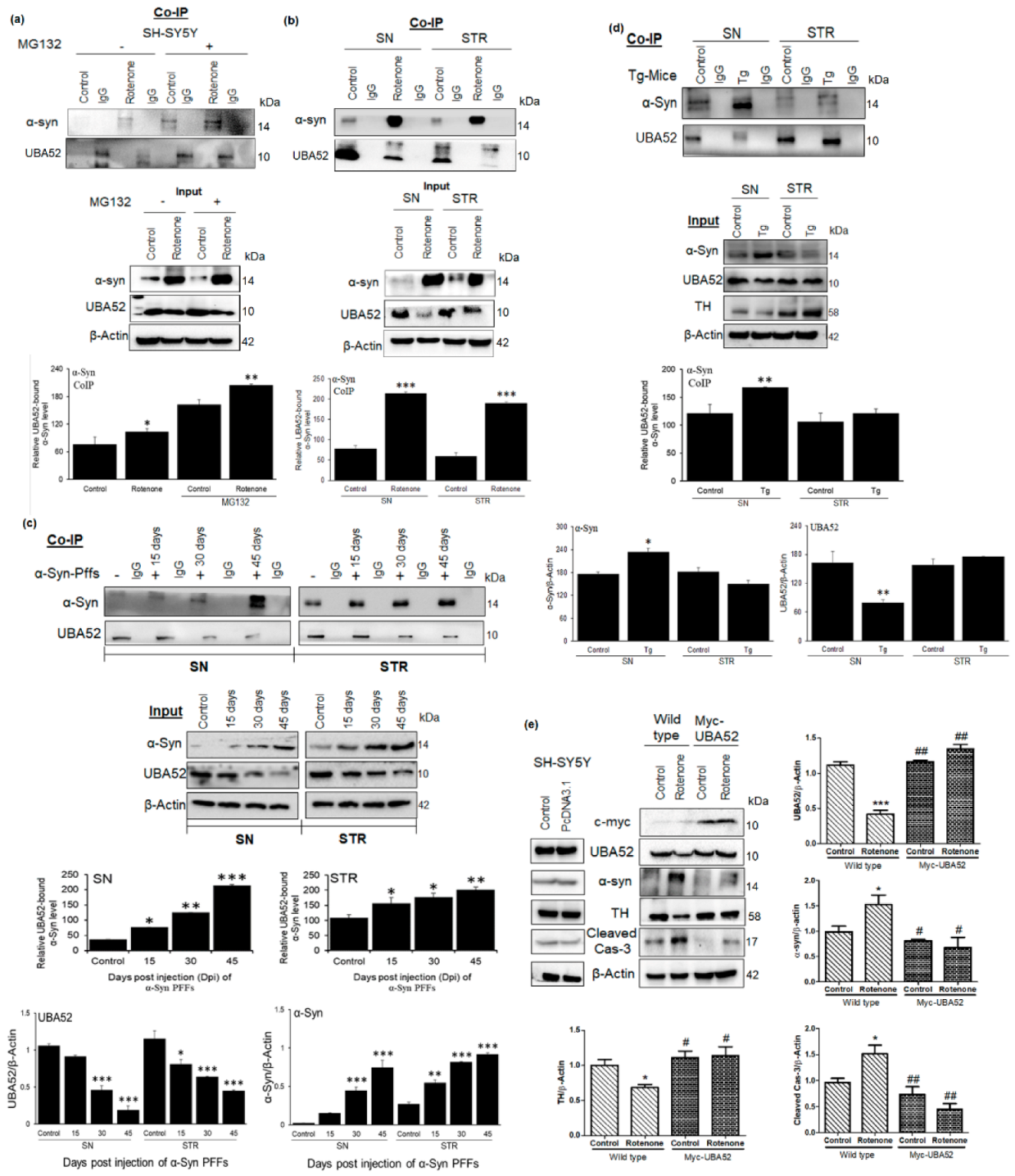
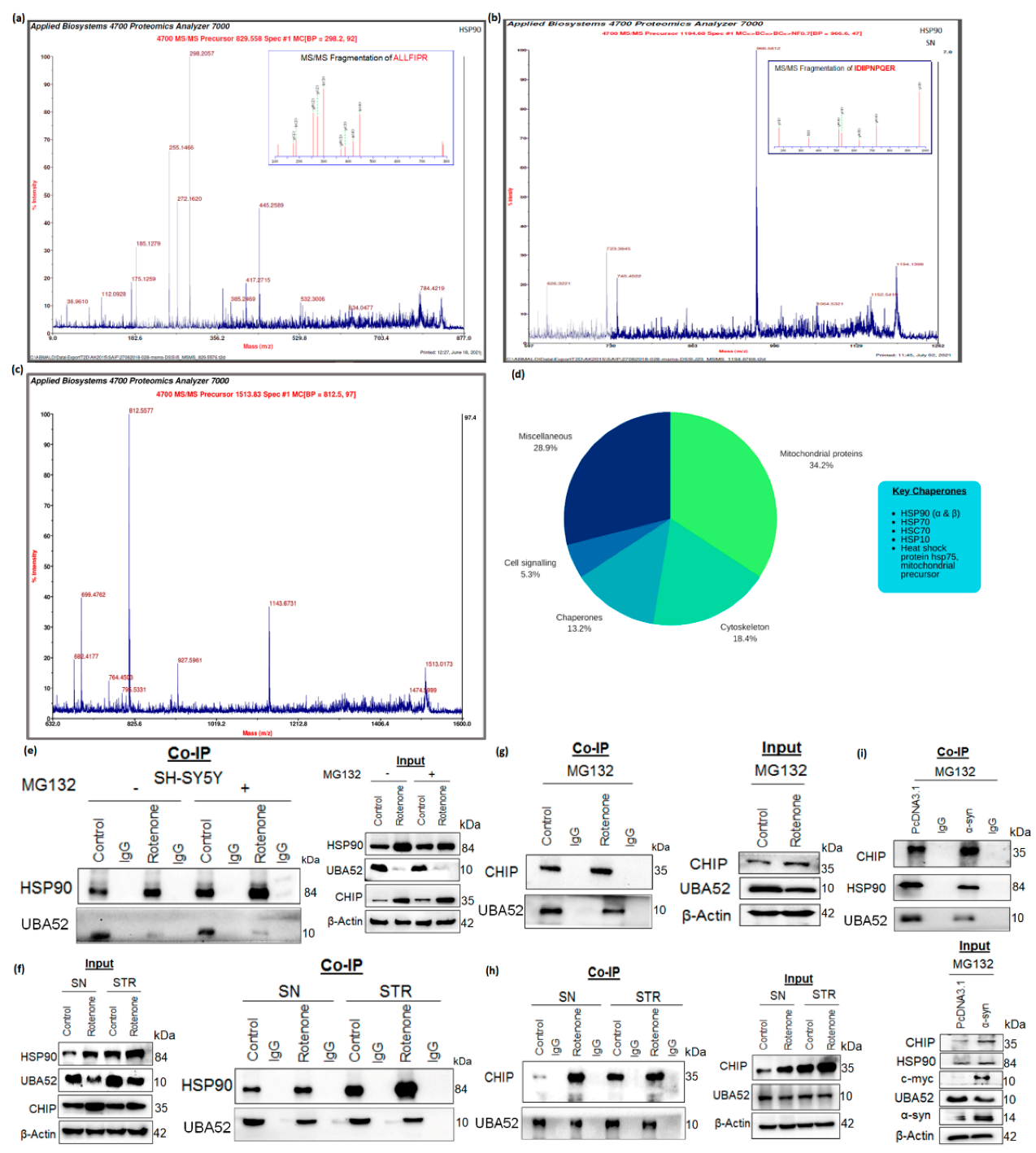
3.5. Tuning the Interaction between HSP90 and CHIP by UBA52
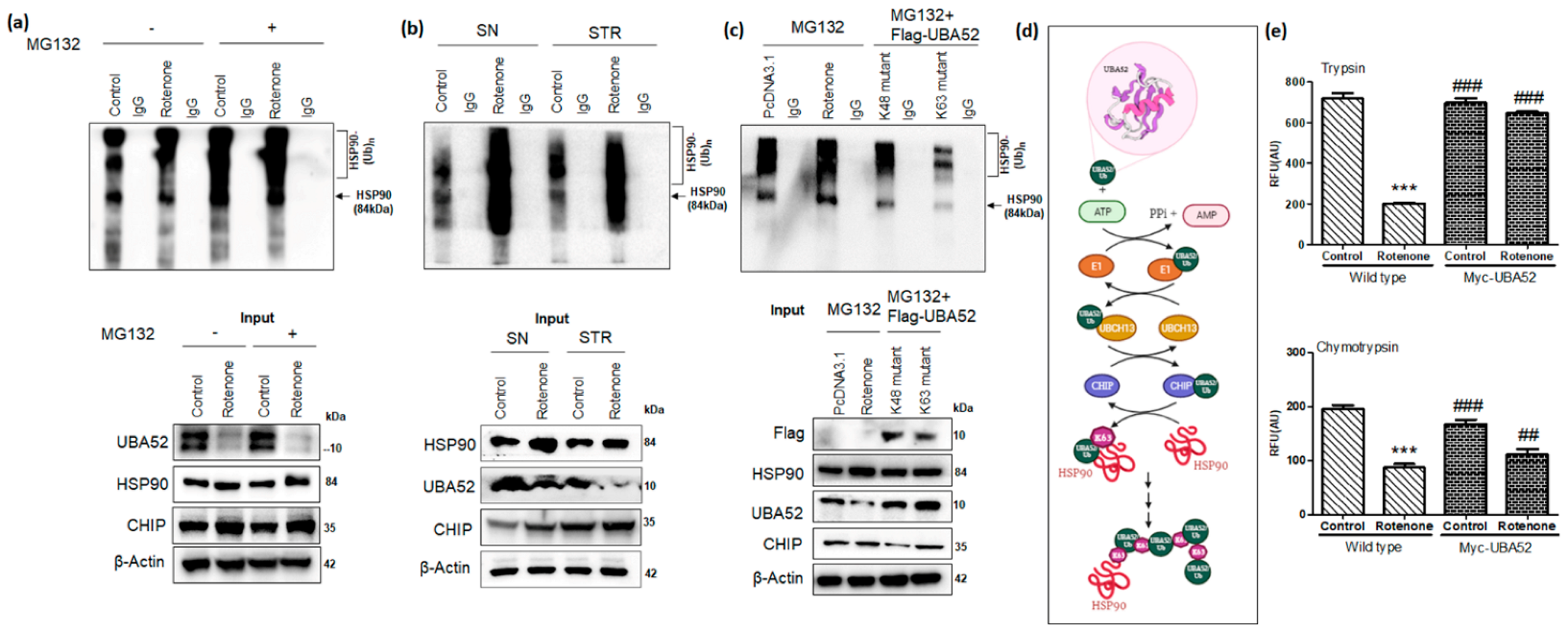
3.6. Role of UBA52 in HSP90 Ubiquitylation during Parkinson’s Disease
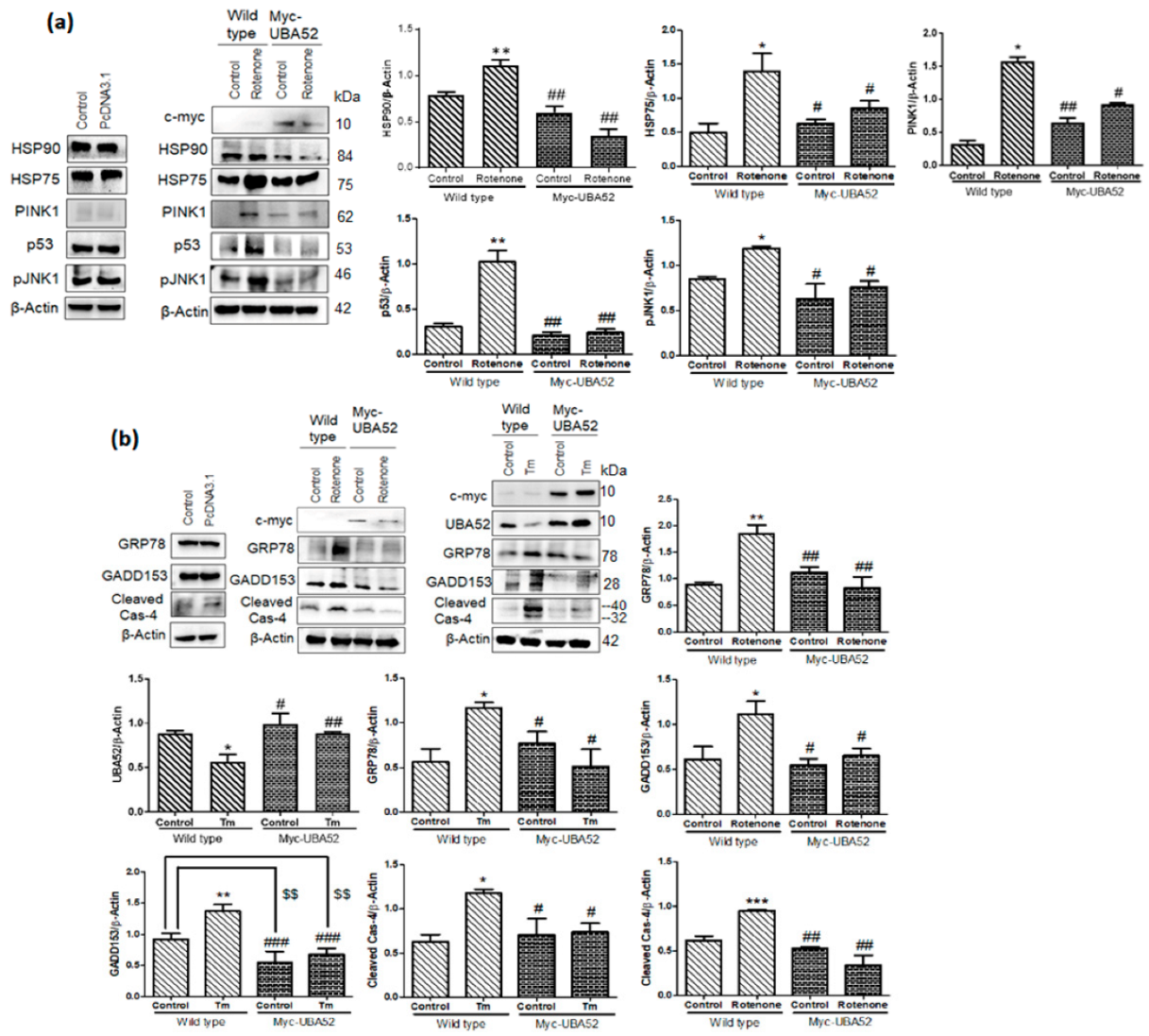
3.7. Effect of UBA52 on Client Proteins of HSP90 and ER Stress-Related Pathophysiological State
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PD | Parkinson’s disease |
| SN | Substantia nigra |
| STR | Striatum |
| TH | Tyrosine hydroxylase |
| UPS | Ubiquitin–proteasome table system |
| ERS | Endoplasmic reticulum stress |
| UBA52 | Ubiquitin-60S ribosomal protein L40 |
| UBB | Polyubiquitin-B |
| UBC | Polyubiquitin-C |
| RPS27a | Ubiquitin-40S ribosomal protein S27a |
| RT-qPCR | Real-time quantitative PCR |
| MS | Mass spectrometry |
| MALDI-TOF | Matrix-assisted laser desorption ionization-time of flight |
| ACN | Acetonitrile |
| ABC | Ammonium Bicarbonate |
| TFA | Trifluro acetic acid |
| IgG: | Immunoglobulin G |
| PQC | Protein quality control |
| PFFs | Preformed fibrils |
| PSOPIA | Prediction server of protein–protein interaction |
| HSP90 | Heat shock protein 90 |
| CHIP | C-terminus of HSC-70 |
| K48R | Lysine residue mutated to arginine at position-48 of UBA52 |
| K63R | Lysine residue mutated to arginine at position-63 of UBA52 |
| pJNK1 | phospho- c-Jun N-terminal kinase 1 |
| AKT1 | RAC-alpha serine/threonine-protein kinase 1 |
| PINK1 | PTEN-induced kinase 1 |
| HSP75 | Heat shock protein 75 |
References
- Lang, A.E.; Lozano, A.M. Parkinson’s Disease. First of Two Parts. N. Engl. J. Med. 1998, 339, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Singh, S. Reciprocal Upshot of Nitric Oxide, Endoplasmic Reticulum Stress, and Ubiquitin Proteasome System in Parkinson’s Disease Pathology. Neuroscientist 2021, 27, 340–354. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Dikshit, M. Apoptotic Neuronal Death in Parkinson’s Disease: Involvement of Nitric Oxide. Brain Res. Rev. 2007, 54, 233–250. [Google Scholar] [CrossRef]
- Goswami, P.; Gupta, S.; Biswas, J.; Sharma, S.; Singh, S. Endoplasmic Reticulum Stress Instigates the Rotenone Induced Oxidative Apoptotic Neuronal Death: A Study in Rat Brain. Mol. Neurobiol. 2016, 53, 5384–5400. [Google Scholar] [CrossRef]
- da Costa, C.A.; el Manaa, W.; Duplan, E.; Checler, F. The Endoplasmic Reticulum Stress/Unfolded Protein Response and Their Contributions to Parkinson’s Disease Physiopathology. Cells 2020, 9, 2495. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, V.; Saarma, M. Endoplasmic Reticulum Stress Regulators: New Drug Targets for Parkinson’s Disease. J. Park. Dis. 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Holtz, W.A.; O’Malley, K.L. Parkinsonian Mimetics Induce Aspects of Unfolded Protein Response in Death of Dopaminergic Neurons. J. Biol. Chem. 2003, 278, 19367–19377. [Google Scholar] [CrossRef] [PubMed]
- Greenamyre, J.T.; Hastings, T.G. Parkinsons-Divergent Causes Convergent Mechanisms. Science (1979) 2004, 304, 1120–1122. [Google Scholar] [CrossRef]
- Ryu, E.J.; Harding, H.P.; Angelastro, J.M.; Vitolo, O.v.; Ron, D.; Greene, L.A. Endoplasmic Reticulum Stress and the Unfolded Protein Response in Cellular Models of Parkinson’s Disease. J. Neurosci. 2002, 22, 10690. [Google Scholar] [CrossRef] [PubMed]
- Cimato, T.R.; Ettinger, M.J.; Zhou, X.; Aletta, J.M. Nerve Growth Factor–Specific Regulation of Protein Methylation during Neuronal Differentiation of PC12 Cells. J. Cell Biol. 1997, 138, 1089. [Google Scholar] [CrossRef]
- Gupta, S.; Mishra, A.; Singh, S. Cardinal Role of Eukaryotic Initiation Factor 2 (EIF2α) in Progressive Dopaminergic Neuronal Death & DNA Fragmentation: Implication of PERK:IRE1α:ATF6 Axis in Parkinson’s Pathology. Cell. Signal. 2021, 81, 109922. [Google Scholar] [CrossRef]
- Singh, A.; Yadawa, A.K.; Chaturvedi, S.; Wahajuddin, M.; Mishra, A.; Singh, S. Mechanism for AntiParkinsonian Effect of Resveratrol: Involvement of Transporters, Synaptic Proteins, Dendrite Arborization, Biochemical Alterations, ER Stress and Apoptosis. Food Chem. Toxicol. 2021, 155, 112433. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Du, X.; Jiao, Q.; Chen, X.; Jiang, H. Expanding the Role of Proteasome Homeostasis in Parkinson’s Disease: Beyond Protein Breakdown. Cell Death Dis. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Liu, J.; McCabe, J.T. Ubiquitin and Ubiquitin-Conjugated Protein Expression in the Rat Cerebral Cortex and Hippocampus Following Traumatic Brain Injury (TBI). Brain Res. 2007, 1182, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Oshima, S.; Maeyashiki, C.; Nibe, Y.; Otsubo, K.; Matsuzawa, Y.; Nemoto, Y.; Nagaishi, T.; Okamoto, R.; Tsuchiya, K.; et al. The Ubiquitin Hybrid Gene UBA52 Regulates Ubiquitination of Ribosome and Sustains Embryonic Development. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Fernández-Pevida, A.; Rodríguez-Galán, O.; Díaz-Quintana, A.; Kressler, D.; de La Cruz, J. Yeast Ribosomal Protein L40 Assembles Late into Precursor 60 S Ribosomes and Is Required for Their Cytoplasmic Maturation. J. Biol. Chem. 2012, 287, 38390–38407. [Google Scholar] [CrossRef]
- Jan, A.; Jansonius, B.; Delaidelli, A.; Bhanshali, F.; An, Y.A.; Ferreira, N.; Smits, L.M.; Negri, G.L.; Schwamborn, J.C.; Jensen, P.H.; et al. Activity of Translation Regulator Eukaryotic Elongation Factor-2 Kinase Is Increased in Parkinson Disease Brain and Its Inhibition Reduces Alpha Synuclein Toxicity. Acta Neuropathol. Commun. 2018, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Uryu, K.; Richter-Landsberg, C.; Welch, W.; Sun, E.; Goldbaum, O.; Norris, E.H.; Pham, C.T.; Yazawa, I.; Hilburger, K.; Micsenyi, M.; et al. Convergence of Heat Shock Protein 90 with Ubiquitin in Filamentous α-Synuclein Inclusions of α-Synucleinopathies. Am. J. Pathol. 2006, 168, 947–961. [Google Scholar] [CrossRef]
- Burmann, B.M.; Gerez, J.A.; Matečko-Burmann, I.; Campioni, S.; Kumari, P.; Ghosh, D.; Mazur, A.; Aspholm, E.E.; Šulskis, D.; Wawrzyniuk, M.; et al. Regulation of α-Synuclein by Chaperones in Mammalian Cells. Nature 2020, 577, 127–132. [Google Scholar] [CrossRef]
- Friesen, E.L.; de Snoo, M.L.; Rajendran, L.; Kalia, L.v.; Kalia, S.K. Chaperone-Based Therapies for Disease Modification in Parkinson’s Disease. Park. Dis. 2017, 2017. [Google Scholar] [CrossRef]
- Zhang, M.; Windheim, M.; Roe, S.M.; Peggie, M.; Cohen, P.; Prodromou, C.; Pearl, L.H. Chaperoned Ubiquitylation—Crystal Structures of the CHIP U Box E3 Ubiquitin Ligase and a CHIP-Ubc13-Uev1a Complex. Mol. Cell. 2005, 20, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.K.K.; Dawson, V.L.; Dawson, T.M. The Role of the Ubiquitin-Proteasomal Pathway in Parkinson ’ s Disease and Other Neurodegenerative Disorders Resulted in the Discovery of a Pathway That Provides. Trends Neurosci. 2001, 24, 7–14. [Google Scholar] [CrossRef]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin Is Phosphorylated by PINK1 to Activate Parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Kame, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, Paraquat, and Parkinson’s Disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y Cell Line in Parkinson’s Disease Research: A Systematic Review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.; Xing, V.; Wang, T.T.; Bureau, S.C.; Link, G.A.; Fortin, T.; Zhang, H.; Hayley, S.; Sun, H. Alleviating Toxic α-Synuclein Accumulation by Membrane Depolarization: Evidence from an in Vitro Model of Parkinson’s Disease. Mol. Brain 2020, 13. [Google Scholar] [CrossRef]
- Cheung, Y.T.; Lau, W.K.W.; Yu, M.S.; Lai, C.S.W.; Yeung, S.C.; So, K.F.; Chang, R.C.C. Effects of All-Trans-Retinoic Acid on Human SH-SY5Y Neuroblastoma as in Vitro Model in Neurotoxicity Research. Neurotoxicology 2009, 30, 127–135. [Google Scholar] [CrossRef] [PubMed]
- S Narasimhan, K.K.; Devarajan, A.; Karan, G.; Sundaram, S.; Wang, Q.; van Groen, T.; del Monte, F.; Rajasekaran, N.S. Reductive Stress Promotes Protein Aggregation and Impairs Neurogenesis. Redox Biol. 2020, 37, 101739. [Google Scholar] [CrossRef]
- Borland, M.K.; Trimmer, P.A.; Rubinstein, J.D.; Keeney, P.M.; Mohanakumar, K.P.; Liu, L.; Bennett, J.P. Chronic, Low-Dose Rotenone Reproduces Lewy Neurites Found in Early Stages of Parkinson’s Disease, Reduces Mitochondrial Movement and Slowly Kills Differentiated SH-SY5Y Neural Cells. Mol. Neurodegener. 2008, 3. [Google Scholar] [CrossRef]
- Verma, D.K.; Gupta, S.; Biswas, J.; Joshi, N.; Singh, A.; Gupta, P.; Tiwari, S.; Sivarama Raju, K.; Chaturvedi, S.; Wahajuddin, M.; et al. New Therapeutic Activity of Metabolic Enhancer Piracetam in Treatment of Neurodegenerative Disease: Participation of Caspase Independent Death Factors, Oxidative Stress, Inflammatory Responses and Apoptosis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2078–2096. [Google Scholar] [CrossRef] [PubMed]
- Richfield, E.K.; Thiruchelvam, M.J.; Cory-Slechta, D.A.; Wuertzer, C.; Gainetdinov, R.R.; Caron, M.G.; di Monte, D.A.; Federoff, H.J. Behavioral and Neurochemical Effects of Wild-Type and Mutated Human Alpha-Synuclein in Transgenic Mice. Exp. Neurol. 2002, 175, 35–48. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, P.; Tan, J.; Li, M.; Xu, X.; Shao, X.; Fang, F.; Zou, Z.; Zhou, Y.; Tian, B. Cdk5 Phosphorylation-Induced SIRT2 Nuclear Translocation Promotes the Death of Dopaminergic Neurons in Parkinson’s Disease. NPJ Park. Dis. 2022, 8. [Google Scholar] [CrossRef]
- Paxinos, g.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2006; Available online: https://www.elsevier.com/books/the-rat-brain-in-stereotaxic-coordinates/paxinos/978-0-12-374121-9 (accessed on 4 May 2021).
- Paumier, K.L.; Luk, K.C.; Manfredsson, F.P.; Kanaan, N.M.; Lipton, J.W.; Collier, T.J.; Steece-Collier, K.; Kemp, C.J.; Celano, S.; Schulz, E.; et al. Intrastriatal Injection of Pre-Formed Mouse α-Synuclein Fibrils into Rats Triggers α-Synuclein Pathology and Bilateral Nigrostriatal Degeneration. Neurobiol. Dis. 2015, 82, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E.; Ajamieh, H.H.; Sam, S.; Martínez, G.; Leoón Fernaández, O.S. Nimesulide Limits Kainate-Induced Oxidative Damage in the Rat Hippocampus. Eur. J. Pharmacol. 2000, 390, 295–298. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Monville, C.; Torres, E.M.; Dunnett, S.B. Comparison of Incremental and Accelerating Protocols of the Rotarod Test for the Assessment of Motor Deficits in the 6-OHDA Model. J. Neurosci. Methods 2006, 158, 219–223. [Google Scholar] [CrossRef]
- Tiwari, S.; Gupta, P.; Singh, A.; Chaturvedi, S.; Wahajuddin, M.; Mishra, A.; Singh, S. 4-Phenylbutyrate Mitigates the Motor Impairment and Dopaminergic Neuronal Death During Parkinson’s Disease Pathology via Targeting VDAC1 Mediated Mitochondrial Function and Astrocytes Activation. Neurochem. Res. 2022, 47, 3385–3401. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Goswami, P.; Biswas, J.; Joshi, N.; Sharma, S.; Nath, C.; Singh, S. 6-Hydroxydopamine and Lipopolysaccharides Induced DNA Damage in Astrocytes: Involvement of Nitric Oxide and Mitochondria. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 778, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Gupta, P.; Tiwari, S.; Mishra, A.; Singh, S. Guanabenz Mitigates the Neuropathological Alterations and Cell Death in Alzheimer’s Disease. Cell Tissue Res. 2022, 388, 239–258. [Google Scholar] [CrossRef]
- Gupta, P.; Tiwari, S.; Singh, A.; Pal, A.; Mishra, A.; Singh, S. Rivastigmine Attenuates the Alzheimer’s Disease Related Protein Degradation and Apoptotic Neuronal Death Signalling. Biochem. J. 2021, 478, 1435–1451. [Google Scholar] [CrossRef]
- Simunovic, F.; Yi, M.; Wang, Y.; Macey, L.; Brown, L.T.; Krichevsky, A.M.; Andersen, S.L.; Stephens, R.M.; Benes, F.M.; Sonntag, K.C. Gene Expression Profiling of Substantia Nigra Dopamine Neurons: Further Insights into Parkinson’s Disease Pathology. Brain 2009, 132, 1795–1809. [Google Scholar] [CrossRef]
- Gentier, R.J.; van Leeuwen, F.W. Misframed Ubiquitin and Impaired Protein Quality Control: An Early Event in Alzheimer’s Disease. Front. Mol. Neurosci. 2015, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Lindsten, K.; de Vrij, F.M.S.; Verhoef, L.G.G.C.; Fischer, D.F.; van Leeuwen, F.W.; Hol, E.M.; Masucci, M.G.; Dantuma, N.P. Mutant Ubiquitin Found in Neurodegenerative Disorders Is a Ubiquitin Fusion Degradation Substrate That Blocks Proteasomal Degradation. J. Cell Biol. 2002, 157, 417–427. [Google Scholar] [CrossRef]
- Fischer, D.F.; Vos, R.A.I.; Dijk, R.; Vrij, F.M.S.; Proper, E.A.; Sonnemans, M.A.F.; Verhage, M.C.; Sluijs, J.A.; Hobo, B.; Zouambia, M.; et al. Disease-specific Accumulation of Mutant Ubiquitin as a Marker for Proteasomal Dysfunction in the Brain. FASEB J. 2003, 17, 2014–2024. [Google Scholar] [CrossRef]
- Sherer, T.B.; Kim, J.H.; Betarbet, R.; Greenamyre, J.T. Subcutaneous Rotenone Exposure Causes Highly Selective Dopaminergic Degeneration and α-Synuclein Aggregation. Exp. Neurol. 2003, 179, 9–16. [Google Scholar] [CrossRef]
- Alam, M.; Schmidt, W.J. Rotenone Destroys Dopaminergic Neurons and Induces Parkinsonian Symptoms in Rats. Behav. Brain Res. 2002, 136, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.v.; Greenamyre, J.T. Chronic Systemic Pesticide Exposure Reproduces Features of Parkinson’s Disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Faull, R.L.M.; Laverty, R. Changes in Dopamine Levels in the Corpus Striatum Following Lesions in the Substantia Nigra. Exp. Neurol. 1969, 23, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Bové, J.; Prou, D.; Perier, C.; Przedborski, S. Toxin-Induced Models of Parkinson’s Disease. NeuroRx 2005, 2, 484. [Google Scholar] [CrossRef]
- Murakami, Y.; Mizuguchi, K. Homology-Based Prediction of Interactions between Proteins Using Averaged One-Dependence Estimators. BMC Bioinform. 2014, 15, 213. [Google Scholar] [CrossRef] [PubMed]
- Chesselet, M.F. In Vivo Alpha-Synuclein Overexpression in Rodents: A Useful Model of Parkinson’s Disease? Exp. Neurol. 2008, 209, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Ko, H.S.; Dawson, V.L. Genetic Animal Models of Parkinson’s Disease. Neuron 2010, 66, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Kundrat, L.; Regan, L. Identification of Residues on Hsp70 and Hsp90 Ubiquitinated by the Cochaperone CHIP. J. Mol. Biol. 2010, 395, 587–594. [Google Scholar] [CrossRef]
- Alam, Q.; Alam, M.Z.; Wali Sait, K.H.; Anfinan, N.; Noorwali, A.W.; Kamal, M.A.; Ahmad Khan, M.S.; Haque, A. Translational Shift of HSP90 as a Novel Therapeutic Target from Cancer to Neurodegenerative Disorders: An Emerging Trend in the Cure of Alzheimer’s and Parkinson’s Diseases. Curr. Drug Metab. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.C.; Helgason, E.; Yu, C.; Phu, L.; Arnott, D.P.; Bosanac, I.; Compaan, D.M.; Huang, O.W.; Fedorova, A.v.; Kirkpatrick, D.S.; et al. Preparation of Distinct Ubiquitin Chain Reagents of High Purity and Yield. Structure 2011, 19, 1053–1063. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Kim, H.T.; Hourez, R.; Jung, J.W.; Kim, K.P.; Goldberg, A.L. Ubiquitin Ligase Nedd4 Promotes α-Synuclein Degradation by the Endosomal-Lysosomal Pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 17004–17009. [Google Scholar] [CrossRef]
- Tai, H.C.; Schuman, E.M. Ubiquitin, the Proteasome and Protein Degradation in Neuronal Function and Dysfunction. Nat. Rev. Neurosci. 2008, 9, 826–838. [Google Scholar] [CrossRef]
- Park, S.J.; Kostic, M.; Dyson, H.J. Dynamic Interaction of Hsp90 with Its Client Protein P53. J. Mol. Biol. 2011, 411, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative Analysis of Hsp90-Client Interactions Reveals Principles of Substrate Recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Morishima, Y.; Gestwicki, J.E.; Lieberman, A.P.; Osawa, Y. A Model in Which Heat Shock Protein 90 Targets Protein-Folding Clefts: Rationale for a New Approach to Neuroprotective Treatment of Protein Folding Diseases. Exp. Biol. Med. 2014, 239, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-I.; Oh, Y.J. Tumor Necrosis Factor-Associated Protein 1 (TRAP1) Is Released from the Mitochondria Following 6-Hydroxydopamine Treatment. Exp. Neurobiol. 2014, 23, 65–76. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The Co-Chaperone Carboxyl Terminus of Hsp70-Interacting Protein (CHIP) Mediates Alpha-Synuclein Degradation Decisions between Proteasomal and Lysosomal Pathways. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef]
- Mund, T.; Masuda-Suzukake, M.; Goedert, M.; Pelham, H.R. Ubiquitination of Alpha-Synuclein Filaments by Nedd4 Ligases. PLoS ONE 2018, 13, e0200763. [Google Scholar] [CrossRef] [PubMed]
- Rohé, C.F.; Montagna, P.; Breedveld, G.; Cortelli, P.; Oostra, B.A.; Bonifati, V. Homozygous PINK1 C-Terminus Mutation Causing Early-Onset Parkinsonism. Ann. Neurol. 2004, 56, 427–431. [Google Scholar] [CrossRef]
- Wauer, T.; Simicek, M.; Schubert, A.; Komander, D. Mechanism of Phospho-Ubiquitin-Induced PARKIN Activation. Nature 2015, 524, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, Parkin, and Mitochondrial Quality Control: What Can We Learn about Parkinson’s Disease Pathobiology? J. Park. Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Yu, H.; Mim, C.; Matouschek, A. Regulated Protein Turnover: Snapshots of the Proteasome in Action. Nat. Rev. Mol. Cell. Biol. 2014, 15, 122–133. [Google Scholar] [CrossRef]
- Erpapazoglou, Z.; Walker, O.; Haguenauer-Tsapis, R. Versatile Roles of K63-Linked Ubiquitin Chains in Trafficking. Cells 2014, 3, 1027–1088. [Google Scholar] [CrossRef]
- Lim, K.L.; Lim, G.G.Y. K63-Linked Ubiquitination and Neurodegeneration. Neurobiol. Dis. 2011, 43, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Dong, Y.; Liu, M.; Li, Y.; Zhang, Y. Clinical Significance of UbA52 Level in the Urine of Patients with Type 2 Diabetes Mellitus and Diabetic Kidney Disease. Nefrología 2021, 41, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Wahlster, L.; McLean, P.J. Protein Degradation Pathways in Parkinson’s Disease: Curse or Blessing. Acta Neuropathol. 2012, 124, 153–172. [Google Scholar] [CrossRef]
- Pridgeon, J.W.; Olzmann, J.A.; Chin, L.S.; Li, L. PINK1 Protects against Oxidative Stress by Phosphorylating Mitochondrial Chaperone TRAP1. PLoS Biol. 2007, 5, e172. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Weidenauer, L.; Luengo, T.M.; Pieters, E.C.; Echeverría, P.C.; Bernasconi, L.; Wider, D.; Sadian, Y.; Koopman, M.B.; Villemin, M.; et al. The Hsp70-Hsp90 Co-Chaperone Hop/Stip1 Shifts the Proteostatic Balance from Folding towards Degradation. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiwari, S.; Singh, A.; Gupta, P.; Singh, S. UBA52 Is Crucial in HSP90 Ubiquitylation and Neurodegenerative Signaling during Early Phase of Parkinson’s Disease. Cells 2022, 11, 3770. https://doi.org/10.3390/cells11233770
Tiwari S, Singh A, Gupta P, Singh S. UBA52 Is Crucial in HSP90 Ubiquitylation and Neurodegenerative Signaling during Early Phase of Parkinson’s Disease. Cells. 2022; 11(23):3770. https://doi.org/10.3390/cells11233770
Chicago/Turabian StyleTiwari, Shubhangini, Abhishek Singh, Parul Gupta, and Sarika Singh. 2022. "UBA52 Is Crucial in HSP90 Ubiquitylation and Neurodegenerative Signaling during Early Phase of Parkinson’s Disease" Cells 11, no. 23: 3770. https://doi.org/10.3390/cells11233770
APA StyleTiwari, S., Singh, A., Gupta, P., & Singh, S. (2022). UBA52 Is Crucial in HSP90 Ubiquitylation and Neurodegenerative Signaling during Early Phase of Parkinson’s Disease. Cells, 11(23), 3770. https://doi.org/10.3390/cells11233770