
The Role of Bone Morphogenetic Protein Receptor Type 2 (BMPR2) and the Prospects of Utilizing Induced Pluripotent Stem Cells (iPSCs) in Pulmonary Arterial Hypertension Disease Modeling


Abstract
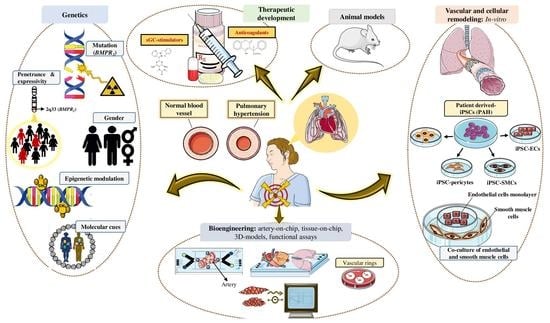
1. Introduction
2. Vascular Remodeling in PAH
3. The Genetics and Gender Prevalence behind Pulmonary Arterial Hypertension

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | References |
|---|---|---|
| BMPR2 | Bone morphogenetic protein receptor type 2 | [24,25] |
| ENG | Endoglin | [31] |
| ALK1 | Activin receptor-like kinase 1 | [32] |
| BMPR1B (ALK6) | Bone morphogenetic protein receptor type 1B | [33] |
| SMAD4 | SMAD family member 4 | [34] |
| SMAD9 | SMAD family member 9 | [34] |
| GDF2 (BMP9) | Growth differentiation factor 2 | [35,36] |
| CAV1 | Caveolin-1 | [37] |
| KCNK3 | Potassium two pore domain channel subfamily K member 3 | [38] |
| TBX4 | T-Box 4 | [39] |
| EIF2AK4 | Eukaryotic initiation translation factor 2 alpha kinase 4 | [40] |
| ATP13A3 | ATPase 13A3 | [36] |
| AQP1 | Aquaporin 1 | [36] |
| SOX17 | SRY-Box 17 | [36] |
4. Bone Morphogenetic Proteins (BMPs) and Their Receptors
4.1. The BMP Type II Receptor (BMPR2)
4.2. The Bone Morphogenetic Protein Receptor Signaling
5. Challenges Involved in Investigating Pulmonary Arterial Hypertension
6. Human Pluripotent Stem Cell (hPSC) Utility for Disease Modeling Systems
6.1. Induced Pluripotent Stem Cell Model Systems for Pulmonary Arterial Hypertension
6.2. Limitations for Currently Available iPSC Model Systems for Pulmonary Arterial Hypertension
6.2.1. Gaps in Knowledge
6.2.2. Gaps in Methodology
7. Emerging Therapeutic Modalities for Pulmonary Arterial Hypertension Targeting the BMPR2 Signaling Pathway
- a.
- Targeted Delivery of Exogenous BMPR2
- b.
- Ataluren/PTC124 Acting Via the Regulation of Ribosomal Translational Read-through
- c.
- Tacrolimus/FK506 Acting Via the Indirect Cytoplasmic FKBP12 Blockade
- d.
- Sotatercept/ActR2a-Fc directed towards the abnormal BMP/activin/growth and differential factors (GDF) signaling via cell membrane:
- e.
- Etanercept Targeting Inflammation That affects BMPR2 Via Cell Membrane
- f.
- Hydroxychloroquine Promoting Degradation and Autophagy Via Lysosome
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Investig. 2012, 122, 4306–4313. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.C.; Potoka, K.C.; Champion, H.C.; Mora, A.L.; Gladwin, M.T. Pulmonary arterial hypertension: The clinical syndrome. Circ. Res. 2014, 115, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [CrossRef] [PubMed]
- Ruopp, N.F.; Farber, H.W. The New World Symposium on Pulmonary Hypertension Guidelines: Should Twenty-One Be the New Twenty-Five? Circulation 2019, 140, 1134–1136. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Gomez Sanchez, M.A.; Krishna Kumar, R.; Landzberg, M.; Machado, R.F.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Trembath, R.C. Genetics of pulmonary hypertension: From bench to bedside. Eur. Respir. J. 2002, 20, 741–749. [Google Scholar] [CrossRef]
- Simonneau, G.; Robbins, I.M.; Beghetti, M.; Channick, R.N.; Delcroix, M.; Denton, C.P.; Elliott, C.G.; Gaine, S.P.; Gladwin, M.T.; Jing, Z.C.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2009, 54, 43–54. [Google Scholar] [CrossRef]
- Morrell, N.W. Role of bone morphogenetic protein receptors in the development of pulmonary arterial hypertension. Adv. Exp. Med. Biol. 2010, 661, 251–264. [Google Scholar] [CrossRef]
- West, J.; Cogan, J.; Geraci, M.; Robinson, L.; Newman, J.; Phillips, J.A.; Lane, K.; Meyrick, B.; Loyd, J. Gene expression in BMPR2 mutation carriers with and without evidence of pulmonary arterial hypertension suggests pathways relevant to disease penetrance. BMC Med. Genom. 2008, 1, 45. [Google Scholar] [CrossRef] [PubMed]
- Soubrier, F.; Chung, W.K.; Machado, R.; Grunig, E.; Aldred, M.; Geraci, M.; Loyd, J.E.; Elliott, C.G.; Trembath, R.C.; Newman, J.H.; et al. Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2013, 62, 13–21. [Google Scholar] [CrossRef]
- Andre, P.; Joshi, S.R.; Briscoe, S.D.; Alexander, M.J.; Li, G.; Kumar, R. Therapeutic Approaches for Treating Pulmonary Arterial Hypertension by Correcting Imbalanced TGF-beta Superfamily Signaling. Front. Med. 2021, 8, 814222. [Google Scholar] [CrossRef] [PubMed]
- Montani, D.; Gunther, S.; Dorfmuller, P.; Perros, F.; Girerd, B.; Garcia, G.; Jais, X.; Savale, L.; Artaud-Macari, E.; Price, L.C.; et al. Pulmonary arterial hypertension. Orphanet. J. Rare Dis. 2013, 8, 97. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O.; Simonneau, G. Novel therapeutic perspectives in pulmonary arterial hypertension. Eur. Respir. J. 2003, 22, 193–194. [Google Scholar] [CrossRef]
- Misra, S.; Fu, A.A.; Misra, K.D.; Shergill, U.M.; Leof, E.B.; Mukhopadhyay, D. Hypoxia-induced phenotypic switch of fibroblasts to myofibroblasts through a matrix metalloproteinase 2/tissue inhibitor of metalloproteinase-mediated pathway: Implications for venous neointimal hyperplasia in hemodialysis access. J. Vasc. Interv. Radiol. 2010, 21, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 13–24. [Google Scholar] [CrossRef]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thebaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef] [PubMed]
- Balabanian, K.; Foussat, A.; Dorfmuller, P.; Durand-Gasselin, I.; Capel, F.; Bouchet-Delbos, L.; Portier, A.; Marfaing-Koka, A.; Krzysiek, R.; Rimaniol, A.C.; et al. CX(3)C chemokine fractalkine in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2002, 165, 1419–1425. [Google Scholar] [CrossRef]
- Sitbon, O.; Channick, R.; Chin, K.M.; Frey, A.; Gaine, S.; Galie, N.; Ghofrani, H.A.; Hoeper, M.M.; Lang, I.M.; Preiss, R.; et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 2522–2533. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, H.; Simonneau, G.; Galie, N.; Higenbottam, T.; Naeije, R.; Rubin, L.J.; Nikkho, S.; Speich, R.; Hoeper, M.M.; Behr, J.; et al. Inhaled iloprost for severe pulmonary hypertension. N. Engl. J. Med. 2002, 347, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.J. Cyclic AMP and mechanisms of vasodilation. Pharmacol. Ther. 1990, 47, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; D’Armini, A.M.; Grimminger, F.; Hoeper, M.M.; Jansa, P.; Kim, N.H.; Mayer, E.; Simonneau, G.; Wilkins, M.R.; Fritsch, A.; et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N. Engl. J. Med. 2013, 369, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galie, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I.; et al. Bosentan therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2002, 346, 896–903. [Google Scholar] [CrossRef]
- Deng, Z.; Morse, J.H.; Slager, S.L.; Cuervo, N.; Moore, K.J.; Venetos, G.; Kalachikov, S.; Cayanis, E.; Fischer, S.G.; Barst, R.J.; et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am. J. Hum. Genet. 2000, 67, 737–744. [Google Scholar] [CrossRef]
- International, P.P.H.C.; Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., 3rd; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef]
- Best, D.H.; Austin, E.D.; Chung, W.K.; Elliott, C.G. Genetics of pulmonary hypertension. Curr Opin Cardiol 2014, 29, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; Southgate, L.; Eichstaedt, C.A.; Aldred, M.A.; Austin, E.D.; Best, D.H.; Chung, W.K.; Benjamin, N.; Elliott, C.G.; Eyries, M.; et al. Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum. Mutat. 2015, 36, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; Aldred, M.A.; James, V.; Harrison, R.E.; Patel, B.; Schwalbe, E.C.; Gruenig, E.; Janssen, B.; Koehler, R.; Seeger, W.; et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum. Mutat. 2006, 27, 121–132. [Google Scholar] [CrossRef]
- Larkin, E.K.; Newman, J.H.; Austin, E.D.; Hemnes, A.R.; Wheeler, L.; Robbins, I.M.; West, J.D.; Phillips, J.A., 3rd; Hamid, R.; Loyd, J.E. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 892–896. [Google Scholar] [CrossRef]
- Morrell, N.W.; Adnot, S.; Archer, S.L.; Dupuis, J.; Lloyd Jones, P.; MacLean, M.R.; McMurtry, I.F.; Stenmark, K.R.; Thistlethwaite, P.A.; Weissmann, N.; et al. Cellular and molecular basis of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, 20–31. [Google Scholar] [CrossRef]
- Harrison, R.E.; Flanagan, J.A.; Sankelo, M.; Abdalla, S.A.; Rowell, J.; Machado, R.D.; Elliott, C.G.; Robbins, I.M.; Olschewski, H.; McLaughlin, V.; et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J. Med. Genet. 2003, 40, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Chaouat, A.; Coulet, F.; Favre, C.; Simonneau, G.; Weitzenblum, E.; Soubrier, F.; Humbert, M. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax 2004, 59, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Chida, A.; Shintani, M.; Nakayama, T.; Furutani, Y.; Hayama, E.; Inai, K.; Saji, T.; Nonoyama, S.; Nakanishi, T. Missense mutations of the BMPR1B (ALK6) gene in childhood idiopathic pulmonary arterial hypertension. Circ. J. 2012, 76, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Nasim, M.T.; Ogo, T.; Ahmed, M.; Randall, R.; Chowdhury, H.M.; Snape, K.M.; Bradshaw, T.Y.; Southgate, L.; Lee, G.J.; Jackson, I.; et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum. Mutat. 2011, 32, 1385–1389. [Google Scholar] [CrossRef]
- Wang, G.; Fan, R.; Ji, R.; Zou, W.; Penny, D.J.; Varghese, N.P.; Fan, Y. Novel homozygous BMP9 nonsense mutation causes pulmonary arterial hypertension: A case report. BMC Pulm. Med. 2016, 16, 17. [Google Scholar] [CrossRef]
- Graf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef]
- Austin, E.D.; Ma, L.; LeDuc, C.; Berman Rosenzweig, E.; Borczuk, A.; Phillips, J.A., 3rd; Palomero, T.; Sumazin, P.; Kim, H.R.; Talati, M.H.; et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ. Cardiovasc. Genet. 2012, 5, 336–343. [Google Scholar] [CrossRef]
- Ma, L.; Roman-Campos, D.; Austin, E.D.; Eyries, M.; Sampson, K.S.; Soubrier, F.; Germain, M.; Tregouet, D.A.; Borczuk, A.; Rosenzweig, E.B.; et al. A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 351–361. [Google Scholar] [CrossRef]
- Kerstjens-Frederikse, W.S.; Bongers, E.M.; Roofthooft, M.T.; Leter, E.M.; Douwes, J.M.; Van Dijk, A.; Vonk-Noordegraaf, A.; Dijk-Bos, K.K.; Hoefsloot, L.H.; Hoendermis, E.S.; et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J. Med. Genet. 2013, 50, 500–506. [Google Scholar] [CrossRef]
- Best, D.H.; Sumner, K.L.; Smith, B.P.; Damjanovich-Colmenares, K.; Nakayama, I.; Brown, L.M.; Ha, Y.; Paul, E.; Morris, A.; Jama, M.A.; et al. EIF2AK4 Mutations in Patients Diagnosed With Pulmonary Arterial Hypertension. Chest 2017, 151, 821–828. [Google Scholar] [CrossRef]
- Wang, E.A.; Rosen, V.; Cordes, P.; Hewick, R.M.; Kriz, M.J.; Luxenberg, D.P.; Sibley, B.S.; Wozney, J.M. Purification and characterization of other distinct bone-inducing factors. Proc. Natl. Acad. Sci. USA 1988, 85, 9484–9488. [Google Scholar] [CrossRef] [PubMed]
- Hogan, B.L. Bone morphogenetic proteins: Multifunctional regulators of vertebrate development. Genes Dev. 1996, 10, 1580–1594. [Google Scholar] [CrossRef] [PubMed]
- Barron, M.; Gao, M.; Lough, J. Requirement for BMP and FGF signaling during cardiogenic induction in non-precardiac mesoderm is specific, transient, and cooperative. Dev. Dyn. 2000, 218, 383–393. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [PubMed]
- de Caestecker, M. The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 2004, 15, 1–11. [Google Scholar] [CrossRef]
- Miyazono, K.; Kamiya, Y.; Morikawa, M. Bone morphogenetic protein receptors and signal transduction. J. Biochem. 2010, 147, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.B.; Beppu, H.; Kawai, N.; Li, E.; Bloch, K.D. Bone morphogenetic protein (BMP) type II receptor deletion reveals BMP ligand-specific gain of signaling in pulmonary artery smooth muscle cells. J. Biol. Chem. 2005, 280, 24443–24450. [Google Scholar] [CrossRef]
- Foletta, V.C.; Lim, M.A.; Soosairajah, J.; Kelly, A.P.; Stanley, E.G.; Shannon, M.; He, W.; Das, S.; Massague, J.; Bernard, O. Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J. Cell Biol. 2003, 162, 1089–1098. [Google Scholar] [CrossRef]
- Machado, R.D.; Rudarakanchana, N.; Atkinson, C.; Flanagan, J.A.; Harrison, R.; Morrell, N.W.; Trembath, R.C. Functional interaction between BMPR-II and Tctex-1, a light chain of Dynein, is isoform-specific and disrupted by mutations underlying primary pulmonary hypertension. Hum. Mol. Genet. 2003, 12, 3277–3286. [Google Scholar] [CrossRef]
- Chan, M.C.; Nguyen, P.H.; Davis, B.N.; Ohoka, N.; Hayashi, H.; Du, K.; Lagna, G.; Hata, A. A novel regulatory mechanism of the bone morphogenetic protein (BMP) signaling pathway involving the carboxyl-terminal tail domain of BMP type II receptor. Mol. Cell Biol. 2007, 27, 5776–5789. [Google Scholar] [CrossRef]
- Yang, J.; Li, X.; Li, Y.; Southwood, M.; Ye, L.; Long, L.; Al-Lamki, R.S.; Morrell, N.W. Id proteins are critical downstream effectors of BMP signaling in human pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Pregizer, S.; Mortlock, D.P. Control of BMP gene expression by long-range regulatory elements. Cytokine Growth Factor Rev. 2009, 20, 509–515. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cogan, J.; Austin, E.; Hedges, L.; Womack, B.; West, J.; Loyd, J.; Hamid, R. Role of BMPR2 alternative splicing in heritable pulmonary arterial hypertension penetrance. Circulation 2012, 126, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- ten Dijke, P.; Yamashita, H.; Ichijo, H.; Franzen, P.; Laiho, M.; Miyazono, K.; Heldin, C.H. Characterization of type I receptors for transforming growth factor-beta and activin. Science 1994, 264, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Macias-Silva, M.; Hoodless, P.A.; Tang, S.J.; Buchwald, M.; Wrana, J.L. Specific activation of Smad1 signaling pathways by the BMP7 type I receptor, ALK2. J. Biol. Chem. 1998, 273, 25628–25636. [Google Scholar] [CrossRef]
- Yamashita, H.; ten Dijke, P.; Huylebroeck, D.; Sampath, T.K.; Andries, M.; Smith, J.C.; Heldin, C.H.; Miyazono, K. Osteogenic protein-1 binds to activin type II receptors and induces certain activin-like effects. J. Cell Biol. 1995, 130, 217–226. [Google Scholar] [CrossRef]
- Ebisawa, T.; Tada, K.; Kitajima, I.; Tojo, K.; Sampath, T.K.; Kawabata, M.; Miyazono, K.; Imamura, T. Characterization of bone morphogenetic protein-6 signaling pathways in osteoblast differentiation. J. Cell Sci. 1999, 112, 3519–3527. [Google Scholar] [CrossRef]
- Brown, M.A.; Zhao, Q.; Baker, K.A.; Naik, C.; Chen, C.; Pukac, L.; Singh, M.; Tsareva, T.; Parice, Y.; Mahoney, A.; et al. Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J. Biol. Chem. 2005, 280, 25111–25118. [Google Scholar] [CrossRef]
- David, L.; Mallet, C.; Mazerbourg, S.; Feige, J.J.; Bailly, S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007, 109, 1953–1961. [Google Scholar] [CrossRef]
- Alt, A.; Miguel-Romero, L.; Donderis, J.; Aristorena, M.; Blanco, F.J.; Round, A.; Rubio, V.; Bernabeu, C.; Marina, A. Structural and functional insights into endoglin ligand recognition and binding. PLoS ONE 2012, 7, e29948. [Google Scholar] [CrossRef]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, T.; Watabe, T. Bone Morphogenetic Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021899. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Lebrin, F.; Larsson, J.; Mummery, C.; Karlsson, S.; ten Dijke, P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell 2003, 12, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.C.; Randall, R.A.; Hill, C.S. Transforming growth factor beta-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol. Cell Biol. 2008, 28, 6889–6902. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, X.F. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Kameda, T.; Koike, C.; Saitoh, K.; Kuroiwa, A.; Iba, H. Developmental patterning in chondrocytic cultures by morphogenic gradients: BMP induces expression of indian hedgehog and noggin. Genes Cells 1999, 4, 175–184. [Google Scholar] [CrossRef]
- Pereira, R.C.; Economides, A.N.; Canalis, E. Bone morphogenetic proteins induce gremlin, a protein that limits their activity in osteoblasts. Endocrinology 2000, 141, 4558–4563. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marques, G.; Musacchio, M.; Shimell, M.J.; Wunnenberg-Stapleton, K.; Cho, K.W.; O’Connor, M.B. Production of a DPP activity gradient in the early Drosophila embryo through the opposing actions of the SOG and TLD proteins. Cell 1997, 91, 417–426. [Google Scholar] [CrossRef]
- Piccolo, S.; Agius, E.; Lu, B.; Goodman, S.; Dale, L.; De Robertis, E.M. Cleavage of Chordin by Xolloid metalloprotease suggests a role for proteolytic processing in the regulation of Spemann organizer activity. Cell 1997, 91, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Binder, O.; Wu, Y.; Aitsebaomo, J.; Ren, R.; Bode, C.; Bautch, V.L.; Conlon, F.L.; Patterson, C. BMPER, a novel endothelial cell precursor-derived protein, antagonizes bone morphogenetic protein signaling and endothelial cell differentiation. Mol. Cell. Biol. 2003, 23, 5664–5679. [Google Scholar] [CrossRef]
- Halbrooks, P.J.; Ding, R.; Wozney, J.M.; Bain, G. Role of RGM coreceptors in bone morphogenetic protein signaling. J. Mol. Signal 2007, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Takase, M.; Nishihara, A.; Oeda, E.; Hanai, J.; Kawabata, M.; Miyazono, K. Smad6 inhibits signalling by the TGF-beta superfamily. Nature 1997, 389, 622–626. [Google Scholar] [CrossRef]
- Heldin, C.H.; Moustakas, A. Role of Smads in TGFbeta signaling. Cell Tissue Res. 2012, 347, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Kavsak, P.; Abdollah, S.; Wrana, J.L.; Thomsen, G.H. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 1999, 400, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Murakami, G.; Watabe, T.; Takaoka, K.; Miyazono, K.; Imamura, T. Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol. Biol. Cell 2003, 14, 2809–2817. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.I.; Mardaryev, A.N.; Lewis, C.J.; Sharov, A.A.; Botchkareva, N.V. MicroRNA-21 is an important downstream component of BMP signalling in epidermal keratinocytes. J. Cell Sci. 2011, 124, 3399–3404. [Google Scholar] [CrossRef]
- Kang, H.; Louie, J.; Weisman, A.; Sheu-Gruttadauria, J.; Davis-Dusenbery, B.N.; Lagna, G.; Hata, A. Inhibition of microRNA-302 (miR-302) by bone morphogenetic protein 4 (BMP4) facilitates the BMP signaling pathway. J. Biol. Chem. 2012, 287, 38656–38664. [Google Scholar] [CrossRef]
- Spiekerkoetter, E.; Tian, X.; Cai, J.; Hopper, R.K.; Sudheendra, D.; Li, C.G.; El-Bizri, N.; Sawada, H.; Haghighat, R.; Chan, R.; et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Invest. 2013, 123, 3600–3613. [Google Scholar] [CrossRef]
- West, J.D.; Austin, E.D.; Gaskill, C.; Marriott, S.; Baskir, R.; Bilousova, G.; Jean, J.C.; Hemnes, A.R.; Menon, S.; Bloodworth, N.C.; et al. Identification of a common Wnt-associated genetic signature across multiple cell types in pulmonary arterial hypertension. Am. J. Physiol. Cell Physiol. 2014, 307, 415–430. [Google Scholar] [CrossRef]
- Sa, S.; Gu, M.; Chappell, J.; Shao, N.Y.; Ameen, M.; Elliott, K.A.; Li, D.; Grubert, F.; Li, C.G.; Taylor, S.; et al. Induced Pluripotent Stem Cell Model of Pulmonary Arterial Hypertension Reveals Novel Gene Expression and Patient Specificity. Am. J. Respir. Crit. Care Med. 2017, 195, 930–941. [Google Scholar] [CrossRef]
- Gu, M.; Shao, N.Y.; Sa, S.; Li, D.; Termglinchan, V.; Ameen, M.; Karakikes, I.; Sosa, G.; Grubert, F.; Lee, J.; et al. Patient-Specific iPSC-Derived Endothelial Cells Uncover Pathways that Protect against Pulmonary Hypertension in BMPR2 Mutation Carriers. Cell Stem Cell 2017, 20, 490–504. [Google Scholar] [CrossRef]
- Kiskin, F.N.; Chang, C.H.; Huang, C.J.Z.; Kwieder, B.; Cheung, C.; Dunmore, B.J.; Serrano, F.; Sinha, S.; Morrell, N.W.; Rana, A.A. Contributions of BMPR2 Mutations and Extrinsic Factors to Cellular Phenotypes of Pulmonary Arterial Hypertension Revealed by Induced Pluripotent Stem Cell Modeling. Am. J. Respir. Crit. Care Med. 2018, 198, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Shamblott, M.J.; Axelman, J.; Wang, S.; Bugg, E.M.; Littlefield, J.W.; Donovan, P.J.; Blumenthal, P.D.; Huggins, G.R.; Gearhart, J.D. Derivation of pluripotent stem cells from cultured human primordial germ cells. Proc. Natl. Acad. Sci. USA 1998, 95, 13726–13731. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Amato, P.; Sparman, M.; Gutierrez, N.M.; Tippner-Hedges, R.; Ma, H.; Kang, E.; Fulati, A.; Lee, H.S.; Sritanaudomchai, H.; et al. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell 2013, 153, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Johannesson, B.; Sagi, I.; Burnett, L.C.; Kort, D.H.; Prosser, R.W.; Paull, D.; Nestor, M.W.; Freeby, M.; Greenberg, E.; et al. Human oocytes reprogram adult somatic nuclei of a type 1 diabetic to diploid pluripotent stem cells. Nature 2014, 510, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Kaebisch, C.; Schipper, D.; Babczyk, P.; Tobiasch, E. The role of purinergic receptors in stem cell differentiation. Comput Struct Biotechnol. J. 2015, 13, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Lensch, M.W.; Schlaeger, T.M.; Zon, L.I.; Daley, G.Q. Teratoma formation assays with human embryonic stem cells: A rationale for one type of human-animal chimera. Cell Stem Cell 2007, 1, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced pluripotent stem cells generated without viral integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.M.; Ratanasirintrawoot, S.; Park, I.H.; Manos, P.D.; Loh, Y.H.; Huo, H.; Miller, J.D.; Hartung, O.; Rho, J.; Ince, T.A.; et al. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat. Biotechnol. 2009, 27, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Robinton, D.A.; Daley, G.Q. The promise of induced pluripotent stem cells in research and therapy. Nature 2012, 481, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Goff, S.P. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 2007, 131, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Yu, J.; Rose, F.F., Jr.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef]
- Soldner, F.; Hockemeyer, D.; Beard, C.; Gao, Q.; Bell, G.W.; Cook, E.G.; Hargus, G.; Blak, A.; Cooper, O.; Mitalipova, M.; et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 2009, 136, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.D.; Yu, J.; Smuga-Otto, K.; Salvagiotto, G.; Rehrauer, W.; Vodyanik, M.; Thomson, J.; Slukvin, I. Hematopoietic and endothelial differentiation of human induced pluripotent stem cells. Stem Cells 2009, 27, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhan, H.; Mali, P.; Dowey, S.; Williams, D.M.; Jang, Y.Y.; Dang, C.V.; Spivak, J.L.; Moliterno, A.R.; Cheng, L. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood 2009, 114, 5473–5480. [Google Scholar] [CrossRef] [PubMed]
- Taura, D.; Noguchi, M.; Sone, M.; Hosoda, K.; Mori, E.; Okada, Y.; Takahashi, K.; Homma, K.; Oyamada, N.; Inuzuka, M.; et al. Adipogenic differentiation of human induced pluripotent stem cells: Comparison with that of human embryonic stem cells. FEBS Lett. 2009, 583, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Soldner, F.; Cook, E.G.; Gao, Q.; Mitalipova, M.; Jaenisch, R. A drug-inducible system for direct reprogramming of human somatic cells to pluripotency. Cell Stem Cell 2008, 3, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Maherali, N.; Ahfeldt, T.; Rigamonti, A.; Utikal, J.; Cowan, C.; Hochedlinger, K. A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell 2008, 3, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.G.; Sun, L.; Christensen, K.; et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015, 17, 994–1003. [Google Scholar] [CrossRef]
- Cheung, C.; Bernardo, A.S.; Trotter, M.W.; Pedersen, R.A.; Sinha, S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat. Biotechnol. 2012, 30, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- Devendran, A.; Bailey, R.; Kar, S.; Stillitano, F.; Turnbull, I.; Fish, K.; Dubois, N.; Wickramasinghe, N.; Hajjar, R. Elucidating and Characterizing the Molecular Mechanistic Role of Phospholamban L39 Stop in the Pathophysiology of Cardiomyopathy Using Patient-derived Human Induced Pluripotent Stem Cells and Humanized Knock-in Mouse Model Systems. Circ. Res. 2021, 129, ap482. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Jaenisch, R. Induced Pluripotent Stem Cells Meet Genome Editing. Cell Stem Cell 2016, 18, 573–586. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Soldner, F.; Beard, C.; Gao, Q.; Mitalipova, M.; DeKelver, R.C.; Katibah, G.E.; Amora, R.; Boydston, E.A.; Zeitler, B.; et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009, 27, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Cong, L.; Zhou, Y.; Cunniff, M.M.; Feng, G.; Zhang, F. A transcription activator-like effector toolbox for genome engineering. Nat. Protoc. 2012, 7, 171–192. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Murai, K.; Sun, G.; Ye, P.; Tian, E.; Yang, S.; Cui, Q.; Sun, G.; Trinh, D.; Sun, O.; Hong, T.; et al. The TLX-miR-219 cascade regulates neural stem cell proliferation in neurodevelopment and schizophrenia iPSC model. Nat. Commun. 2016, 7, 10965. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Talbot, C.C., Jr.; Liu, Q.; Jing, Z.C.; Damico, R.L.; Tuder, R.; Barnes, K.C.; Hassoun, P.M.; Gao, L. Identifying microRNAs targeting Wnt/beta-catenin pathway in end-stage idiopathic pulmonary arterial hypertension. J. Mol. Med. 2016, 94, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Laumanns, I.P.; Fink, L.; Wilhelm, J.; Wolff, J.C.; Mitnacht-Kraus, R.; Graef-Hoechst, S.; Stein, M.M.; Bohle, R.M.; Klepetko, W.; Hoda, M.A.; et al. The noncanonical WNT pathway is operative in idiopathic pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2009, 40, 683–691. [Google Scholar] [CrossRef]
- Diebold, I.; Hennigs, J.K.; Miyagawa, K.; Li, C.G.; Nickel, N.P.; Kaschwich, M.; Cao, A.; Wang, L.; Reddy, S.; Chen, P.I.; et al. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. 2015, 21, 596–608. [Google Scholar] [CrossRef]
- Usman, A.; Haase, A.; Merkert, S.; Gohring, G.; Hansmann, G.; Gall, H.; Schermuly, R.; Martin, U.; Olmer, R. Generation of pulmonary arterial hypertension patient-specific induced pluripotent stem cell lines from three unrelated patients with a heterozygous missense mutation in exon 12, a heterozygous in-frame deletion in exon 3 and a missense mutation in exon 11 of the BMPR2 gene. Stem Cell Res. 2021, 55, 102488. [Google Scholar] [CrossRef]
- Orlova, V.V.; van den Hil, F.E.; Petrus-Reurer, S.; Drabsch, Y.; Ten Dijke, P.; Mummery, C.L. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat. Protoc. 2014, 9, 1514–1531. [Google Scholar] [CrossRef]
- Dye, B.R.; Hill, D.R.; Ferguson, M.A.; Tsai, Y.H.; Nagy, M.S.; Dyal, R.; Wells, J.M.; Mayhew, C.N.; Nattiv, R.; Klein, O.D.; et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife 2015, 4, e05098. [Google Scholar] [CrossRef]
- Leibel, S.L.; Tseu, I.; Zhou, A.; Hodges, A.; Yin, J.; Bilodeau, C.; Goltsis, O.; Post, M. Metabolomic profiling of human pluripotent stem cell differentiation into lung progenitors. iScience 2022, 25, 103797. [Google Scholar] [CrossRef] [PubMed]
- Al-Hilal, T.A.; Keshavarz, A.; Kadry, H.; Lahooti, B.; Al-Obaida, A.; Ding, Z.; Li, W.; Kamm, R.; McMurtry, I.F.; Lahm, T.; et al. Pulmonary-arterial-hypertension (PAH)-on-a-chip: Fabrication, validation and application. Lab Chip 2020, 20, 3334–3345. [Google Scholar] [CrossRef] [PubMed]
- Hye, T.; Dwivedi, P.; Li, W.; Lahm, T.; Nozik-Grayck, E.; Stenmark, K.R.; Ahsan, F. Newer insights into the pathobiological and pharmacological basis of the sex disparity in patients with pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 320, L1025–L1037. [Google Scholar] [CrossRef] [PubMed]
- Colter, J.; Murari, K.; Biernaskie, J.; Kallos, M.S. Induced pluripotency in the context of stem cell expansion bioprocess development, optimization, and manufacturing: A roadmap to the clinic. NPJ Regen. Med. 2021, 6, 72. [Google Scholar] [CrossRef]
- Rich, S.; Kaufmann, E.; Levy, P.S. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N. Engl. J. Med. 1992, 327, 76–81. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Galie, N.; Grimminger, F.; Grunig, E.; Humbert, M.; Jing, Z.C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef]
- Rubin, L.J.; Galie, N.; Grimminger, F.; Grunig, E.; Humbert, M.; Jing, Z.C.; Keogh, A.; Langleben, D.; Fritsch, A.; Menezes, F.; et al. Riociguat for the treatment of pulmonary arterial hypertension: A long-term extension study (PATENT-2). Eur. Respir. J. 2015, 45, 1303–1313. [Google Scholar] [CrossRef]
- McLaughlin, V.V.; Shillington, A.; Rich, S. Survival in primary pulmonary hypertension: The impact of epoprostenol therapy. Circulation 2002, 106, 1477–1482. [Google Scholar] [CrossRef]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galie, N.; Ghofrani, H.A.; Jansa, P.; Jing, Z.C.; Le Brun, F.O.; Mehta, S.; et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef]
- McMurtry, M.S.; Moudgil, R.; Hashimoto, K.; Bonnet, S.; Michelakis, E.D.; Archer, S.L. Overexpression of human bone morphogenetic protein receptor 2 does not ameliorate monocrotaline pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.M.; Xia, W.; Holmes, M.D.; Hodge, S.J.; Danilov, S.; Curiel, D.T.; Morrell, N.W.; Reynolds, P.N. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.M.; Holmes, M.D.; Danilov, S.M.; Reynolds, P.N. Targeted gene delivery of BMPR2 attenuates pulmonary hypertension. Eur. Respir. J. 2012, 39, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Harper, R.L.; Reynolds, A.M.; Bonder, C.S.; Reynolds, P.N. BMPR2 gene therapy for PAH acts via Smad and non-Smad signalling. Respirology 2016, 21, 727–733. [Google Scholar] [CrossRef]
- Harper, R.L.; Maiolo, S.; Ward, R.J.; Seyfang, J.; Cockshell, M.P.; Bonder, C.S.; Reynolds, P.N. BMPR2-expressing bone marrow-derived endothelial-like progenitor cells alleviate pulmonary arterial hypertension in vivo. Respirology 2019, 24, 1095–1103. [Google Scholar] [CrossRef]
- Aliotta, J.M.; Pereira, M.; Wen, S.; Dooner, M.S.; Del Tatto, M.; Papa, E.; Goldberg, L.R.; Baird, G.L.; Ventetuolo, C.E.; Quesenberry, P.J.; et al. Exosomes induce and reverse monocrotaline-induced pulmonary hypertension in mice. Cardiovasc. Res. 2016, 110, 319–330. [Google Scholar] [CrossRef]
- Isken, O.; Maquat, L.E. Quality control of eukaryotic mRNA: Safeguarding cells from abnormal mRNA function. Genes Dev. 2007, 21, 1833–1856. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef]
- Nagy, E.; Maquat, L.E. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem. Sci. 1998, 23, 198–199. [Google Scholar] [CrossRef]
- Lappalainen, T.; Sammeth, M.; Friedlander, M.R.; t Hoen, P.A.; Monlong, J.; Rivas, M.A.; Gonzalez-Porta, M.; Kurbatova, N.; Griebel, T.; Ferreira, P.G.; et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013, 501, 506–511. [Google Scholar] [CrossRef]
- Clancy, J.P.; Bebok, Z.; Ruiz, F.; King, C.; Jones, J.; Walker, L.; Greer, H.; Hong, J.; Wing, L.; Macaluso, M.; et al. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2001, 163, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Wilschanski, M.; Yahav, Y.; Yaacov, Y.; Blau, H.; Bentur, L.; Rivlin, J.; Aviram, M.; Bdolah-Abram, T.; Bebok, Z.; Shushi, L.; et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N. Engl. J. Med. 2003, 349, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Linde, L.; Kerem, B. Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008, 24, 552–563. [Google Scholar] [CrossRef]
- Namy, O.; Hatin, I.; Rousset, J.P. Impact of the six nucleotides downstream of the stop codon on translation termination. EMBO Rep. 2001, 2, 787–793. [Google Scholar] [CrossRef]
- Nasim, M.T.; Ghouri, A.; Patel, B.; James, V.; Rudarakanchana, N.; Morrell, N.W.; Trembath, R.C. Stoichiometric imbalance in the receptor complex contributes to dysfunctional BMPR-II mediated signalling in pulmonary arterial hypertension. Hum. Mol. Genet. 2008, 17, 1683–1694. [Google Scholar] [CrossRef] [PubMed]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Wilschanski, M.; Miller, L.L.; Shoseyov, D.; Blau, H.; Rivlin, J.; Aviram, M.; Cohen, M.; Armoni, S.; Yaakov, Y.; Pugatsch, T.; et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur. Respir. J. 2011, 38, 59–69. [Google Scholar] [CrossRef]
- Campbell, C.; Shieh, P.; Sejersen, T.; Luo, X.; Elfring, G.; Kroger, H.; Riebling, P.; Ong, T.; Spiegel, R.; Peltz, S.; et al. Safety and Tolerability of Ataluren in a Phase 3 Study of Patients with Nonsense Mutation Duchenne Muscular Dystrophy (P3.164). Neurology 2016, 86, P3.164. [Google Scholar]
- Drake, K.M.; Dunmore, B.J.; McNelly, L.N.; Morrell, N.W.; Aldred, M.A. Correction of nonsense BMPR2 and SMAD9 mutations by ataluren in pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 49, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Yang, X.; Southwood, M.; Moore, S.; Crosby, A.; Upton, P.D.; Dunmore, B.J.; Morrell, N.W. Targeting translational read-through of premature termination mutations in BMPR2 with PTC124 for pulmonary arterial hypertension. Pulm. Circ. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Albinana, V.; Sanz-Rodriguez, F.; Recio-Poveda, L.; Bernabeu, C.; Botella, L.M. Immunosuppressor FK506 increases endoglin and activin receptor-like kinase 1 expression and modulates transforming growth factor-beta1 signaling in endothelial cells. Mol. Pharmacol. 2011, 79, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Bill, M.; Aldred, M.A.; van de Veerdonk, M.C.; Vonk Noordegraaf, A.; Long-Boyle, J.; Dash, R.; Yang, P.C.; et al. Low-Dose FK506 (Tacrolimus) in End-Stage Pulmonary Arterial Hypertension. Am. J. Respir. Crit Care Med. 2015, 192, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Scott, V.; Del Rosario, P.; Bill, M.; Haddad, F.; Long-Boyle, J.; Hedlin, H.; Zamanian, R.T. Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur. Respir. J. 2017, 50, 1602449. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Crosby, A.; Yang, X.; Southwood, M.; Upton, P.D.; Kim, D.K.; Morrell, N.W. Altered bone morphogenetic protein and transforming growth factor-beta signaling in rat models of pulmonary hypertension: Potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation 2009, 119, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Yndestad, A.; Larsen, K.O.; Oie, E.; Ueland, T.; Smith, C.; Halvorsen, B.; Sjaastad, I.; Skjonsberg, O.H.; Pedersen, T.M.; Anfinsen, O.G.; et al. Elevated levels of activin A in clinical and experimental pulmonary hypertension. J. Appl. Physiol. 2009, 106, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Kudryashova, T.V.; Shen, Y.; Pena, A.; Cronin, E.; Okorie, E.; Goncharov, D.A.; Goncharova, E.A. Inhibitory Antibodies against Activin A and TGF-beta Reduce Self-Supported, but Not Soluble Factors-Induced Growth of Human Pulmonary Arterial Vascular Smooth Muscle Cells in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 2957. [Google Scholar] [CrossRef]
- Sun, N.; Chen, Y.; Yu, F.; Zhixin, F.; Lin, J.; Sun, B.; Yu, B.; Cheng, X.; Zheng, X.; Wu, B. Monocrotaline pyrrole enhanced bone morphogenetic protein 7 signaling transduced by alternative activin A receptor type 2A in pulmonary arterial smooth muscle cells. Eur. J. Pharmacol. 2019, 863, 172679. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.A.; Dunmore, B.J.; Long, L.; Crosby, A.; Al-Lamki, R.; Deighton, J.; Southwood, M.; Yang, X.; Nikolic, M.Z.; Herrera, B.; et al. TNFalpha drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun. 2017, 8, 14079. [Google Scholar] [CrossRef] [PubMed]
- Upton, P.D.; Davies, R.J.; Trembath, R.C.; Morrell, N.W. Bone morphogenetic protein (BMP) and activin type II receptors balance BMP9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J. Biol. Chem. 2009, 284, 15794–15804. [Google Scholar] [CrossRef] [PubMed]
- Yung, L.M.; Yang, P.; Joshi, S.; Augur, Z.M.; Kim, S.S.J.; Bocobo, G.A.; Dinter, T.; Troncone, L.; Chen, P.S.; McNeil, M.E.; et al. ACTRIIA-Fc rebalances activin/GDF versus BMP signaling in pulmonary hypertension. Sci. Transl. Med. 2020, 12, aaz5660. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Porter, J.; Origa, R.; Forni, G.L.; Voskaridou, E.; Galacteros, F.; Taher, A.T.; Arlet, J.B.; Ribeil, J.A.; Garbowski, M.; et al. Sotatercept, a novel transforming growth factor beta ligand trap, improves anemia in beta-thalassemia: A phase II, open-label, dose-finding study. Haematologica 2019, 104, 477–484. [Google Scholar] [CrossRef]
- Komrokji, R.; Garcia-Manero, G.; Ades, L.; Prebet, T.; Steensma, D.P.; Jurcic, J.G.; Sekeres, M.A.; Berdeja, J.; Savona, M.R.; Beyne-Rauzy, O.; et al. Sotatercept with long-term extension for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes: A phase 2, dose-ranging trial. Lancet Haematol 2018, 5, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.B.; Ghofrani, H.A.; Escribano Subias, P.; et al. Sotatercept for the treatment of pulmonary arterial hypertension: PULSAR open-label extension. Eur. Respir. J. 2022, 60, 2201347. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Escribano Subias, P.; Feldman, J.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Groves, B.; Badesch, D.B.; Voelkel, N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 1994, 144, 275–285. [Google Scholar] [PubMed]
- Sakamaki, F.; Kyotani, S.; Nagaya, N.; Sato, N.; Oya, H.; Satoh, T.; Nakanishi, N. Increased plasma P-selectin and decreased thrombomodulin in pulmonary arterial hypertension were improved by continuous prostacyclin therapy. Circulation 2000, 102, 2720–2725. [Google Scholar] [CrossRef] [PubMed]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Savale, L.; Tu, L.; Rideau, D.; Izziki, M.; Maitre, B.; Adnot, S.; Eddahibi, S. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir. Res. 2009, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Phan, C.; Tu, L.; Le Hiress, M.; Thuillet, R.; Jutant, E.M.; Fadel, E.; Savale, L.; Huertas, A.; Humbert, M.; et al. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J. Clin. Invest. 2018, 128, 1956–1970. [Google Scholar] [CrossRef]
- Hernandez-Sanchez, J.; Harlow, L.; Church, C.; Gaine, S.; Knightbridge, E.; Bunclark, K.; Gor, D.; Bedding, A.; Morrell, N.; Corris, P.; et al. Clinical trial protocol for TRANSFORM-UK: A therapeutic open-label study of tocilizumab in the treatment of pulmonary arterial hypertension. Pulm. Circ. 2018, 8, 1–8. [Google Scholar] [CrossRef]
- Simpson, C.E.; Chen, J.Y.; Damico, R.L.; Hassoun, P.M.; Martin, L.J.; Yang, J.; Nies, M.; Griffiths, M.; Vaidya, R.D.; Brandal, S.; et al. Cellular sources of interleukin-6 and associations with clinical phenotypes and outcomes in pulmonary arterial hypertension. Eur. Respir. J. 2020, 55, 1901761. [Google Scholar] [CrossRef]
- Toshner, M.; Rothman, A. IL-6 in pulmonary hypertension: Why novel is not always best. Eur. Respir. J. 2020, 55, 2000314. [Google Scholar] [CrossRef]
- Ford, A.C.; Sandborn, W.J.; Khan, K.J.; Hanauer, S.B.; Talley, N.J.; Moayyedi, P. Efficacy of biological therapies in inflammatory bowel disease: Systematic review and meta-analysis. Am. J. Gastroenterol. 2011, 106, 644–659. [Google Scholar] [CrossRef] [PubMed]
- Moreland, L.W.; Schiff, M.H.; Baumgartner, S.W.; Tindall, E.A.; Fleischmann, R.M.; Bulpitt, K.J.; Weaver, A.L.; Keystone, E.C.; Furst, D.E.; Mease, P.J.; et al. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann. Intern. Med. 1999, 130, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J. Tumour necrosis factor (TNF) in psoriatic arthritis: Pathophysiology and treatment with TNF inhibitors. Ann. Rheum. Dis. 2002, 61, 298–304. [Google Scholar] [CrossRef]
- Bozkurt, B.; Torre-Amione, G.; Warren, M.S.; Whitmore, J.; Soran, O.Z.; Feldman, A.M.; Mann, D.L. Results of targeted anti-tumor necrosis factor therapy with etanercept (ENBREL) in patients with advanced heart failure. Circulation 2001, 103, 1044–1047. [Google Scholar] [CrossRef] [PubMed]
- Lis, K.; Kuzawinska, O.; Balkowiec-Iskra, E. Tumor necrosis factor inhibitors—State of knowledge. Arch. Med. Sci. 2014, 10, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Lu, J.; Li, M.T.; Wang, Q.; Zeng, X.F. Preventive and remedial application of etanercept attenuate monocrotaline-induced pulmonary arterial hypertension. Int. J. Rheum. Dis. 2016, 19, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Cool, C.D.; Rai, P.R.; Yeager, M.E.; Hernandez-Saavedra, D.; Serls, A.E.; Bull, T.M.; Geraci, M.W.; Brown, K.K.; Routes, J.M.; Tuder, R.M.; et al. Expression of human herpesvirus 8 in primary pulmonary hypertension. N. Engl. J. Med. 2003, 349, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Katano, H.; Ito, K.; Shibuya, K.; Saji, T.; Sato, Y.; Sata, T. Lack of human herpesvirus 8 infection in lungs of Japanese patients with primary pulmonary hypertension. J. Infect. Dis. 2005, 191, 743–745. [Google Scholar] [CrossRef][Green Version]
- Henke-Gendo, C.; Mengel, M.; Hoeper, M.M.; Alkharsah, K.; Schulz, T.F. Absence of Kaposi’s sarcoma-associated herpesvirus in patients with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2005, 172, 1581–1585. [Google Scholar] [CrossRef]
- Coscoy, L.; Ganem, D. A viral protein that selectively downregulates ICAM-1 and B7-2 and modulates T cell costimulation. J. Clin. Invest. 2001, 107, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, M.; Douglas, J.; Rose, P.P.; Gouveia, K.; Thomas, G.; Means, R.E.; Moses, A.V.; Fruh, K. Kaposi sarcoma herpesvirus K5 removes CD31/PECAM from endothelial cells. Blood 2006, 108, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Durrington, H.J.; Upton, P.D.; Hoer, S.; Boname, J.; Dunmore, B.J.; Yang, J.; Crilley, T.K.; Butler, L.M.; Blackbourn, D.J.; Nash, G.B.; et al. Identification of a lysosomal pathway regulating degradation of the bone morphogenetic protein receptor type II. J. Biol. Chem. 2010, 285, 37641–37649. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zvi, I.; Kivity, S.; Langevitz, P.; Shoenfeld, Y. Hydroxychloroquine: From malaria to autoimmunity. Clin. Rev. Allergy Immunol. 2012, 42, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Yang, X.; Southwood, M.; Lu, J.; Marciniak, S.J.; Dunmore, B.J.; Morrell, N.W. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ. Res. 2013, 112, 1159–1170. [Google Scholar] [CrossRef]
- Dunmore, B.J.; Drake, K.M.; Upton, P.D.; Toshner, M.R.; Aldred, M.A.; Morrell, N.W. The lysosomal inhibitor, chloroquine, increases cell surface BMPR-II levels and restores BMP9 signalling in endothelial cells harbouring BMPR-II mutations. Hum. Mol. Genet. 2013, 22, 3667–3679. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Puerto, M.C.; van Zuijen, I.; Huang, C.J.; Szulcek, R.; Pan, X.; van Dinther, M.A.; Kurakula, K.; Wiesmeijer, C.C.; Goumans, M.J.; Bogaard, H.J.; et al. Autophagy contributes to BMP type 2 receptor degradation and development of pulmonary arterial hypertension. J. Pathol. 2019, 249, 356–367. [Google Scholar] [CrossRef]
- Orriols, M.; Gomez-Puerto, M.C.; Ten Dijke, P. BMP type II receptor as a therapeutic target in pulmonary arterial hypertension. Cell Mol. Life Sci. 2017, 74, 2979–2995. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, A.; Rudarakanchana, N.; Upton, P.D.; Yang, J.; Crilley, T.K.; Trembath, R.C.; Morrell, N.W. Failure of bone morphogenetic protein receptor trafficking in pulmonary arterial hypertension: Potential for rescue. Hum. Mol. Genet. 2008, 17, 3180–3190. [Google Scholar] [CrossRef]
- Long, L.; Ormiston, M.L.; Yang, X.; Southwood, M.; Graf, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Mutschler, D.; Wikstrom, G.; Lind, L.; Larsson, A.; Lagrange, A.; Eriksson, M. Etanercept reduces late endotoxin-induced pulmonary hypertension in the pig. J. Interferon. Cytokine Res. 2006, 26, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Abdulkadyrov, K.M.; Salogub, G.N.; Khuazheva, N.K.; Sherman, M.L.; Laadem, A.; Barger, R.; Knight, R.; Srinivasan, S.; Terpos, E. Sotatercept in patients with osteolytic lesions of multiple myeloma. Br. J. Haematol. 2014, 165, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Meloche, J.; Pflieger, A.; Vaillancourt, M.; Paulin, R.; Potus, F.; Zervopoulos, S.; Graydon, C.; Courboulin, A.; Breuils-Bonnet, S.; Tremblay, E.; et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 2014, 129, 786–797. [Google Scholar] [CrossRef]
- Guo, X.X.; Wu, H.L.; Shi, H.Y.; Su, L.; Zhang, X. The efficacy and safety of olaparib in the treatment of cancers: A meta-analysis of randomized controlled trials. Cancer Manag. Res. 2018, 10, 2553–2562. [Google Scholar] [CrossRef] [PubMed]
- Frump, A.L.; Lowery, J.W.; Hamid, R.; Austin, E.D.; de Caestecker, M. Abnormal trafficking of endogenously expressed BMPR2 mutant allelic products in patients with heritable pulmonary arterial hypertension. PLoS ONE 2013, 8, e80319. [Google Scholar] [CrossRef] [PubMed]
- Dunmore, B.J.; Yang, X.; Crosby, A.; Moore, S.; Long, L.; Huang, C.; Southwood, M.; Austin, E.D.; Rana, A.; Upton, P.D.; et al. 4PBA Restores Signaling of a Cysteine-substituted Mutant BMPR2 Receptor Found in Patients with Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2020, 63, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Koyama, M.; Furuhashi, M.; Ishimura, S.; Mita, T.; Fuseya, T.; Okazaki, Y.; Yoshida, H.; Tsuchihashi, K.; Miura, T. Reduction of endoplasmic reticulum stress by 4-phenylbutyric acid prevents the development of hypoxia-induced pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1314–H1323. [Google Scholar] [CrossRef]
- Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Sutendra, G.; Michelakis, E.D. Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation 2013, 127, 115–125. [Google Scholar] [CrossRef]
- Danilov, S.M. Conformational Fingerprinting Using Monoclonal Antibodies (on the Example of Angiotensin I-Converting Enzyme-ACE). Mol. Biol. 2017, 51, 1046–1061. (In Russian) [Google Scholar] [CrossRef] [PubMed]








| Drug/Compound | Route of Administration | Mechanism of Action | Clinical Trial | Pre-Clinical Animal Studies | Pre-Clinical Human Studies | References |
|---|---|---|---|---|---|---|
| Ataluren/PTC124 (Translama) | Oral | Regulates translational read-through of premature stop codons in BMPR2 mutants | -- | Bmpr2 R584X mice | PAECs and BOECs, Bmpr2 R584X PASMCs | [149,150,191] |
| Exogenous BMPR2 | Delivery of wild-type BMPR2 via viral like particles (VLP), exosomes, or nanoparticles | -- | Chronic hypoxia rats; Chronic hypoxia and MCT rats | Non-transformed mouse mammary gland epithelial cells (NMuMGs) and human umbilical vein endothelial cells (HUVECs), PMECs, BM-ELPCs | [132,133,134,135] | |
| BMP9/10 | Regulates BMP signaling and improved expression of BMPR2 | MCT rats, SU-5416 hypoxia rats, and Bmpr2 R899X mice | PAECs and BOECs | [159,192] | ||
| Etanercept (Embrel) | Subcutaneous injection | Obstructs TNF-alpha-induced inflammation and the downregulation of BMPR2 by performing as a TNF-alpha ligand trap | MCT rats; endotoxaemic pigs; SU-416 hypoxia rats | PAECs and PASMCs | [158,175,179,193] | |
| Tacrolimus/FK506 | Oral | Reestablishes BMP signaling by the inhibition of FKBP12 signaling blockage. | Low-dose FK506 at end-stage PAH; transform PAH— NCT01647945 at Phase II | MCT rats, SU-5416 hypoxia rats, and EC-Bmpr2−/− mice | PAECs | [78,152,153] |
| Sotatercept (ACE-011) | Subcutaneous injection | Inhibits TGF beta signaling by serving as an activin ligand trap | PULSAR—NCT03496207 (Phase II) SPECTRA—NCT03738150 (Phase IIa) | MCT rats and SU-5416 hypoxia rats | PMECs and PAMSCs129 | [160,161,162,194] |
| Hydroxychloroquine sulfate (Plaquenil) | Oral | Inhibits the lysosomal damage of BMPR2 | MCT rats | PASMCs and PAECs; PAECs and BOECs; PAECs, PASMCs, and HMEC | [186,187,188,189] | |
| Olaparib (Lynparza) | Oral | Prevents PARP1-induced DNA restoration in the deficiency of BRCA1 expression | Olaparib for PAH— NCT03782818 (Phase Ib) | MCT rats and SU-5416 hypoxia rats | PASMCs | [195,196] |
| Sodium 4-phenylbutyrate/4PBA or glycerol phenyl butyrate (Ravicti) | Oral | Discharges endoplasmic-reticulum-confined BMPR2 | Bmpr2 ΔEx2 mice; Bmpr2 C118W mice; chronic hypoxia mice; chronic hypoxia mice and MCT rats | HeLas and MRC-5s; Bmpr2 ΔEx2 PMECs; BMPR2 C118W HDFs and Bmpr2 C118W PASMCs | [191,197,198,199,200,201] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devendran, A.; Kar, S.; Bailey, R.; Trivieri, M.G. The Role of Bone Morphogenetic Protein Receptor Type 2 (BMPR2) and the Prospects of Utilizing Induced Pluripotent Stem Cells (iPSCs) in Pulmonary Arterial Hypertension Disease Modeling. Cells 2022, 11, 3823. https://doi.org/10.3390/cells11233823
Devendran A, Kar S, Bailey R, Trivieri MG. The Role of Bone Morphogenetic Protein Receptor Type 2 (BMPR2) and the Prospects of Utilizing Induced Pluripotent Stem Cells (iPSCs) in Pulmonary Arterial Hypertension Disease Modeling. Cells. 2022; 11(23):3823. https://doi.org/10.3390/cells11233823
Chicago/Turabian StyleDevendran, Anichavezhi, Sumanta Kar, Rasheed Bailey, and Maria Giovanna Trivieri. 2022. "The Role of Bone Morphogenetic Protein Receptor Type 2 (BMPR2) and the Prospects of Utilizing Induced Pluripotent Stem Cells (iPSCs) in Pulmonary Arterial Hypertension Disease Modeling" Cells 11, no. 23: 3823. https://doi.org/10.3390/cells11233823
APA StyleDevendran, A., Kar, S., Bailey, R., & Trivieri, M. G. (2022). The Role of Bone Morphogenetic Protein Receptor Type 2 (BMPR2) and the Prospects of Utilizing Induced Pluripotent Stem Cells (iPSCs) in Pulmonary Arterial Hypertension Disease Modeling. Cells, 11(23), 3823. https://doi.org/10.3390/cells11233823