


Delivery of Active AKT1 to Human Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Molecular Cloning and Gene Synthesis
2.2. Protein Production and Purification
2.3. Affinity Column Chromatography
2.4. Size Exclusion Chromatography
2.5. Anion Exchange Chromatography
2.6. AKT1 Kinase Activity Assay
2.7. Protein Incubation with Cells
2.8. Transfection of HEK 293T Cells
2.9. Western Blotting
2.10. Microscopy and Cell Imaging
2.11. Cytotoxicity Assay
2.12. Trypan Blue and Sytox Blue Assays
2.13. Quantification and Statistical Analysis
3. Results
3.1. Production and Purification of Recombinant AKT1 and Site-Specifically Phosphorylated AKT1 Variants
3.2. Enzymatic Activity of AKT1 and TAT-AKT1 Variants
3.3. Delivery of TAT-AKT1 Variants to Human Cells
3.4. Selective Phosphorylation of GSK-3α by TAT-pAKT1T308
3.5. TAT-pAKT1T308 Stimulates Downstream AKT1 Signaling to Ribosomal Protein S6
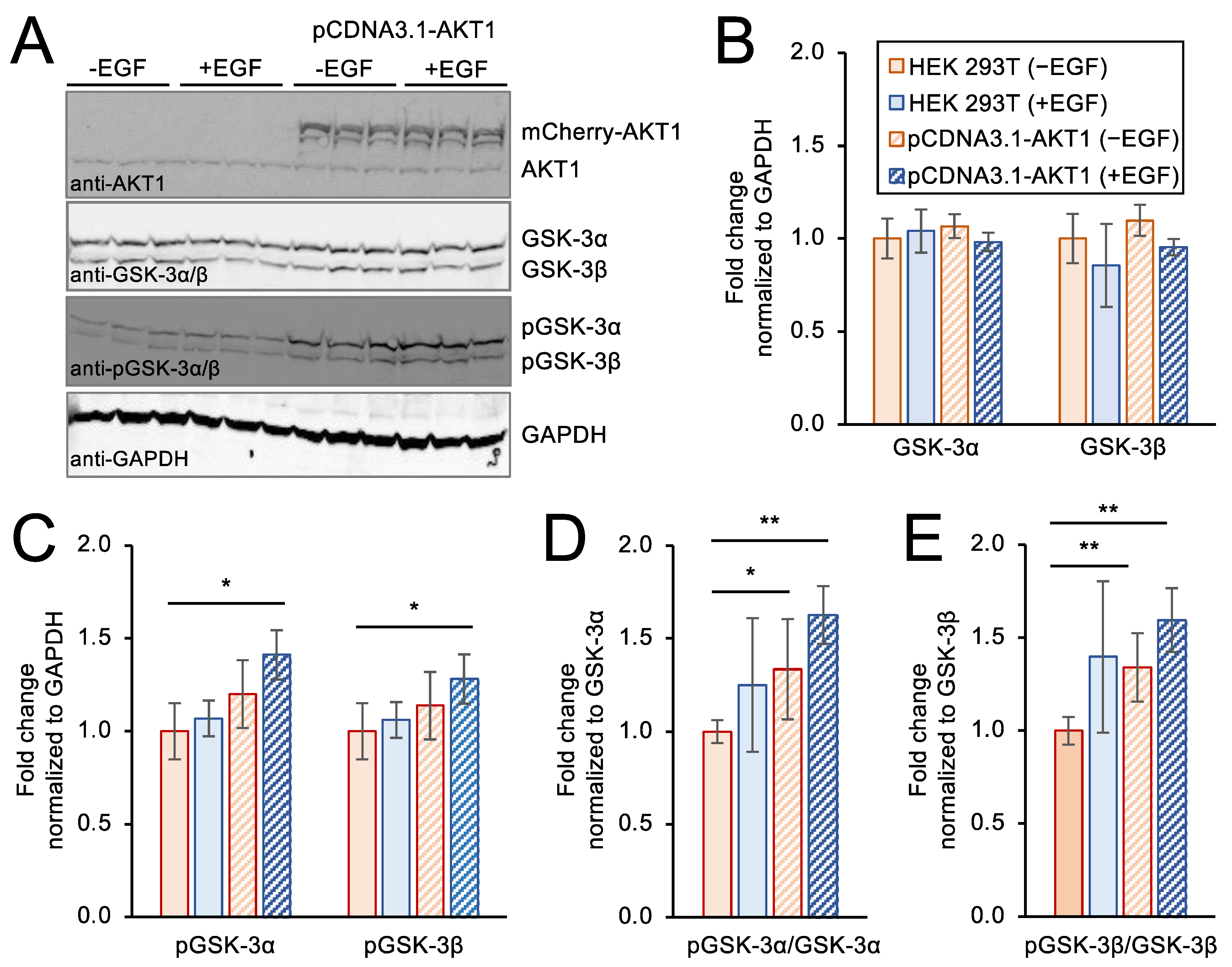
3.6. Genetic Over-Expression Model of AKT1 Activity in Stimulated Cells
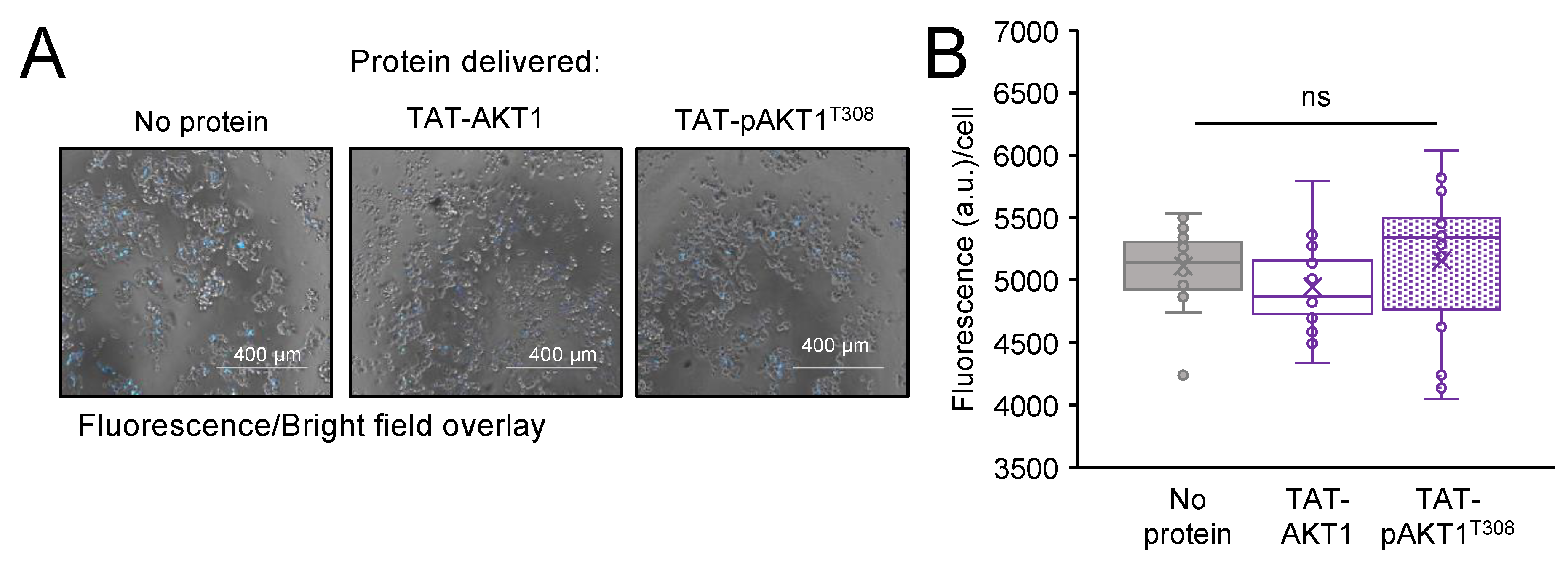
3.7. Delivery Efficiency and Localization of TAT-Tagged AKT1
3.8. Cellular Viability and Toxicity
4. Discussion
4.1. Fusing AKT1 with the Cell Penetrating Peptide TAT
4.2. Impact of TAT-Tagged Protein Delivery on Cell Fitness
4.3. Delivery of Active TAT-pAKT1T308 Stimulates AKT1 Signaling in Human Cells
4.4. TAT-Fusion of Peptides and Proteins to Modulate AKT1 Signaling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vogiatzi, P.; Giordano, A. Following the tracks of AKT1 gene. Cancer Biol. Ther. 2007, 6, 1521–1524. [Google Scholar] [CrossRef]
- Chen, W.S.; Xu, P.-Z.; Gottlob, K.; Chen, M.-L.; Sokol, K.; Shiyanova, T.; Roninson, I.; Weng, W.; Suzuki, R.; Tobe, K. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001, 15, 2203–2208. [Google Scholar] [CrossRef]
- Karege, F.; Perroud, N.; Schürhoff, F.; Meary, A.; Marillier, G.; Burkhardt, S.; Ballmann, E.; Fernandez, R.; Jamain, S.; Leboyer, M. Association of AKT1 gene variants and protein expression in both schizophrenia and bipolar disorder. Genes Brain Behav. 2010, 9, 503–511. [Google Scholar] [PubMed]
- Santi, S.A.; Lee, H. The Akt isoforms are present at distinct subcellular locations. Am. J. Physiol. -Cell Physiol. 2010, 298, C580–C591. [Google Scholar] [CrossRef]
- Staal, S.P. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef]
- Bellacosa, A.; De Feo, D.; Godwin, A.K.; Bell, D.W.; Cheng, J.Q.; Altomare, D.A.; Wan, M.; Dubeau, L.; Scambia, G.; Masciullo, V. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int. J. Cancer 1995, 64, 280–285. [Google Scholar] [CrossRef]
- Cheng, J.Q.; Ruggeri, B.; Klein, W.M.; Sonoda, G.; Altomare, D.A.; Watson, D.K.; Testa, J.R. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc. Natl. Acad. Sci. USA 1996, 93, 3636–3641. [Google Scholar] [CrossRef]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef]
- Gao, T.; Furnari, F.; Newton, A.C. PHLPP: A phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 2005, 18, 13–24. [Google Scholar] [CrossRef]
- Ugi, S.; Imamura, T.; Maegawa, H.; Egawa, K.; Yoshizaki, T.; Shi, K.; Obata, T.; Ebina, Y.; Kashiwagi, A.; Olefsky, J.M. Protein phosphatase 2A negatively regulates insulin’s metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol. Cell. Biol. 2004, 24, 8778–8789. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.C.; Madison, V. AKT crystal structure and AKT-specific inhibitors. Oncogene 2005, 24, 7493–7501. [Google Scholar] [CrossRef] [PubMed]
- Andjelkovic, M.; Alessi, D.R.; Meier, R.; Fernandez, A.; Lamb, N.J.; Frech, M.; Cron, P.; Cohen, P.; Lucocq, J.M.; Hemmings, B.A. Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 1997, 272, 31515–31524. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase–AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Balasuriya, N.; Davey, N.E.; Johnson, J.L.; Liu, H.; Biggar, K.K.; Cantley, L.C.; Li, S.S.-C.; O’Donoghue, P. Phosphorylation-dependent substrate selectivity of protein kinase B (AKT1). J. Biol. Chem. 2020, 295, 8120–8134. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Stambolic, V.; Woodgett, J.R. Functional distinctions of protein kinase B/Akt isoforms defined by their influence on cell migration. Trends Cell Biol. 2006, 16, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.; Hengstschlager, M. Nucleocytoplasmic localization of p70 S6K1, but not of its isoforms p85 and p31, is regulated by TSC2/mTOR. Oncogene 2011, 30, 4509–4522. [Google Scholar] [CrossRef]
- Rosner, M.; Hengstschlager, M. Evidence for cell cycle-dependent, rapamycin-resistant phosphorylation of ribosomal protein S6 at S240/244. Amino Acids 2010, 39, 1487–1492. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, A.; Kanno, T.; Nishizaki, T. PI3 kinase directly phosphorylates Akt1/2 at Ser473/474 in the insulin signal transduction pathway. J. Endocrinol. 2014, 220, 49. [Google Scholar] [CrossRef] [PubMed]
- Okano, J.-i.; Gaslightwala, I.; Birnbaum, M.J.; Rustgi, A.K.; Nakagawa, H. Akt/protein kinase B isoforms are differentially regulated by epidermal growth factor stimulation. J. Biol. Chem. 2000, 275, 30934–30942. [Google Scholar] [CrossRef] [PubMed]
- App, H.; Hazan, R.; Zilberstein, A.; Ullrich, A.; Schlessinger, J.; Rapp, U. Epidermal growth factor (EGF) stimulates association and kinase activity of Raf-1 with the EGF receptor. Mol. Cell. Biol. 1991, 11, 913–919. [Google Scholar] [PubMed]
- Wu, J.; Dent, P.; Jelinek, T.; Wolfman, A.; Weber, M.J.; Sturgill, T.W. Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3′,5′-monophosphate. Science 1993, 262, 1065–1069. [Google Scholar] [CrossRef]
- Schlessinger, J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science 2004, 306, 1506–1507. [Google Scholar] [CrossRef]
- Komurov, K.; Padron, D.; Cheng, T.; Roth, M.; Rosenblatt, K.P.; White, M.A. Comprehensive mapping of the human kinome to epidermal growth factor receptor signaling. J. Biol. Chem. 2010, 285, 21134–21142. [Google Scholar] [CrossRef]
- Frederick, M.I.; Siddika, T.; Zhang, P.; Balasuriya, N.; Turk, M.A.; O’Donoghue, P.; Heinemann, I.U. miRNA-Dependent Regulation of AKT1 Phosphorylation. Cells 2022, 11, 821. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.; Balasuriya, N.; Zhong, S.; Li, S.S.; O’Donoghue, P. Phospho-Form Specific Substrates of Protein Kinase B (AKT1). Front. Bioeng. Biotechnol. 2020, 8, 619252. [Google Scholar] [CrossRef] [PubMed]
- Balasuriya, N.; Kunkel, M.T.; Liu, X.; Biggar, K.K.; Li, S.S.-C.; Newton, A.C.; O’Donoghue, P. Genetic code expansion and live cell imaging reveal that Thr-308 phosphorylation is irreplaceable and sufficient for Akt1 activity. J. Biol. Chem. 2018, 293, 10744–10756. [Google Scholar] [CrossRef] [PubMed]
- Balasuriya, N.; McKenna, M.; Liu, X.; Li, S.S.; O’Donoghue, P. Phosphorylation-dependent inhibition of Akt1. Genes 2018, 9, 450. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, H.; Vocero-Akbani, A.M.; Snyder, E.L.; Ho, A.; Latham, D.G.; Lissy, N.A.; Becker-Hapak, M.; Ezhevsky, S.A.; Dowdy, S.F. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 1998, 4, 1449–1452. [Google Scholar] [CrossRef]
- Truernit, E.; Haseloff, J. A simple way to identify non-viable cells within living plant tissue using confocal microscopy. Plant Methods 2008, 4, 1–6. [Google Scholar] [CrossRef]
- Wright, D.E.; Siddika, T.; Heinemann, I.U.; O’Donoghue, P. Delivery of the selenoprotein thioredoxin reductase 1 to mammalian cells. Front. Mol. Biosci. 2022, 9, 1031756. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Kurrikoff, K.; Vunk, B.; Langel, U. Status update in the use of cell-penetrating peptides for the delivery of macromolecular therapeutics. Expert Opin. Biol. Ther. 2021, 21, 361–370. [Google Scholar] [CrossRef]
- Fawell, S.; Seery, J.; Daikh, Y.; Moore, C.; Chen, L.L.; Pepinsky, B.; Barsoum, J. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. USA 1994, 91, 664–668. [Google Scholar] [CrossRef]
- Thomas, P.; Smart, T.G. HEK293 cell line: A vehicle for the expression of recombinant proteins. J. Pharmacol. Toxicol. Methods 2005, 51, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Meier, R.; Alessi, D.R.; Cron, P.; Andjelkovic, M.; Hemmings, B.A. Mitogenic activation, phosphorylation, and nuclear translocation of protein kinase Bbeta. J. Biol. Chem. 1997, 272, 30491–30497. [Google Scholar] [CrossRef] [PubMed]
- Dufner, A.; Andjelkovic, M.; Burgering, B.M.; Hemmings, B.A.; Thomas, G. Protein kinase B localization and activation differentially affect S6 kinase 1 activity and eukaryotic translation initiation factor 4E-binding protein 1 phosphorylation. Mol. Cell. Biol. 1999, 19, 4525–4534. [Google Scholar] [CrossRef] [PubMed]
- Myohanen, T.T.; Mertens, F.; Norrbacka, S.; Cui, H. Deletion or inhibition of prolyl oligopeptidase blocks lithium-induced phosphorylation of GSK3b and Akt by activation of protein phosphatase 2A. Basic Clin. Pharmacol. Toxicol. 2021, 129, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Z.; Balasuriya, N.; Manni, E.; Liu, X.; Li, S.S.; O’Donoghue, P.; Heinemann, I.U. Gld2 activity is regulated by phosphorylation in the N-terminal domain. RNA Biol. 2019, 16, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Z.; Balasuriya, N.; Siddika, T.; Frederick, M.I.; Heinemann, I.U. Gld2 activity and RNA specificity is dynamically regulated by phosphorylation and interaction with QKI-7. RNA Biol. 2021, 18, 397–408. [Google Scholar] [CrossRef]
- Palma, M.; Leroy, C.; Salome-Desnoulez, S.; Werkmeister, E.; Kong, R.; Mongy, M.; Le Hir, H.; Lejeune, F. A role for AKT1 in nonsense-mediated mRNA decay. Nucleic Acids Res. 2021, 49, 11022–11037. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar]
- Burgering, B.M.; Coffer, P.J. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 1995, 376, 599–602. [Google Scholar] [CrossRef]
- Zhang, H.; Zha, X.; Tan, Y.; Hornbeck, P.V.; Mastrangelo, A.J.; Alessi, D.R.; Polakiewicz, R.D.; Comb, M.J. Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J. Biol. Chem. 2002, 277, 39379–39387. [Google Scholar] [CrossRef] [PubMed]
- Meyuhas, O. Ribosomal Protein S6 Phosphorylation: Four Decades of Research. Int. Rev. Cell Mol. Biol. 2015, 320, 41–73. [Google Scholar] [PubMed]
- Gerasimovskaya, E.V.; Tucker, D.A.; Weiser-Evans, M.; Wenzlau, J.M.; Klemm, D.J.; Banks, M.; Stenmark, K.R. Extracellular ATP-induced proliferation of adventitial fibroblasts requires phosphoinositide 3-kinase, Akt, mammalian target of rapamycin, and p70 S6 kinase signaling pathways. J. Biol. Chem. 2005, 280, 1838–1848. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar]
- Kunkel, M.T.; Ni, Q.; Tsien, R.Y.; Zhang, J.; Newton, A.C. Spatio-temporal dynamics of protein kinase B/Akt signaling revealed by a genetically encoded fluorescent reporter. J. Biol. Chem. 2005, 280, 5581–5587. [Google Scholar] [CrossRef]
- Toulany, M.; Maier, J.; Iida, M.; Rebholz, S.; Holler, M.; Grottke, A.; Jüker, M.; Wheeler, D.L.; Rothbauer, U.; Rodemann, H.P. Akt1 and Akt3 but not Akt2 through interaction with DNA-PKcs stimulate proliferation and post-irradiation cell survival of K-RAS-mutated cancer cells. Cell Death Discov. 2017, 3, 1–10. [Google Scholar] [CrossRef]
- Bessière, L.; Todeschini, A.-L.; Auguste, A.; Sarnacki, S.; Flatters, D.; Legois, B.; Sultan, C.; Kalfa, N.; Galmiche, L.; Veitia, R.A. A hot-spot of in-frame duplications activates the oncoprotein AKT1 in juvenile granulosa cell tumors. eBioMedicine 2015, 2, 421–431. [Google Scholar] [CrossRef]
- Tian, Y.; Zhou, S. Advances in cell penetrating peptides and their functionalization of polymeric nanoplatforms for drug delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2021, 13, e1668. [Google Scholar] [CrossRef]
- Yokoo, H.; Oba, M.; Uchida, S. Cell-Penetrating Peptides: Emerging Tools for mRNA Delivery. Pharmaceutics 2021, 14, 78. [Google Scholar] [CrossRef]
- Arya, S.K.; Guo, C.; Josephs, S.F.; Wong-Staal, F. Trans-activator gene of human T-lymphotropic virus type III (HTLV-III). Science 1985, 229, 69–73. [Google Scholar] [CrossRef]
- Klein, S.; Geiger, T.; Linchevski, I.; Lebendiker, M.; Itkin, A.; Assayag, K.; Levitzki, A. Expression and purification of active PKB kinase from Escherichia coli. Protein Expr. Purif. 2005, 41, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.W.; Christison, R.; Bundell, K.; Voyce, C.J.; Brockbank, S.M.; Newham, P.; Lindsay, M.A. Characterisation of cell-penetrating peptide-mediated peptide delivery. Br. J. Pharmacol. 2005, 145, 1093. [Google Scholar] [CrossRef] [PubMed]
- Riss, T.; Niles, A.; Moravec, R.; Karassina, N.; Vidugiriene, J. Cytotoxicity assays: In vitro methods to measure dead cells. In Assay Guidance Manual; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2019. [Google Scholar]
- Harms, J.S.; Khan, M.; Hall, C.; Splitter, G.A.; Homan, E.J.; Bremel, R.D.; Smith, J.A. Brucella peptide cross-reactive major histocompatibility complex class I presentation activates SIINFEKL-specific T cell receptor-expressing T cells. Infect. Immun. 2018, 86, e00281-18. [Google Scholar] [CrossRef] [PubMed]
- Sheng, S.; Qiao, M.; Pardee, A.B. Metastasis and AKT activation. J. Cell. Physiol. 2009, 218, 451–454. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881. [Google Scholar] [PubMed]
- Mailleux, A.A.; Overholtzer, M.; Schmelzle, T.; Bouillet, P.; Strasser, A.; Brugge, J.S. BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev. Cell 2007, 12, 221–234. [Google Scholar] [CrossRef]
- Soutar, M.P.; Kim, W.Y.; Williamson, R.; Peggie, M.; Hastie, C.J.; McLauchlan, H.; Snider, W.D.; Gordon-Weeks, P.R.; Sutherland, C. Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J. Neurochem. 2010, 115, 974–983. [Google Scholar] [CrossRef]
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen synthase kinase 3α-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007, 6, 329–337. [Google Scholar] [CrossRef]
- Hu, J.; Ahuja, L.G.; Meharena, H.S.; Kannan, N.; Kornev, A.P.; Taylor, S.S.; Shaw, A.S. Kinase regulation by hydrophobic spine assembly in cancer. Mol. Cell. Biol. 2015, 35, 264–276. [Google Scholar] [CrossRef]
- Antal, C.E.; Newton, A.C. Spatiotemporal dynamics of phosphorylation in lipid second messenger signaling. Mol. Cell. Proteom. 2013, 12, 3498–3508. [Google Scholar] [CrossRef]
- Liu, R.; Wang, L.; Chen, C.; Liu, Y.; Zhou, P.; Wang, Y.; Wang, X.; Turnbull, J.; Minassian, B.A.; Liu, Y.; et al. Laforin negatively regulates cell cycle progression through glycogen synthase kinase 3beta-dependent mechanisms. Mol. Cell. Biol. 2008, 28, 7236–7244. [Google Scholar] [CrossRef] [PubMed]
- Vernia, S.; Heredia, M.; Criado, O.; Rodriguez de Cordoba, S.; Garcia-Roves, P.M.; Cansell, C.; Denis, R.; Luquet, S.; Foufelle, F.; Ferre, P.; et al. Laforin, a dual specificity phosphatase involved in Lafora disease, regulates insulin response and whole-body energy balance in mice. Hum. Mol. Genet. 2011, 20, 2571–2584. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Y.; Wu, C.; Liu, Y.; Zheng, P. Dimerization of Laforin is required for its optimal phosphatase activity, regulation of GSK3beta phosphorylation, and Wnt signaling. J. Biol. Chem. 2006, 281, 34768–34774. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Hirano, K.; Hirano, M.; Nishimura, J.; Nakano, H.; Kanaide, H. Akt plays a central role in the anti-apoptotic effect of estrogen in endothelial cells. Biochem. Biophys. Res. Commun. 2004, 324, 321–325. [Google Scholar] [CrossRef]
- Yu, J.; Taylor, L.; Mierke, D.; Berg, E.; Shia, M.; Fishman, J.; Sallum, C.; Polgar, P. Limiting angiotensin II signaling with a cell-penetrating peptide mimicking the second intracellular loop of the angiotensin II type-I receptor. Chem. Biol. Drug Des. 2010, 76, 70–76. [Google Scholar] [CrossRef]
- Liu, R.; Xi, L.; Luo, D.; Ma, X.; Yang, W.; Xi, Y.; Wang, H.; Qian, M.; Fan, L.; Xia, X.; et al. Enhanced targeted anticancer effects and inhibition of tumor metastasis by the TMTP1 compound peptide TMTP1-TAT-NBD. J. Control. Release 2012, 161, 893–902. [Google Scholar] [CrossRef]
- Fan, Y.X.; Liang, Z.X.; Liu, Q.Z.; Xiao, H.; Li, K.B.; Wu, J.Z. Cell penetrating peptide of sodium-iodide symporter effect on the I-131 radiotherapy on thyroid cancer. Exp. Ther. Med. 2017, 13, 989–994. [Google Scholar] [CrossRef]
- He, L.; Lai, H.; Chen, T. Dual-function nanosystem for synergetic cancer chemo-/radiotherapy through ROS-mediated signaling pathways. Biomaterials 2015, 51, 30–42. [Google Scholar] [CrossRef]
- Konoeda, H.; Yang, H.; Yang, C.; Gower, A.; Xu, C.; Zhang, W.; Liu, M. Protein Kinase C-delta Inhibitor Peptide Formulation using Gold Nanoparticles. J. Vis. Exp. 2019, e58741. [Google Scholar] [CrossRef]
- Zanin, S.; Sandre, M.; Cozza, G.; Ottaviani, D.; Marin, O.; Pinna, L.A.; Ruzzene, M. Chimeric peptides as modulators of CK2-dependent signaling: Mechanism of action and off-target effects. Biochim. Biophys. Acta (BBA) -Proteins Proteom. 2015, 1854, 1694–1707. [Google Scholar] [CrossRef]
- Ekokoski, E.; Aitio, O.; Tornquist, K.; Yli-Kauhaluoma, J.; Tuominen, R.K. HIV-1 Tat-peptide inhibits protein kinase C and protein kinase A through substrate competition. Eur. J. Pharm. Sci. 2010, 40, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.G.; Sayers, E.J.; He, L.; Narayan, R.; Williams, T.L.; Mills, E.M.; Allemann, R.K.; Luk, L.Y.P.; Jones, A.T.; Tsai, Y.H. Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green florescent protein in different cell lines. Sci. Rep. 2019, 9, 6298. [Google Scholar] [CrossRef] [PubMed]
- El-Andaloussi, S.; Jarver, P.; Johansson, H.J.; Langel, U. Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: A comparative study. Biochem. J. 2007, 407, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Potocky, T.B.; Menon, A.K.; Gellman, S.H. Cytoplasmic and nuclear delivery of a TAT-derived peptide and a beta-peptide after endocytic uptake into HeLa cells. J. Biol. Chem. 2003, 278, 50188–50194. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siddika, T.; Balasuriya, N.; Frederick, M.I.; Rozik, P.; Heinemann, I.U.; O’Donoghue, P. Delivery of Active AKT1 to Human Cells. Cells 2022, 11, 3834. https://doi.org/10.3390/cells11233834
Siddika T, Balasuriya N, Frederick MI, Rozik P, Heinemann IU, O’Donoghue P. Delivery of Active AKT1 to Human Cells. Cells. 2022; 11(23):3834. https://doi.org/10.3390/cells11233834
Chicago/Turabian StyleSiddika, Tarana, Nileeka Balasuriya, Mallory I. Frederick, Peter Rozik, Ilka U. Heinemann, and Patrick O’Donoghue. 2022. "Delivery of Active AKT1 to Human Cells" Cells 11, no. 23: 3834. https://doi.org/10.3390/cells11233834
APA StyleSiddika, T., Balasuriya, N., Frederick, M. I., Rozik, P., Heinemann, I. U., & O’Donoghue, P. (2022). Delivery of Active AKT1 to Human Cells. Cells, 11(23), 3834. https://doi.org/10.3390/cells11233834