
Stem-Cell-Derived β-Like Cells with a Functional PTPN2 Knockout Display Increased Immunogenicity
 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. hPSC Culture and Differentiation of Stem-Cell-Derived Beta-Like Cells (sBC)
2.2. CRISPR-Cas9 Genome Engineering—PTPN2 KO
2.3. Immunofluorescence
2.4. Flow Cytometry
2.5. GFP+ sBC Sorting
2.6. Content Analysis
2.7. Western Blotting
2.8. Bulk RNA Sequencing
2.9. Differential Expression Analysis
2.10. Gene Set Enrichment Analysis (GSEA)
2.11. T Cell Stimulation Assay
2.12. Statistical Analysis
2.13. Data and Resource Availability
3. Results
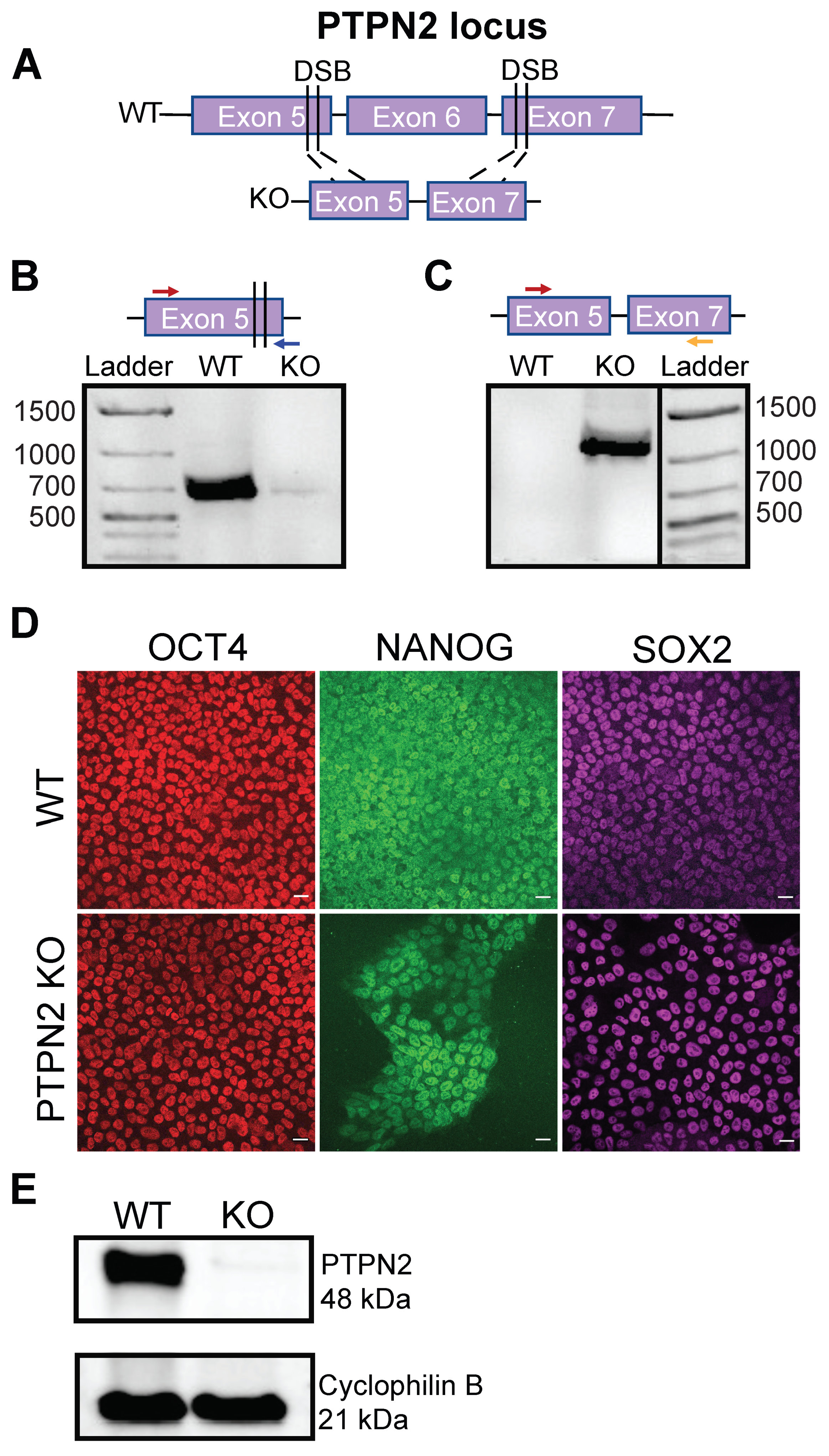
3.1. Generation of a CRISPR-Cas9-Mediated PTPN2 Knockout hPSC
3.2. PTPN2 KO hPSC Efficiently Differentiate into Pancreatic Stem-Cell-Derived Beta Cells
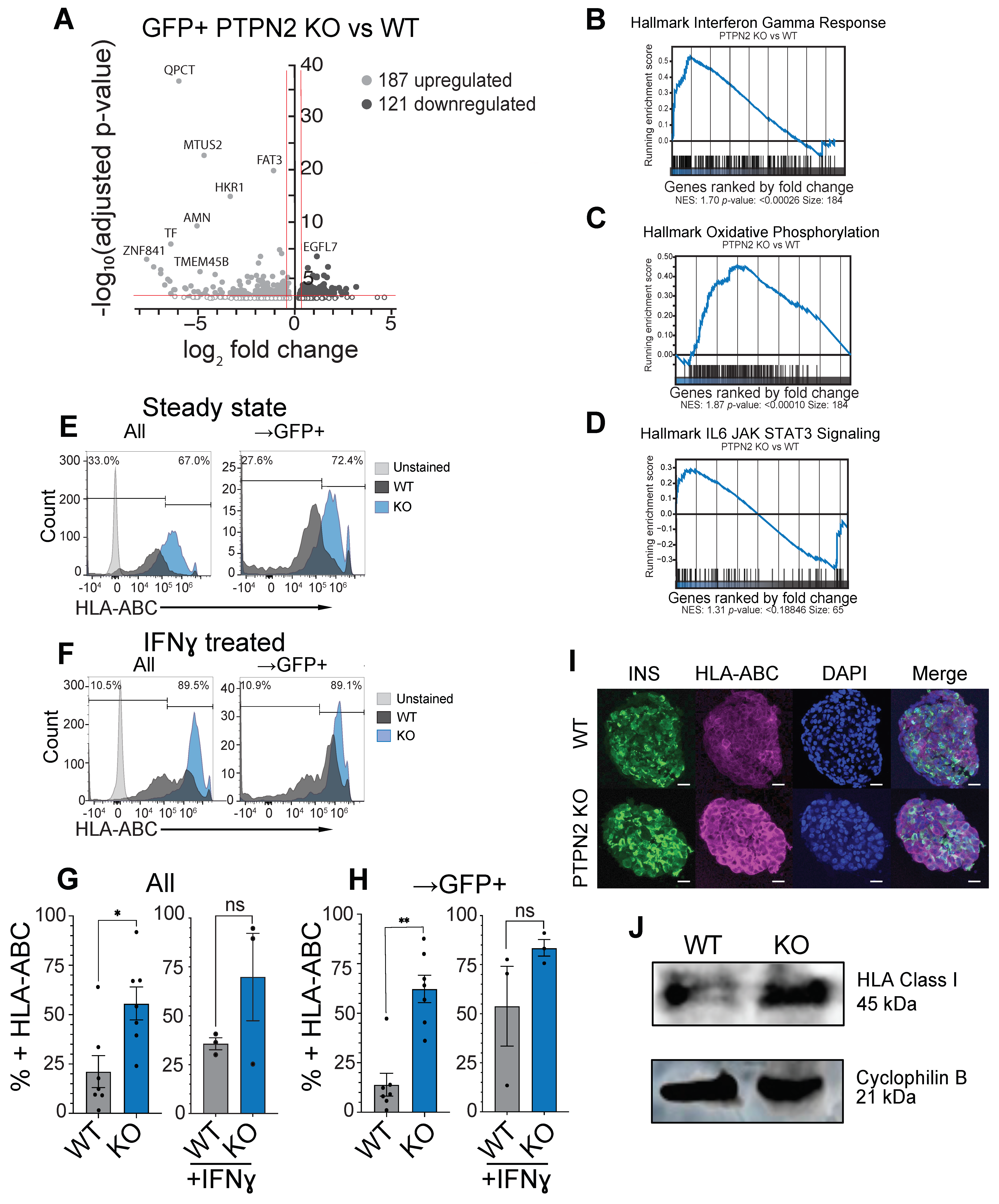
3.3. PTPN2 KO sBC Display Increased HLA Class I Molecules Compared to WT sBCs
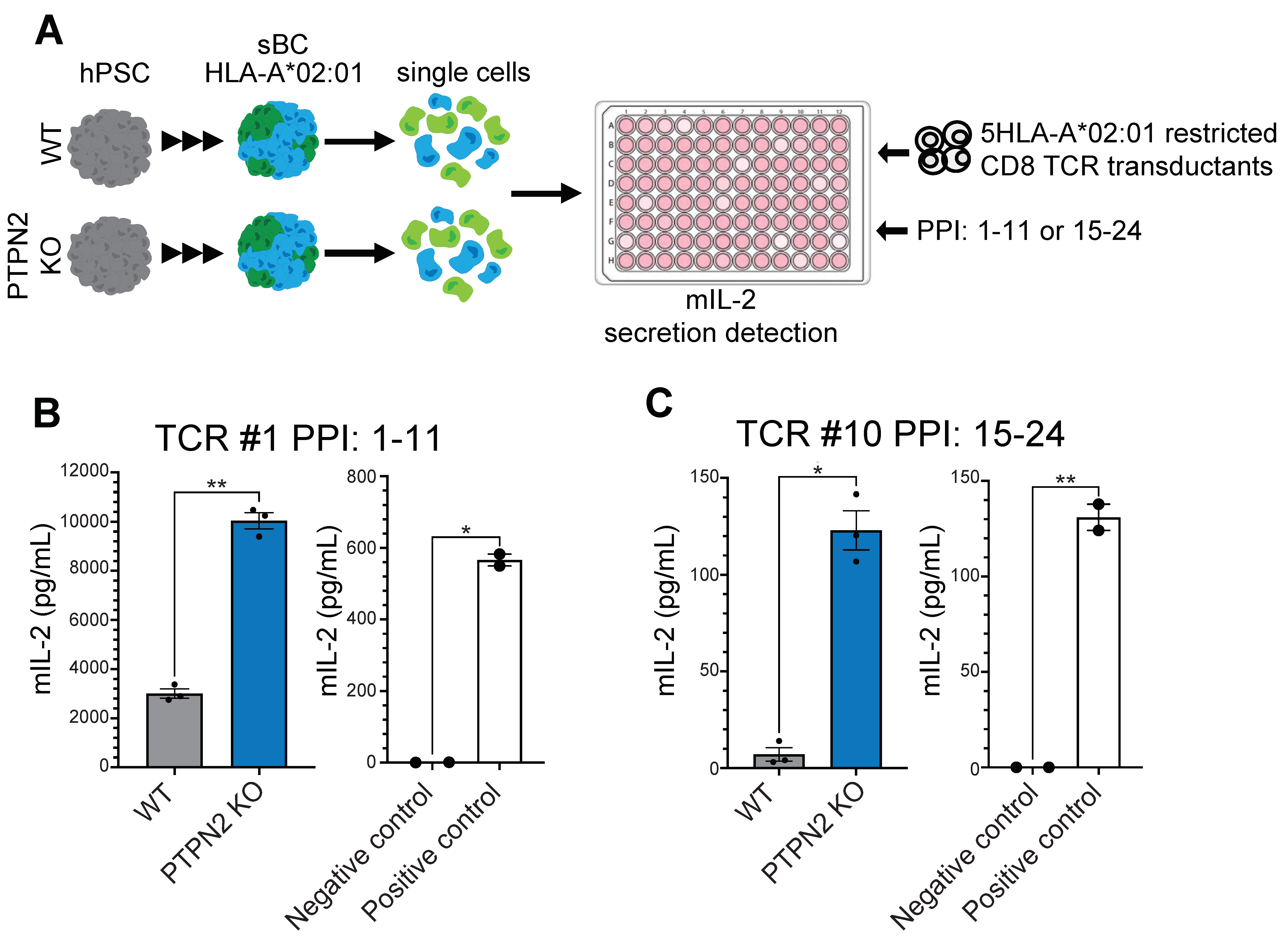
3.4. PTPN2 KO sBC Display Increased Stimulation of Autoreactive TCR Transductants Compared to WT sBC
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bonifacio, E. Predicting type 1 diabetes using biomarkers. Diabetes Care 2015, 38, 989–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, M.A.; Roep, B.O.; Posgai, A.; Wheeler, D.C.; Peakman, M. The challenge of modulating β-cell autoimmunity in type 1 diabetes. Lancet Diabetes Endocrinol. 2019, 7, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Gitelman, S.E.; Ehlers, M.R.; Gottlieb, P.A.; Greenbaum, C.J.; Hagopian, W.; Boyle, K.D.; Keyes-Elstein, L.; Aggarwal, S.; Phippard, D.; et al. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: Metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 2013, 62, 3766–3774. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, M.; Ahmed, S.; Anderson, M.S.; Atkinson, M.A.; Becker, D.; Bingley, P.J.; Bosi, E.; Brusko, T.M.; DiMeglio, L.A.; Evans-Molina, C.; et al. Introducing the Endotype Concept to Address the Challenge of Disease Heterogeneity in Type 1 Diabetes. Diabetes Care 2020, 43, 5–12. [Google Scholar] [CrossRef]
- Santin, I.; Eizirik, D.L. Candidate genes for type 1 diabetes modulate pancreatic islet inflammation and beta-cell apoptosis. Diabetes Obes. Metab. 2013, 15 (Suppl. S3), 71–81. [Google Scholar] [CrossRef]
- Steck, A.K.; Rewers, M.J. Genetics of type 1 diabetes. Clin. Chem. 2011, 57, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Concannon, P.; Rich, S.S.; Nepom, G.T. Genetics of type 1A diabetes. N. Engl. J. Med. 2009, 360, 1646–1654. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Sammeth, M.; Bouckenooghe, T.; Bottu, G.; Sisino, G.; Igoillo-Esteve, M.; Ortis, F.; Santin, I.; Colli, M.L.; Barthson, J.; et al. The human pancreatic islet transcriptome: Expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012, 8, e1002552. [Google Scholar] [CrossRef]
- Chiou, J.; Geusz, R.J.; Okino, M.-L.; Han, J.Y.; Miller, M.; Melton, R.; Beebe, E.; Benaglio, P.; Huang, S.; Korgaonkar, K.; et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 2021, 594, 398–402. [Google Scholar] [CrossRef]
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Pagliuca, F.W.; Millman, J.R.; Gürtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic β cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef] [Green Version]
- Velazco-Cruz, L.; Song, J.; Maxwell, K.G.; Goedegebuure, M.M.; Augsornworawat, P.; Hogrebe, N.J.; Millman, J.R. Acquisition of dynamic function in human stem cell-derived β cells. Stem Cell Rep. 2019, 12, 351–365. [Google Scholar] [CrossRef] [Green Version]
- Docherty, F.M.; Riemondy, K.A.; Castro-Gutierrez, R.; Dwulet, J.M.; Shilleh, A.H.; Hansen, M.S.; Williams, S.P.M.; Armitage, L.H.; Santostefano, K.E.; Wallet, M.A.; et al. ENTPD3 Marks Mature Stem Cell-Derived β-Cells Formed by Self-Aggregation In Vitro. Diabetes 2021, 70, 2554–2567. [Google Scholar] [CrossRef] [PubMed]
- Nair, G.G.; Liu, J.S.; Russ, H.A.; Tran, S.; Saxton, M.S.; Chen, R.; Juang, C.; Li, M.-l.; Nguyen, V.Q.; Giacometti, S.; et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived beta cells. Nat. Cell. Biol. 2019, 21, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.R.; Pettus, J.; Wilensky, J.; Shapiro, J.; Senior, P.A.; Roep, B.; Wang, R.; Kroon, E.J.; Scott, M.; D’Amour, K.; et al. Initial Clinical Evaluation of VC-01TM Combination Product—A Stem Cell–Derived Islet Replacement for Type 1 Diabetes (T1D). Diabetes 2018, 67 (Suppl. S1), 138-OR. [Google Scholar] [CrossRef]
- Russ, H.A.; Shilleh, A.H.; Sussel, L. From the dish to humans: A stem cell recipe for success. Cell Metab. 2022, 34, 193–196. [Google Scholar] [CrossRef]
- Frederiksen, B.; Liu, E.; Romanos, J.; Steck, A.K.; Yin, X.; Kroehl, M.; Fingerlin, T.E.; Erlich, E.; Eisenbarth, G.S.; Rewers, M.; et al. Investigation of the vitamin D receptor gene (VDR) and its interaction with protein tyrosine phosphatase, non-receptor type 2 gene (PTPN2) on risk of islet autoimmunity and type 1 diabetes: The Diabetes Autoimmunity Study in the Young (DAISY). J. Steroid. Biochem. Mol. Biol. 2013, 133, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Espino-Paisan, L.; de la Calle, H.; Fernández-Arquero, M.; Figueredo, M.Á.; de la Concha, E.G.; Urcelay, E.; Santiago, J.L. A polymorphism in PTPN2 gene is associated with an earlier onset of type 1 diabetes. Immunogenetics 2011, 63, 255–258. [Google Scholar] [CrossRef]
- Kleppe, M.; Lahortiga, I.; El Chaar, T.; De Keersmaecker, K.; Mentens, N.; Graux, C.; Van Roosbroeck, K.; Ferrando, A.A.; Langerak, A.W.; Meijerink, J.P.P.; et al. Deletion of the protein tyrosine phosphatase gene PTPN2 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2010, 42, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Wiede, F.; Shields, B.J.; Chew, S.H.; Kyparissoudis, K.; van Vliet, C.; Galic, S.; Tremblay, M.L.; Russell, S.M.; Godfrey, D.I.; Tiganis, T. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J. Clin. Investig. 2011, 121, 4758–4774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, I.; Veillette, A. Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat. Immunol. 2012, 13, 439–447. [Google Scholar] [CrossRef]
- Hinman, R.M.; Smith, M.J.; Cambier, J.C. B cells and type 1 diabetes… in mice and men. Immunol. Lett. 2014, 160, 128–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santin, I.; Moore, F.; Colli, M.L.; Gurzov, E.N.; Marselli, L.; Marchetti, P.; Eizirik, D.L. PTPN2, a candidate gene for type 1 diabetes, modulates pancreatic β-cell apoptosis via regulation of the BH3-only protein Bim. Diabetes 2011, 60, 3279–3288. [Google Scholar] [CrossRef] [Green Version]
- Moore, F.; Colli, M.L.; Cnop, M.; Esteve, M.I.; Cardozo, A.K.; Cunha, D.A.; Bugliani, M.; Marchetti, P.; Eizirik, D.L. PTPN2, a candidate gene for type 1 diabetes, modulates interferon-gamma-induced pancreatic beta-cell apoptosis. Diabetes 2009, 58, 1283–1291. [Google Scholar] [CrossRef] [Green Version]
- Elvira, B.; Vandenbempt, V.; Bauzá-Martinez, J.; Crutzen, R.; Negueruela, J.; Ibrahim, H.; Winder, M.L.; Brahma, M.K.; Vekeriotaite, B.; Martens, P.-J.; et al. PTPN2 Regulates the Interferon Signaling and Endoplasmic Reticulum Stress Response in Pancreatic β-Cells in Autoimmune Diabetes. Diabetes 2022, 71, 653–668. [Google Scholar] [CrossRef]
- Micallef, S.J.; Li, X.; Schiesser, J.V.; Hirst, C.E.; Yu, Q.C.; Lim, S.M.; Nostro, M.C.; Elliott, D.A.; Sarangi, F.; Harrison, L.C.; et al. INS(GFP/w) human embryonic stem cells facilitate isolation of in vitro derived insulin-producing cells. Diabetologia 2012, 55, 694–706. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Gutierrez, R.; Alkanani, A.; Mathews, C.E.; Michels, A.; Russ, H.A. Protecting Stem Cell Derived Pancreatic Beta-Like Cells From Diabetogenic T Cell Recognition. Front. Endocrinol. 2021, 12, 707881. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.M.; Landry, L.G.; Alkanani, A.A.; Pyle, L.; Powers, A.C.; Atkinson, M.A.; Mathews, C.E.; Roep, B.O.; Michels, A.W.; Nakayama, M. Human islet T cells are highly reactive to preproinsulin in type 1 diabetes. Proc. Natl. Acad. Sci. USA 2021, 118, e2107208118. [Google Scholar] [CrossRef]
- Parlato, M.; Nian, Q.; Charbit-Henrion, F.; Ruemmele, F.M.; Rodrigues-Lima, F.; Cerf-Bensussan, N.; Immunobiota Study Group. Loss-of-Function Mutation in PTPN2 Causes Aberrant Activation of JAK Signaling Via STAT and Very Early Onset Intestinal Inflammation. Gastroenterology 2020, 159, 1968–1971.e4. [Google Scholar] [CrossRef]
- Suzuki, N.; Suzuki, S.; Millar, D.G.; Unno, M.; Hara, H.; Calzascia, T.; Yamasaki, S.; Yokosuka, T.; Chen, N.-J.; Elford, A.R.; et al. A Critical Role for the Innate Immune Signaling Molecule IRAK-4 in T Cell Activation. Science 2006, 311, 1927–1932. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.; Bermudez, V.; Palmar, J.; Martínez, M.S.; Olivar, L.C.; Nava, M.; Tomey, D.; Rojas, M.; Salazar, J.; Garicano, C.; et al. Pancreatic Beta Cell Death: Novel Potential Mechanisms in Diabetes Therapy. J. Diabetes Res. 2018, 2018, 9601801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, N.; Suzuki, S.; Duncan, G.S.; Millar, D.G.; Wada, T.; Mirtsos, C.; Takada, H.; Wakeham, A.; Itie, A.; Li, S.; et al. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature 2002, 416, 750–756. [Google Scholar] [CrossRef]
- Mann, S.E.; Zhou, Z.; Landry, L.G.; Anderson, A.M.; Alkanani, A.K.; Fischer, J.; Peakman, M.; Mallone, R.; Campbell, K.; Michels, A.W.; et al. Multiplex T Cell Stimulation Assay Utilizing a T Cell Activation Reporter-Based Detection System. Front. Immunol. 2020, 11, 633. [Google Scholar] [CrossRef] [PubMed]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.M.; Qasem, A.; Naser, S.A. Modulation of PTPN2/22 Function by Spermidine in CRISPR-Cas9-Edited T-Cells Associated with Crohn’s Disease and Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 8883. [Google Scholar] [CrossRef] [PubMed]
- Scharl, M.; McCole, D.F.; Weber, A.; Vavricka, S.R.; Frei, P.; Kellermeier, S.; Pesch, T.; Fried, M.; Rogler, G. Protein tyrosine phosphatase N2 regulates TNFα-induced signalling and cytokine secretion in human intestinal epithelial cells. Gut 2011, 60, 189–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharl, M.; Mwinyi, J.; Fischbeck, A.; Leucht, K.; Eloranta, J.J.; Arikkat, J.; Pesch, T.; Kellermeier, S.; Mair, A.; Kullak-Ublick, G.A.; et al. Crohn’s Disease-associated Polymorphism within the PTPN2 Gene Affects Muramyl-Dipeptide-induced Cytokine Secretion and Autophagy. Inflamm. Bowel Dis. 2011, 18, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Cerosaletti, K.; Buckner, J.H. Protein tyrosine phosphatases and type 1 diabetes: Genetic and functional implications of PTPN2 and PTPN22. Rev. Diabet Stud. 2012, 9, 188–200. [Google Scholar] [CrossRef] [Green Version]
- Cerosaletti, K.; Schneider, A.; Schwedhelm, K.; Frank, I.; Tatum, M.; Wei, S.; Whalen, E.; Greenbaum, C.; Kita, M.; Buckner, J.; et al. Multiple Autoimmune-Associated Variants Confer Decreased IL-2R Signaling in CD4+CD25hi T Cells of Type 1 Diabetic and Multiple Sclerosis Patients. PLoS ONE 2013, 8, e83811. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Triolo, T.M.; Matuschek, J.Q.; Castro-Gutierrez, R.; Shilleh, A.H.; Williams, S.P.M.; Hansen, M.S.; McDaniel, K.; Barra, J.M.; Michels, A.; Russ, H.A. Stem-Cell-Derived β-Like Cells with a Functional PTPN2 Knockout Display Increased Immunogenicity. Cells 2022, 11, 3845. https://doi.org/10.3390/cells11233845
Triolo TM, Matuschek JQ, Castro-Gutierrez R, Shilleh AH, Williams SPM, Hansen MS, McDaniel K, Barra JM, Michels A, Russ HA. Stem-Cell-Derived β-Like Cells with a Functional PTPN2 Knockout Display Increased Immunogenicity. Cells. 2022; 11(23):3845. https://doi.org/10.3390/cells11233845
Chicago/Turabian StyleTriolo, Taylor M., J. Quinn Matuschek, Roberto Castro-Gutierrez, Ali H. Shilleh, Shane P. M. Williams, Maria S. Hansen, Kristen McDaniel, Jessie M. Barra, Aaron Michels, and Holger A. Russ. 2022. "Stem-Cell-Derived β-Like Cells with a Functional PTPN2 Knockout Display Increased Immunogenicity" Cells 11, no. 23: 3845. https://doi.org/10.3390/cells11233845