
Dietary Choline Mitigates High-Fat Diet-Impaired Chylomicrons Assembly via UPRer Modulated by perk DNA Methylation
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Expt. 1: Animals Feeding, Management, and Sampling
2.3. Expt. 2: Cell Culture and Treatments
2.4. Sample Analysis
2.4.1. ORO, H&E, Bodipy 493/503 Staining and Transmission Electron Microscopy (TEM) Observation
2.4.2. Cell Viability, TG Contents, MTP Activity
2.4.3. RNA Isolation and Quantitative Real-Time PCR Analysis (qPCR)
2.4.4. Western Blot
2.4.5. Bisulfite Sequencing of Perk
2.4.6. Chylomicron Isolation
2.4.7. Immunofluorescence Analysis
2.4.8. Small Interference RNA Transfection
2.4.9. Plasmid Construction, Cloning of Promoters, Transfections, and Luciferase Assays
2.4.10. Immunoprecipitation
2.4.11. The Prediction of Perk-Apob48 Structural Protein Models
2.5. Statistical Analysis
3. Results
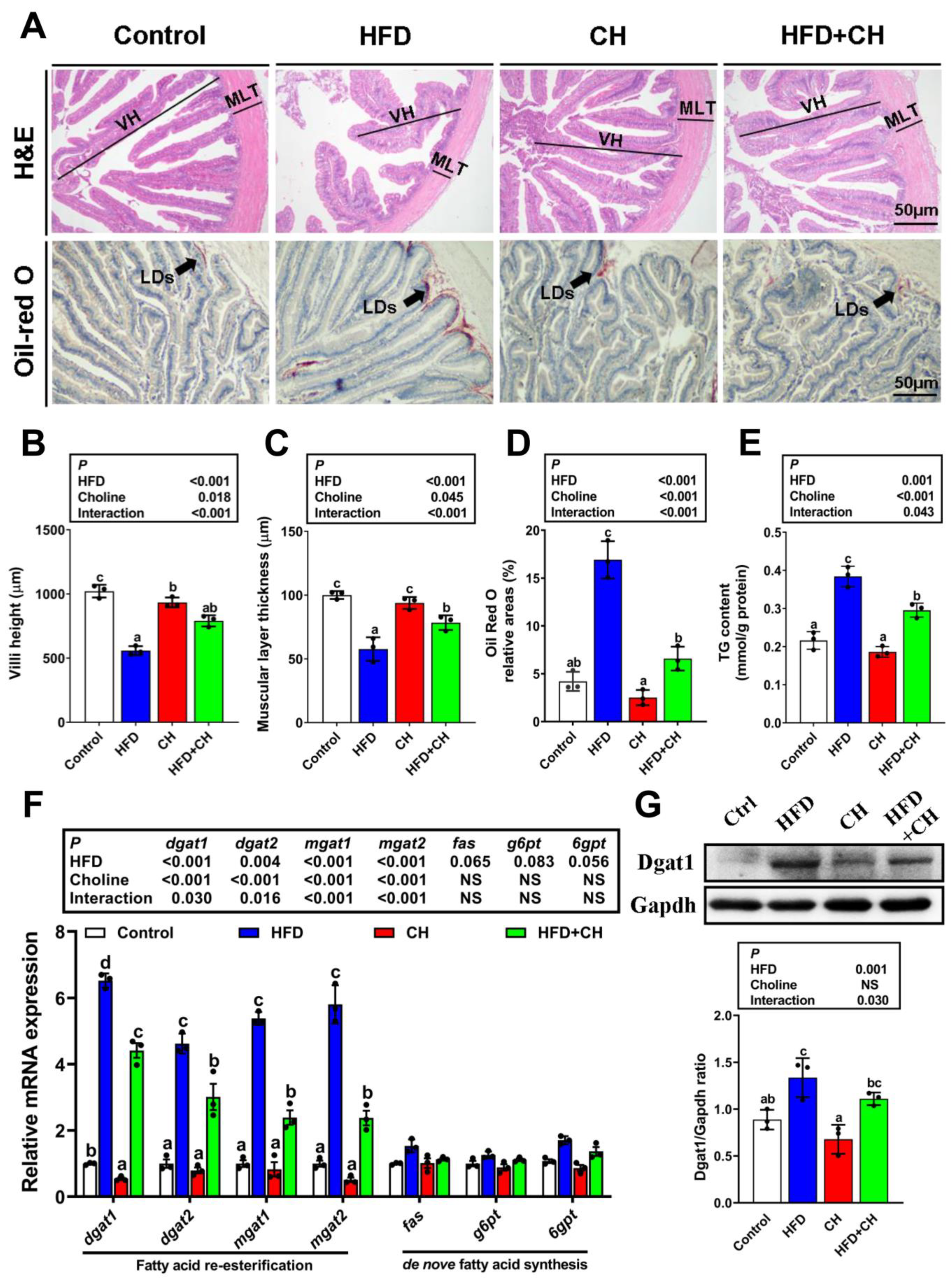
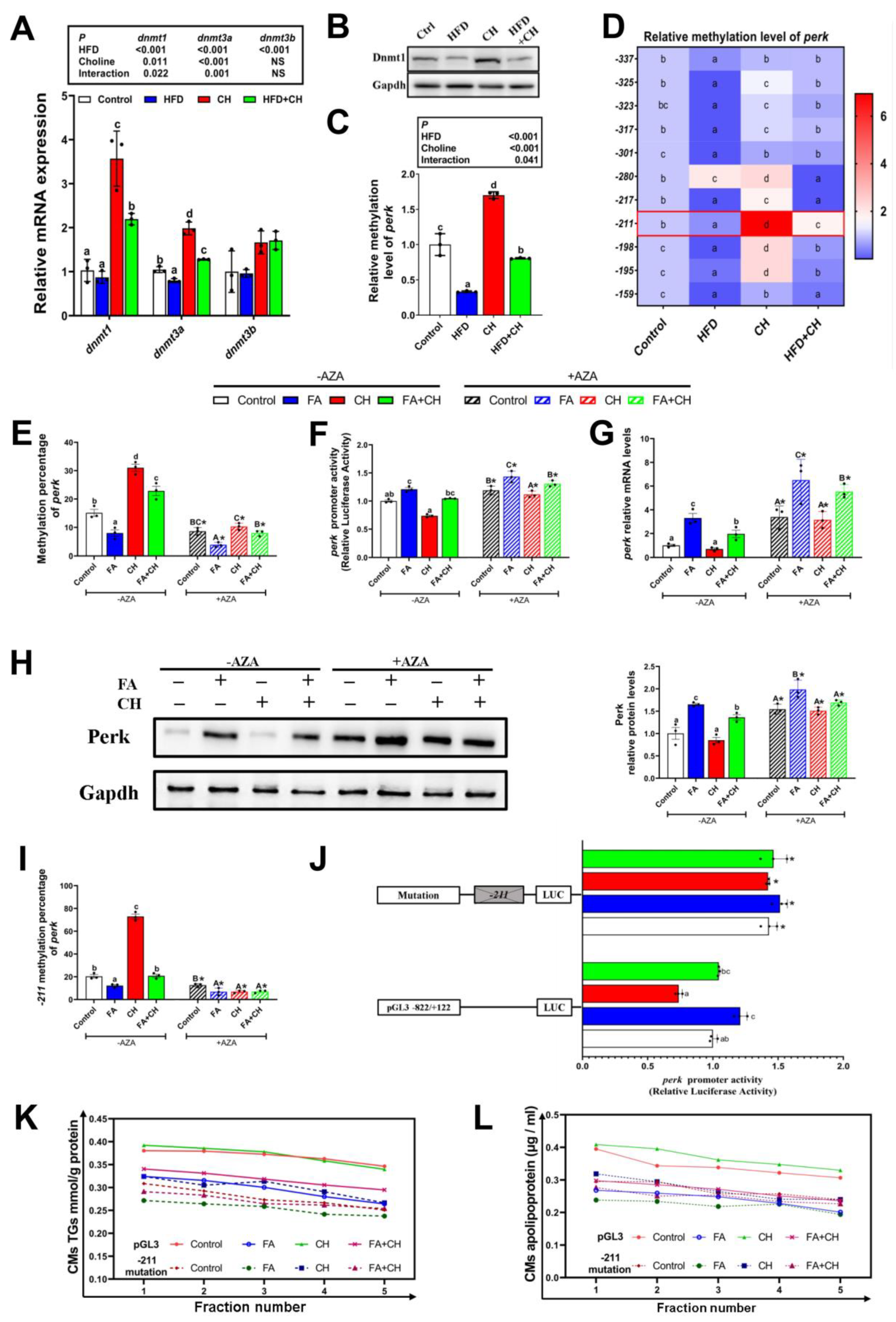
3.1. Dietary Choline Alleviated HFD-Induced Intestinal Lipid Dysregulation
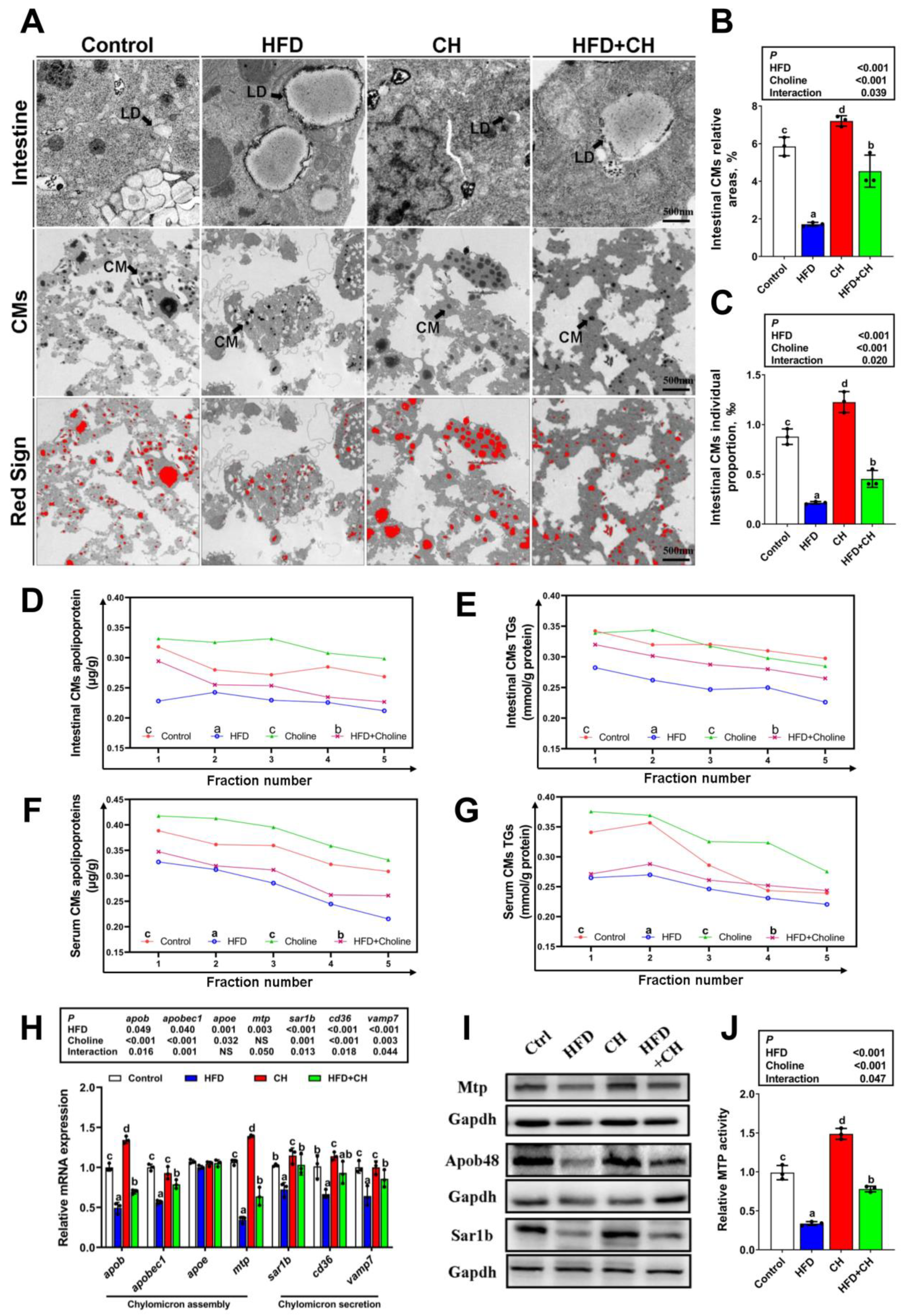
3.2. Alleviated Effects of Dietary Choline on HFD-Induced Impairment of CMs Assembly
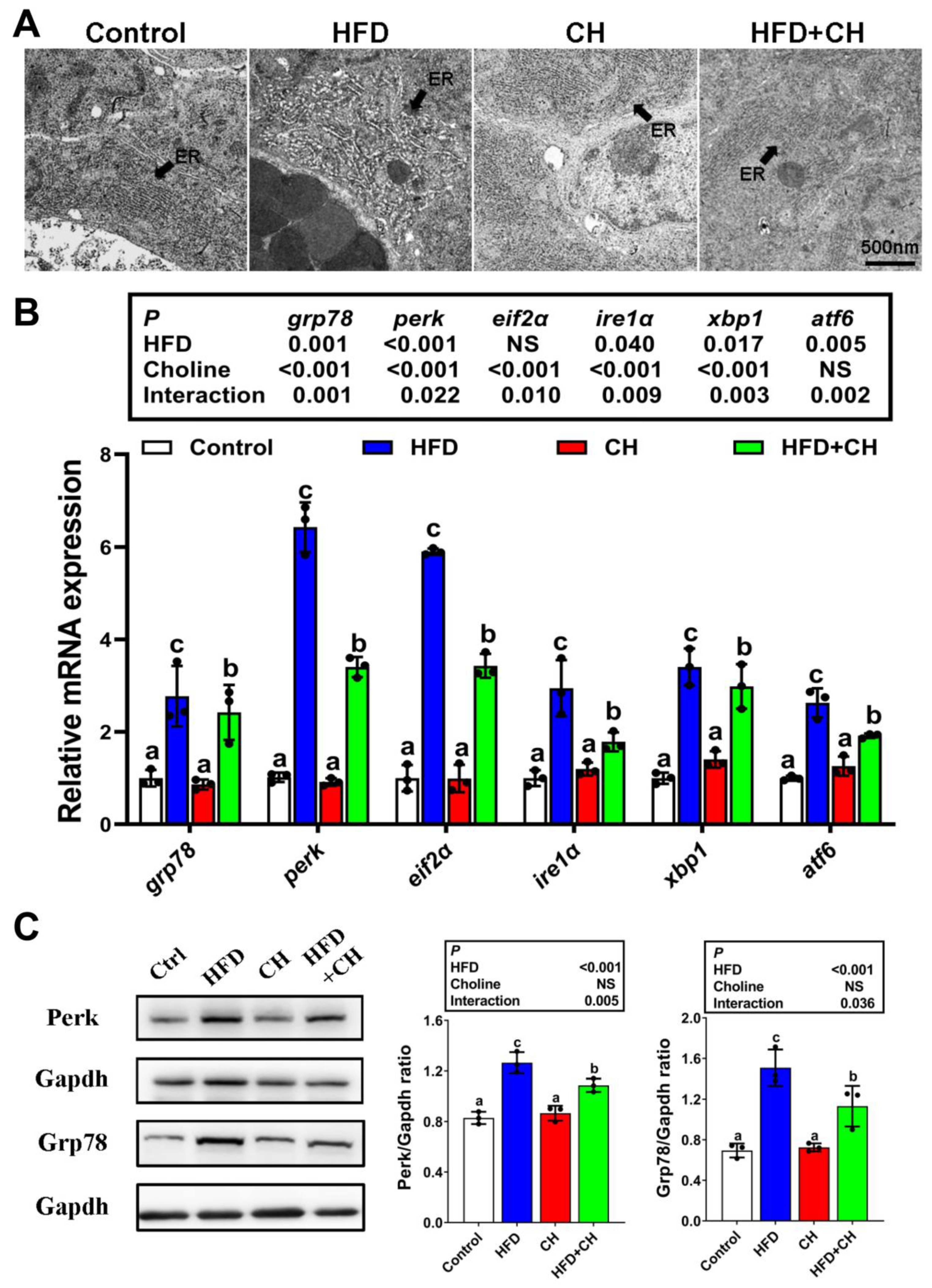
3.3. The Involvement of Perk Signaling-Mediated UPRer in Choline Attenuating HFD-Induced Impairment of CMs Assembly
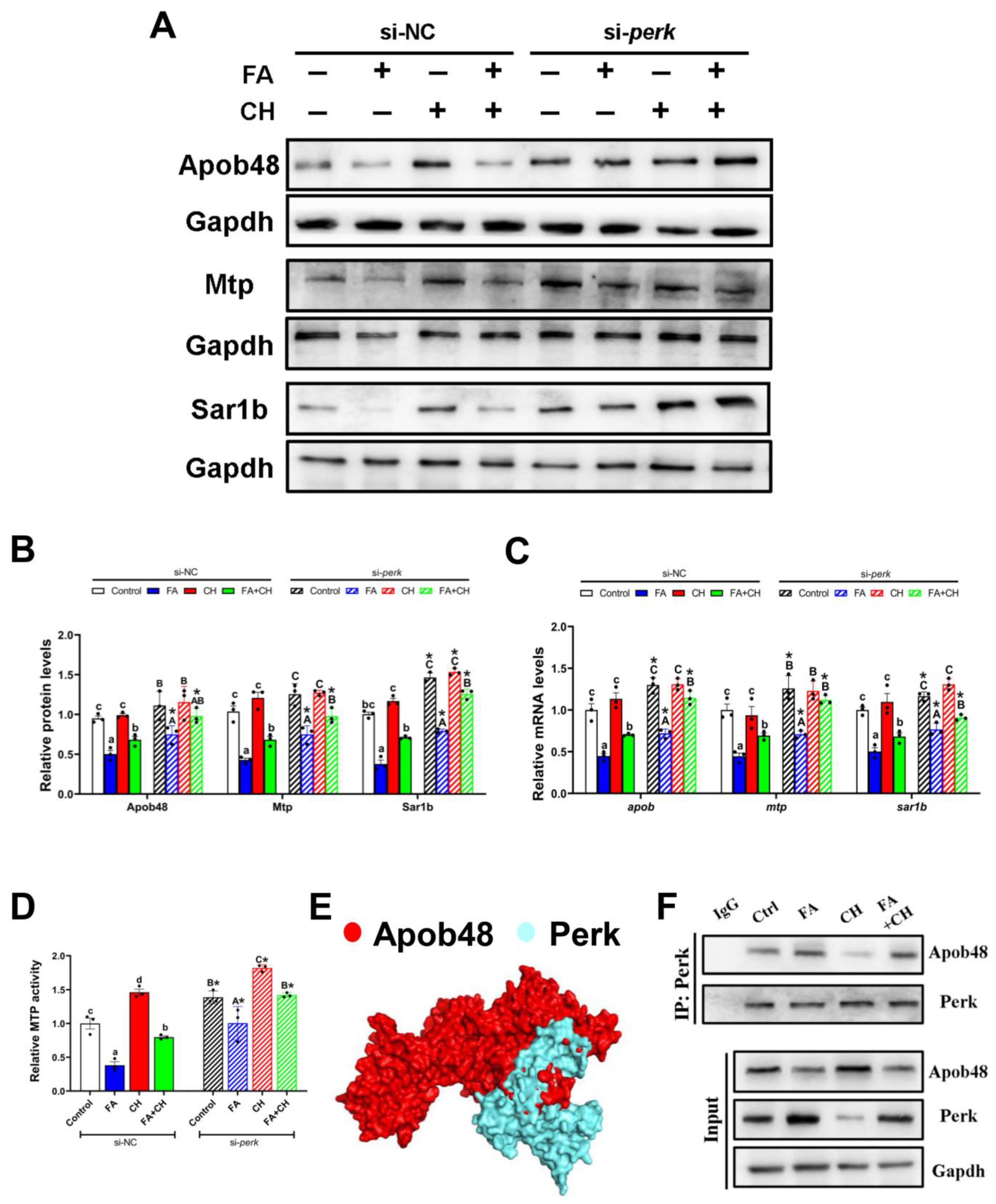
3.4. Perk-Apob48 Interaction Is Required for Perk-Signaling-Mediating CMs Assembly
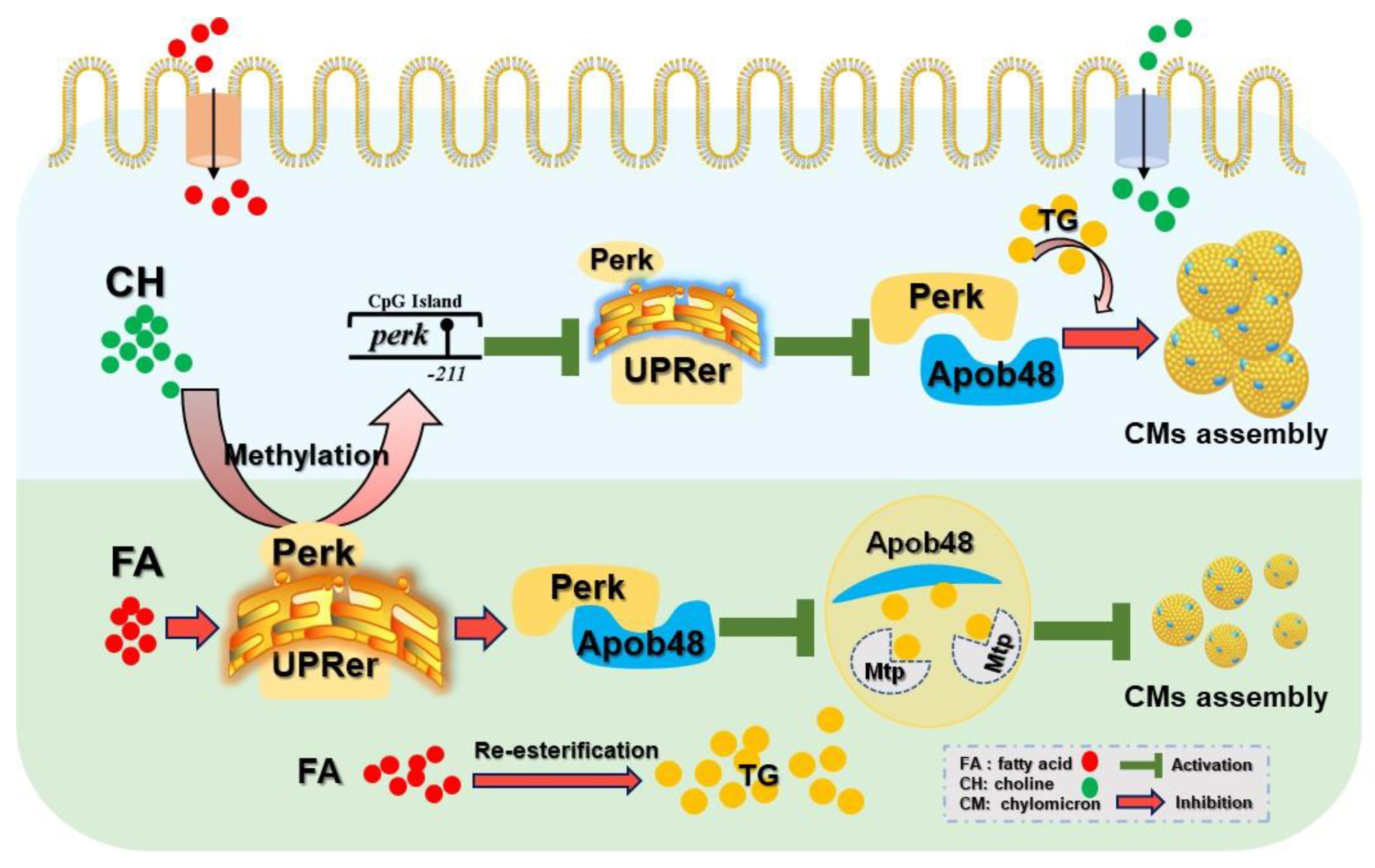
3.5. Choline Down-Regulated Perk Expression Controlled by −211 Site-Specific DNA Methylation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abumrad, N.A.; Davidson, N.O. Role of the gut in lipid homeostasis. Physiol. Rev. 2012, 92, 1061–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, R.K.; Reither, E.N.; Powers, D.A.; Yang, Y.C.; Burger, A.E.; Link, B.G. The impact of obesity on US mortality levels: The importance of age and cohort factors in population estimates. Am. J. Public Health 2013, 103, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Redgrave, T.G. Chylomicron metabolism. Biochem. Soc. Trans. 2004, 32, 79–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, S.; Xiao, C.; Morgantini, C.; Lewis, G.F. New Insights into the Regulation of Chylomicron Production. Annu. Rev. Nutr. 2015, 35, 265–294. [Google Scholar] [CrossRef]
- De Wit, N.; Derrien, M.; Bosch-Vermeulen, H.; Oosterink, E.; Keshtkar, S.; Duval, C.; De Vogel-van den Bosch, J.; Kleerebezem, M.; Müller, M.; Van der Meer, R. Saturated fat stimulates obesity and hepatic steatosis and affects gut microbiota composition by an enhanced overflow of dietary fat to the distal intestine. American journal of physiology. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G589–G599. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Stahel, P.; Lewis, G.F. Regulation of Chylomicron Secretion: Focus on Post-Assembly Mechanisms. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 487–501. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, H.; Fujimoto, K.; Cardelli, J.A.; Nutting, D.F.; Bergstedt, S.; Tso, P. Fat feeding increases size, but not number, of chylomicrons produced by small intestine. Am. J. Physiol. 1990, 259, G709–G719. [Google Scholar] [CrossRef]
- Hernández Vallejo, S.J.; Alqub, M.; Luquet, S.; Cruciani-Guglielmacci, C.; Delerive, P.; Lobaccaro, J.M.; Kalopissis, A.D.; Chambaz, J.; Rousset, M.; Lacorte, J.M. Short-term adaptation of postprandial lipoprotein secretion and intestinal gene expression to a high-fat diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G782–G792. [Google Scholar] [CrossRef] [Green Version]
- Cartwright, I.J.; Higgins, J.A. Direct evidence for a two-step assembly of ApoB48-containing lipoproteins in the lumen of the smooth endoplasmic reticulum of rabbit enterocytes. J. Biol. Chem. 2001, 276, 48048–48057. [Google Scholar] [CrossRef] [Green Version]
- Mansbach, C.M.; Siddiqi, S.A. The biogenesis of chylomicrons. Annu. Rev. Physiol. 2010, 72, 315–333. [Google Scholar] [CrossRef]
- Lo, C.C.; Coschigano, K.T. ApoB48 as an Efficient Regulator of Intestinal Lipid Transport. Front. Physiol. 2020, 11, 796. [Google Scholar] [CrossRef] [PubMed]
- Sané, A.; Ahmarani, L.; Delvin, E.; Auclair, N.; Spahis, S.; Levy, E. SAR1B GTPase is necessary to protect intestinal cells from disorders of lipid homeostasis, oxidative stress, and inflammation. J. Lipid Res. 2019, 60, 1755–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, J.; Dai, K.; Seimon, T.; Jungreis, R.; Oyadomari, M.; Kuriakose, G.; Ron, D.; Tabas, I.; Hussain, M.M. IRE1beta inhibits chylomicron production by selectively degrading MTP mRNA. Cell Metab. 2008, 7, 445–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Van Rijn, J.M.; Van Hoesel, M.; De Heus, C.; Van Vugt, A.; Klumperman, J.; Nieuwenhuis, E.; Houwen, R.; Middendorp, S. DGAT2 partially compensates for lipid-induced ER stress in human DGAT1-deficient intestinal stem cells. J. Lipid Res. 2019, 60, 1787–1800. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Nasiri-Ansari, N.; Nikolopoulou, C.; Papoutsi, K.; Kyrou, I.; Mantzoros, C.S.; Kyriakopoulos, G.; Chatzigeorgiou, A.; Kalotychou, V.; Randeva, M.S.; Chatha, K.; et al. Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE (-/-) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis. Int. J. Mol. Sci. 2021, 22, 818. [Google Scholar] [CrossRef]
- Cheng, L.; Chen, T.; Guo, M.; Liu, P.; Qiao, X.; Wei, Y.; She, J.; Li, B.; Xi, W.; Zhou, J.; et al. Glycoursodeoxycholic acid ameliorates diet-induced metabolic disorders with inhibiting endoplasmic reticulum stress. Clin. Sci. 2021, 135, 1689–1706. [Google Scholar] [CrossRef]
- Ling, S.C.; Wu, K.; Zhang, D.G.; Luo, Z. Endoplasmic Reticulum Stress-Mediated Autophagy and Apoptosis Alleviate Dietary Fat-Induced Triglyceride Accumulation in the Intestine and in Isolated Intestinal Epithelial Cells of Yellow Catfish. J. Nutr. 2019, 149, 1732–1741. [Google Scholar] [CrossRef]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, T.X.; Huang, X.; Li, Y.; Sun, T.; Zang, S.; Guan, K.L.; Xiong, Y.; Liu, J.; Yuan, H.X. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int. 2020, 40, 1378–1394. [Google Scholar] [CrossRef] [PubMed]
- Corbin, K.D.; Zeisel, S.H. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr. Opin. Gastroenterol. 2012, 28, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Wu, Z.; Xu, Y.; Liu, Y.; Liu, W.; Wang, T.; Li, C.; Zhang, C.; Yi, F.; Gao, L.; et al. Increased Tim-3 expression alleviates liver injury by regulating macrophage activation in MCD-induced NASH mice. Cell. Mol. Immunol. 2019, 16, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Su, Q.; Rutledge, A.C.; Zhang, J.; Adeli, K. Glucosamine-induced endoplasmic reticulum stress attenuates apolipoprotein B100 synthesis via PERK signaling. J. Lipid Res. 2009, 50, 1814–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Z.; Wei, C.C.; Ye, H.M.; Zhao, H.P.; Song, Y.F.; Wu, K. Effect of dietary choline levels on growth performance, lipid deposition and metabolism in juvenile yellow catfish Pelteobagrus fulvidraco. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2016, 202, 1–7. [Google Scholar] [CrossRef]
- Jin, M.; Pan, T.; Tocher, D.R.; Betancor, M.B.; Monroig, Ó.; Shen, Y.; Zhu, T.; Sun, P.; Jiao, L.; Zhou, Q. Dietary choline supplementation attenuated high-fat diet-induced inflammation through regulation of lipid metabolism and suppression of NFκB activation in juvenile black seabream (Acanthopagrus schlegelii). J. Nutr. Sci. 2019, 8, e38. [Google Scholar] [CrossRef] [Green Version]
- Park, L.K.; Friso, S.; Choi, S.W. Nutritional influences on epigenetics and age-related disease. Proc. Nutr. Soc. 2012, 71, 75–83. [Google Scholar] [CrossRef]
- Zeisel, S. Choline, Other Methyl-Donors and Epigenetics. Nutrients 2017, 9, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisel, S.H. Dietary choline deficiency causes DNA strand breaks and alters epigenetic marks on DNA and histones. Mutat. Res. Rev. Mutat. Res. 2012, 733, 34–38. [Google Scholar] [CrossRef]
- Yoon, A.; Tammen, S.A.; Park, S.; Han, S.N.; Choi, S.W. Genome-wide hepatic DNA methylation changes in high-fat diet-induced obese mice. Nutr. Res. Pract. 2017, 11, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhang, P.; Luo, J.; Shen, L.; Zhang, S.; Gu, H.; He, J.; Wang, L.; Zhao, X.; Gan, M.; et al. Dietary betaine prevents obesity through gut microbiota-drived microRNA-378a family. Gut Microbes. 2021, 13, 1862612. [Google Scholar] [CrossRef]
- Da Silva, R.P.; Kelly, K.B.; Lewis, E.D.; Leonard, K.A.; Goruk, S.; Curtis, J.M.; Vine, D.F.; Proctor, S.D.; Field, C.J.; Jacobs, R.L. Choline deficiency impairs intestinal lipid metabolism in the lactating rat. J. Nutr. Biochem. 2015, 26, 1077–1083. [Google Scholar] [CrossRef]
- Lee, J.; Ridgway, N.D. Phosphatidylcholine synthesis regulates triglyceride storage and chylomicron secretion by Caco2 cells. J. Lipid Res. 2018, 59, 1940–1950. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Newberry, E.P.; Brunt, E.M.; Ballentine, S.J.; Soleymanjahi, S.; Molitor, E.A.; Davidson, N.O. Inhibition of chylomicron assembly leads to dissociation of hepatic steatosis from inflammation and fibrosis. J. Lipid Res. 2021, 62, 100123. [Google Scholar] [CrossRef]
- Gong, G.; Dan, C.; Xiao, S.; Guo, W.; Huang, P.; Xiong, Y.; Wu, J.; He, Y.; Zhang, J.; Li, X.; et al. Chromosomal-level assembly of yellow catfish genome using third-generation DNA sequencing and Hi-C analysis. GigaScience 2018, 7, giy120. [Google Scholar] [CrossRef]
- Sato, Y.; Hashiguchi, Y.; Nishida, M. Temporal pattern of loss/persistence of duplicate genes involved in signal transduction and metabolic pathways after teleost-specific genome duplication. BMC Evol. Biol. 2009, 9, 127. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.F.; Zheng, H.; Luo, Z.; Hogstrand, C.; Bai, Z.Y.; Wei, X.L. Dietary Choline Alleviates High-Fat Diet-Induced Hepatic Lipid Dysregulation via UPRmt Modulated by SIRT3-Mediated mtHSP70 Deacetylation. Int J Mol Sci. 2022, 23, 4204. [Google Scholar] [CrossRef]
- Zhao, T.; Yang, S.B.; Chen, G.H.; Xu, Y.H.; Xu, Y.C.; Luo, Z. Dietary Glucose Increases Glucose Absorption and Lipid Deposition via SGLT1/2 Signaling and Acetylated ChREBP in the Intestine and Isolated Intestinal Epithelial Cells of Yellow Catfish. J. Nutr. 2020, 150, 1790–1798. [Google Scholar] [CrossRef]
- Wei, C.C.; Luo, Z.; Hogstrand, C.; Xu, Y.H.; Wu, L.X.; Chen, G.H.; Pan, Y.X.; Song, Y.F. Zinc reduces hepatic lipid deposition and activates lipophagy via Zn2+/MTF-1/PPARα and Ca2+/CaMKKβ/AMPK pathways. FASEB J. 2018, 32, 6666–6680. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Xu, Y.C.; Xu, Y.H.; Zhao, T.; Wu, L.X.; Yang, S.B.; Luo, Z. Waterborne Cu exposure increased lipid deposition and lipogenesis by affecting Wnt/β-catenin pathway and the β-catenin acetylation levels of grass carp Ctenopharyngodon idella. Environ. Pollut. 2020, 263, 114420. [Google Scholar] [CrossRef]
- Traber, M.G.; Kayden, H.J.; Rindler, M.J. Polarized secretion of newly synthesized lipoproteins by the Caco-2 human intestinal cell line. J. Lipid Res. 1987, 28, 1350–1363. [Google Scholar] [CrossRef]
- Zhong, C.C.; Zhao, T.; Hogstrand, C.; Chen, F.; Song, C.C.; Luo, Z. Copper (Cu) induced changes of lipid metabolism through oxidative stress-mediated autophagy and Nrf2/PPARγ pathways. J. Nutr. Biochem. 2022, 100, 108883. [Google Scholar] [CrossRef]
- Nakanishi, T.; Fukui, H.; Wang, X.; Nishiumi, S.; Yokota, H.; Makizaki, Y.; Tanaka, Y.; Ohno, H.; Tomita, T.; Oshima, T.; et al. Effect of a High-Fat Diet on the Small-Intestinal Environment and Mucosal Integrity in the Gut-Liver Axis. Cells 2021, 10, 3168. [Google Scholar] [CrossRef]
- Xiao, C.; Stahel, P.; Nahmias, A.; Lewis, G.F. Emerging Role of Lymphatics in the Regulation of Intestinal Lipid Mobilization. Front. Physiol. 2020, 10, 1604. [Google Scholar] [CrossRef]
- Song, Y.F.; Huang, C.; Shi, X.; Pan, Y.X.; Liu, X.; Luo, Z. Endoplasmic reticulum stress and dysregulation of calcium homeostasis mediate Cu-induced alteration in hepatic lipid metabolism of javelin goby Synechogobius hasta. Aquat. Toxicol. 2016, 175, 20–29. [Google Scholar] [CrossRef]
- Sivanesan, S.; Taylor, A.; Zhang, J.; Bakovic, M. Betaine and Choline Improve Lipid Homeostasis in Obesity by Participation in Mitochondrial Oxidative Demethylation. Front. Nutr. 2018, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- McKee, S.E.; Grissom, N.M.; Herdt, C.T.; Reyes, T.M. Methyl donor supplementation alters cognitive performance and motivation in female offspring from high-fat diet-fed dams. FASEB J. 2017, 31, 2352–2363. [Google Scholar] [CrossRef] [Green Version]
- Wankhade, U.D.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Thakali, K.M.; Shankar, K. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS ONE 2017, 12, e0175675. [Google Scholar] [CrossRef]
- Zhu, J.; Lu, T.; Chen, F.; Yan, J.; Chen, F.; Zhang, Q.; Wang, J.; Yan, W.; Yu, T.; Tang, Q.; et al. Choline Protects Against Intestinal Failure-Associated Liver Disease in Parenteral Nutrition-Fed Immature Rats. JPEN J. Parenter. Enteral. Nutr. 2018, 42, 436–445. [Google Scholar] [CrossRef]
- Lyall, M.J.; Cartier, J.; Richards, J.A.; Cobice, D.; Thomson, J.P.; Meehan, R.R.; Anderton, S.M.; Drake, A.J. Methyl donor deficient diets cause distinct alterations in lipid metabolism but are poorly representative of human NAFLD. Wellcome Open Res. 2017, 2, 67. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, Z.-Y.; Zheng, H.; Luo, Z.; Hogstrand, C.; Wang, L.-J.; Song, Y.-F. Dietary Choline Mitigates High-Fat Diet-Impaired Chylomicrons Assembly via UPRer Modulated by perk DNA Methylation. Cells 2022, 11, 3848. https://doi.org/10.3390/cells11233848
Bai Z-Y, Zheng H, Luo Z, Hogstrand C, Wang L-J, Song Y-F. Dietary Choline Mitigates High-Fat Diet-Impaired Chylomicrons Assembly via UPRer Modulated by perk DNA Methylation. Cells. 2022; 11(23):3848. https://doi.org/10.3390/cells11233848
Chicago/Turabian StyleBai, Zhen-Yu, Hua Zheng, Zhi Luo, Christer Hogstrand, Ling-Jiao Wang, and Yu-Feng Song. 2022. "Dietary Choline Mitigates High-Fat Diet-Impaired Chylomicrons Assembly via UPRer Modulated by perk DNA Methylation" Cells 11, no. 23: 3848. https://doi.org/10.3390/cells11233848