Abstract
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a master regulator of the endogenous antioxidant response to reactive oxygen species as well as a controller of Phase II detoxification in response to xenobiotics. This amenity to specific external manipulation exploits the binding affinity of Nrf2 for its constitutive repressor and degradation facilitator Kelch-like erythroid cell-derived protein with CNC homology-associated protein 1 (Keap1). Derived from both natural and synthesized origins, these compounds have been extensively tested without definitive beneficial results. Unfortunately, multiple terminated trials have shown a negative side to Nrf2 with regard to cardiac pathologies while animal-based studies have demonstrated cardiomyocyte hypertrophy and heart failure after chronic Nrf2 upregulation. Putatively based on autophagic control of Nrf2 activity-modulating upstream factors, new evidence of miRNA involvement has added complexity to this mechanism. What follows is an extensive survey of Nrf2-regulating exogenous compounds that may promote cardiomyopathy, clinical trial evidence, and a comparison to exercise-induced factors that also upregulate Nrf2 while preventing cardiac pathologies.
1. Introduction
Reactive oxygen species (ROS) are both a normal byproduct of mitochondrial metabolism and an endproduct of oxidative biochemical reactions in the cell. Balanced levels of subcellular compartmental ROS are important for normal cellular functions, whereas dysregulated ROS, usually caused by relative insufficiency or impairment of the endogenous antioxidant defense system, attack cellular components leading to cellular damage and death, a state referred as to oxidative stress. To maintain cellular redox homeostasis and neutralize uncontrolled ROS, conserved antioxidant defense enzymes are placed under the control of the nuclear factor erythroid 2-related factor 2 (Nrf2) gene which is constitutively expressed in all higher-order animals. This gene, as a master antioxidant transcription factor, is responsible for global antioxidant activity in response to internally and externally sourced ROS threats but also modulates such species to maintain important intracellular second messenger capability. As the inhibitor of Nrf2, Kelch-like erythroid cell-derived protein with CNC homology-associated protein 1 (Keap1), is amenable to attack by exogenous compounds, research has focused its efforts to increase Nrf2 activity via direct interaction with Keap1. These compounds, such as oleanic acid derivative 2-cyano-3,12-dioxoolean-1,9- dien-28-oic acid (CDDO, bardoloxone), have been intensely studied in animal models and human trials as potential defense agents oxidative stress-associated diseases, such as cancer, chronic kidney disease, fatty liver, and endometriosis. Unfortunately, human trials employing CDDO and similar compounds for amelioration of these maladies have met with ambiguous and often disappointing results. Furthermore, multiple trials throughout 2007–2014 were terminated as unforeseen myocardial pathologies resulted. What follows is a survey of Nrf2, its endogenous regulation, action, and potential for exogenous modulation. Additionally, current clinical trial results are presented and analyzed for characteristics of Nrf2 upregulation that result in possible cardiac issues. Finally, comparisons between these pharmaceutical regulators and exercise are made from mechanistic and physiological viewpoints to elucidate the differences between endogenous and exogenous regulatory effects on Nrf2 and cardiac health.
2. Nrf2 Composition
Nrf2 is a basic leucine zipper (bZIP) transcription factor on chromosome 2 in humans, comprised of a common, conserved Cap ‘n’ Collar (CNC) motif of 43 amino acids close to the DNA binding domain [1]. NRF2 consists of 6 exons, encoding 7 Nrf2-ECH homology (Neh) domains, and generates a 67.8 kDa protein from a 605 aa sequence and 2859 bp mRNA strand [2,3,4]. These Neh domains are specific for protein–protein interactions, especially regulatory, degradation, and translocation proteins (Table 1). The half-life of Nrf2 in the cytosol may be as little as 10 min if redox homeostasis is present or as long as 40 min under oxidative stress, relying on a sensitive Neh2-ETGE hinge region and redox-insensitive (but GSK3 interacting) Neh6 region to modulate binding to Keap1 and ubiquitin ligases [5,6]. Nrf2 Neh regions have been extensively illustrated, reviewed, and mapped but Nrf2 crystalline structure without Keap1 binding is sparse, indicating the importance of Keap1 binding in Nrf2 conformation [7,8]. Diverse variants of Nrf2 have been discovered but 9 are predicted to mediate a disease process and 8 mutagenic variants experience either loss of Keap1 binding or function (Table 2) [7].

Table 1.
Neh regions of Nrf2 and their functions [9,10,11,12].
Table 2.
Nrf2 variants associated with disease processes [7].
2.1. Nrf2 Activation Mechanism
The Nrf2 activation mechanism has been canonically divided into 4 stages, highlighted by interactions with ROS-sensitive regulatory elements and translocation machinery. These stages are basal expression/repression, pre-induction, full induction, and post-induction [8].
2.1.1. Basal Expression/Repression of Nrf2: Keap1, ROS, and Autophagy
Like the rest of the CNC family of transcription factors, Nrf2 is activated under stress, namely oxidative stress, and is related to a family of similar stress-response factors (e.g., Nrf1) [1]. Exercise, especially aerobic exercise, is also a potent inducer of Nrf2 (see Section 4.1). It is constitutively expressed and maintains its own 9nt upstream ARE sequence [13]. Levels of Nrf2 are tightly controlled by RONS-sensitive Keap1 through its modulation of K48-linked ubiquitination, together with backup systems such as β-TrCP and Hrd1 (see below).
Keap1 is a 70 kDa protein with a long 12.7 h half-life that localizes to the cytoplasm [7]. It is comprised of a BTB (Broad, Tramtrack and Bric-a-brac region) domain and Kelch repeats that bind directly to Nrf2-Neh2 in a 6-blade, β-propeller configuration that permits dimerization of 2 Keap1 molecules to each Nrf2 molecule in a hinged-capture structure with ETGE and DLG regions on the C-terminal end of Nrf2 to act as pivoting attachment points to Keap1 [4,7]. Under basal conditions, the conformational change induced by the Keap1-Nrf2 complex, NEDD8, and ubiquitin E3 ligase CUL3 exposes lysine residues within Neh2 (and possibly Neh6) to attack by a K48 polyubiquitination complex consisting of CUL3 and ring ligase RBX1 that bind to the BTB region of Keap1 before activation [7,14]. Subsequent proteasomal degradation of K48-polyubiquitinated Nrf2 then occurs within the cytoplasm. The binding affinity of Kesp1 to Nrf2 has been experimentally reported as KD 20 nM and, as such, spontaneous dissociation is unlikely [15]. However, 27 cysteine residues of Keap1 are vulnerable to attack by endogenous and exogenous reactive species, particularly C151 in the homodimerizing BTB region, that dissociate the CUL3 ubiquitin adaptor from the complex, allowing Nrf2 to escape polyubiquitination and begin translocation to the nucleus [4,16,17]. The p62/mTORC-1 dependent machinery, activated by autophagy, can also repress Keap1 by degrading it in the autophagic pathway [18,19]. Other p62-associated molecules, such as TFEB, can also protect Nrf2 by reducing ubiquitination through suppression of E3 ligase complex members (DACF11) while upregulating p62 to inhibit Keap1 binding to Nrf2 [20]. Of note, Nrf2 can also be repressed in a Keap1-independent manner by β-TrCP, which binds to Nrf2-Neh6 in a GSK-3 phosphorylation-dependent manner to facilitate SKP1-CUL1-RBX1/ROC1 ubiquitination [19,21]. E3 ubiquitin protein ligase HRD1 is also involved in Keap1 independent Nrf2 degradation [6]. Constitutively expressed proteins that generate ROS, such as NADPH oxidase-4 (NOX4) are also important in activating Nrf2 translocation [22].
Recent evidence has shown miRNA involvement in post-transcriptional regulation of Nrf2, with miRs -144, -28, -34, and -93 (among others) shown to decrease Nrf2 activity in animal models, while HuR and AUF1 RNA-binding proteins contribute to export and stabilization of the Nrf2 mRNA [3]. Constitutive expression and a short half-life, coupled with exquisitely sensitive, cysteine-based ROS sensors on Keap1 and links to autophagy and GSK-3 pathways, give Nrf2 the speed to react and fluctuate to maintain redox homeostasis under changing conditions.
2.1.2. Nrf2 Action: Pre-Induction
After release, PI3K phosphorylation and Importin a5/B1 binding to specific nuclear localization signals on Nrf2 C- and N-terminal regions (Neh2 aa 42-53 and Neh3 aa 587-593) occurs to facilitate nuclear entry [23,24]. AMPK aids in nuclear accumulation by phosphorylating Ser558 to prevent export [24]. Nrf2 then begins to complex with Maf family members, Creb, and p300 adaptors to form a transcription-initiating complex [10].
2.1.3. Nrf2 Full Induction, Transcription, and Purpose of Target Genes
Once translocation and complex formation are complete, the Nrf2-Maf-Cred-p300 complex binds a wide spectrum of antioxidant response elements (ARE), located 40 to 200 nucleotides upstream of transcription start sites, that encode Phase II detoxification, antioxidant enzyme, energy metabolism, and diverse other genes [13]. These short (9nt) sequences vary by gene and have been extensively reviewed by Raghunath et al. [13]. The Nrf2-Maf-Cred-p300 complex has been reported to control transcription of over 1000 genes and Table 3 shows a selection of genes related to the antioxidant and proliferation responses [25,26]. Of importance are catalases, glutathione S-transferases and cysteine-rich thioredoxins that detoxify xenobiotics, proteins with disulfide bonds, and ingested toxins, as well as ROS from mitochondrial respiration [27].
Of note, Bach1, which competitively binds with ARE sequences in concert with small Maf molecules, has emerged as an important modulator of Nrf2 transcriptional activity since it can directly interact to sense heme and act as a feedback inhibitor for promotion of HO-1 and NQO1 [28,29]. Bach1 is a member of the same CNC family as Nrf2 and is involved in induction of iron-induced immune cell apoptosis (ferroptosis) through prevention of antioxidant genes that counter iron-induced ROS [30].
Not limited to antioxidant defense alone, Nrf2 controls genes from multiple pathways as seen in the recent discovery of Nrf2-mediated cardiac hypertrophy from exogenous upregulators. Nrf2 controls cellular proliferation through PHGDH, PSAT1, SHMT1 and other ser/gly synthesis genes via interaction with ATF4 [25,31,32]. It additionally maintains a favorable redox status to facilitate mRNA translation, upregulates glycolysis/energy metabolism, and also contributes to stem cell viability through ROS regulation plus NOTCH and SIRT1 expression [3,33,34,35,36,37,38]. Thus, Nrf2 is an important co-initiator of the proliferative machinery, energy production, and facilitative redox control needed to prevent ROS damage from increased cellular growth and proliferation. It is these non-ARE effects that may be responsible for the cardiac maladaptation and hypertrophy seen in studies of exogenous Nrf2 upregulators.
Table 3.
Select Genes Controlled by Nrf2 [39].
Table 3.
Select Genes Controlled by Nrf2 [39].
| Function | Gene | Description | Ref. |
|---|---|---|---|
| Detoxification Phase II | AHR | Aryl hydrocarbon receptor | [2] |
| CYP1B1 | Cytochrome P450 Family 1 Subfamily B Member 1 | [2] | |
| ALDH3A2 | Aldehyde Dehydrogenase 3 Family Member A2 | [2] | |
| NQO1 | NAD(P)H Quinone Dehydrogenase 1 | [2] | |
| AKR1C1 | Aldo-Keto Reductase Family 1 Member C1 | [40] | |
| GSTM3 | Glutathione S-Transferase Mu 3 | [40] | |
| Antioxidant Defense | GPX4 | Glutathione Peroxidase 4 | [2] |
| GSR1 | Glutathione reductase, mitochondrial | [2] | |
| TXN1 | Thioredoxin | [2] | |
| PRDX1 | Peroxiredoxin 1 | [2] | |
| SRXN1 | Sulfiredoxin 1 | [2] | |
| SOD1/2 | Superoxide dismutase 1 and 2 | [41] | |
| HO-1 | Heme Oxygenase 1 | [42] | |
| GSTM3 | Glutathione S-Transferase Mu 3 | [40] | |
| Pentose Phosphate Pathway | G6PD | Glucose-6-Phosphate Dehydrogenase | [40] |
| PGD | Phosphogluconate dehydrogenase | [3] | |
| TKT | Transketolase | [3] | |
| Serine/Glycine Biosynthesis | PHGDH | Phosphoglycerate Dehydrogenase | [31] |
| PSAT1 | Phosphoserine Aminotransferase 1 | [31] | |
| SHMT1/2 | Serine Hydroxymethyltransferase 1/2 | [31] | |
| Membrane Trafficking | RAB6B | Ras-related protein Rab-6B | [40] |
| Deubiquitination | UCH-L1 | Ubiquitin C-terminal hydrolase L1 | [40] |
| Zinc Finger Protein | TRIM16L | Tripartite motif-containing protein 16 | [40] |
| Glycolysis/ Glycogen Synthesis | HK1/2 | Hexokinase 1 and 2 | [3] |
| GP11 | Glucose phosphate isomerase 1 | [3] | |
| ALDA | Fructose-bisphosphate aldolase A | [3] | |
| ENO1 | Enolase 1 | [3] | |
| PKM2 | Pyruvate kinase muscle isoform 2 | [3] | |
| GLUT1 | Glucose transporter 1 | [3] |
2.1.4. Nrf2 Post-Induction: Proteasomal Degradation
The 4–5 h window for Nrf2 transcriptional promotion is tightly controlled by phosphorylation, as GSK3 can phosphorylate Nrf2 to reduce its activity and kinases (Fyn and MAPK) prepare Nrf2 for nuclear export [24]. Fyn kinase interacts specifically with Tyr568 on Nrf2 to prepare it for export and another study by Li et al. has also found a leucine-enriched sequence (537-LKKQLSTLYL-546) resident in the Nrf2-Neh1 region that aids in CRM1 interaction for nuclear export [43]. A Neh6 region, containing a GSK3-interacting domain, was reported by McMahon et al. to promote destabilization of Nrf2 in a redox-insensitive manner [5]).
Once exported, the ubiquitin-proteasome complex is free to bind with Nrf2 and degrade it. Recent evidence has also hinted that the nucleus may play a role in degradation through the involvement of promyelocytic leukemia-nuclear bodies (PML-NB), comprised of PML and Sp100 proteins, in a process that sumoylates Nrf2 to render it susceptible to SUMO-targeting ubiquitin ligases [44]. Such regulation has been found to occur at 532-LKDE-535 and putatively at Lys100 (in mice), functioning to stabilize Nrf2 within the nucleus [45]. Thus, multiple degradation domains and pathways, with both redox-sensitive and -insensitive activity, ensures that Nrf2 can be quickly targeted for recycling to maintain rapid turnover and tight control of intracellular Nrf2 protein levels.
3. Effects of Nrf2 in the Heart versus Other Systems
The ability to engage a panoply of antioxidant and pro-growth factors upon ROS challenge, whether from endogenous sources or xenobiotics, makes Nrf2 highly desirable for manipulation to prevent oxidative damage. However, exogenous upregulation of Nrf2 beyond the control of repressive/degradation machinery may be deleterious as seen in studies linking upregulation of Nrf2 to cardiac hypertrophy and immune evasion/chemotherapy resistance in cancers [46,47]. What follows is a brief survey of the role of Nrf2 in the heart with a comparison to the kidneys to evaluate any potential side effects of exogenous Nrf2 enhancers.
3.1. Nrf2 in the Myocardium: Not a Silver Bullet
The heart is obligately aerobic and relies on oxidative phosphorylation to generate the biochemical energy needed for a lifetime of pumping. The coronary arteries supply oxygenated blood to the heart during diastole and increases in oxygen demand by the myocardium are directly related to the heart rate (higher rate = higher oxygen demand and shorter diastole for coronary supply) and saturation of blood by oxygen (to prevent hypoxia). Even at rest, the myocardium consumes 8 to 13 mL of oxygen per 100 g of tissue per minute and ROS from mitochondrial respiration and pro-ROS proteins, such as Nox4, create a pro-oxidative state that requires constant rebalancing to maintain redox homeostasis [48,49]). Xenobiotics may also introduce ROS either by direct chemical action (e.g., nitrosamines from cigarette smoke, fermented foods, or cured meats) or immune response. However, since ROS function as a second-messenger system and have been implicated as crucial regulators of stem cell differentiation and apoptosis/necrosis, tight regulation of the Nrf2-mediated antioxidant response (e.g., via Keap1 direct and Bach1 competitive pathways) is required to maintain such basal messenger activity. Cardiomyocyte differentiation, in particular, is sensitive to ROS, requiring it for progression to maturity, and cardiac-resident stem cells in adults may be similarly affected by imbalanced redox homeostasis, driving them towards hypertrophic or synthetic phenotypes [50].
Of current controversy in cardiac research is the involvement of Nrf2 as a pro-hypertrophic, factor in progressive heart failure. On one side, numerous reports have linked Nrf2 deficiencies to ROS-mediated cardiac hypertrophy related to Angiotensin II, IL-6-mediated inflammation, aortic constriction (TGFβ1/SMAD2 signaling), and obesity-related stress [51,52,53]. Diverse other reports have detailed the role of Nrf2 in preventing cardiomyocyte necrosis, hypertrophy, and fibrosis of the myocardium due to ROS while antioxidant response proteins (e.g., NQO1, SOD1, GPX4) have been found at low expression levels under ischemic cardiomyopathy conditions [54,55]). However, recent evidence that Nrf2 induces progressively maladapted remodeling in the absence of functional autophagy casts doubt on the exploitation of Nrf2 in patients with metabolic disorders or heart disease (Figure 1) [46]. Reports from the Cui research group have indicated that Fyn-mediated nuclear export inhibition is to blame but other yet-discovered factors may also play crucial roles in pathogenesis [46]. Future studies on the effect of autophagy and other regulatory modalities (methylation, sumoylation, etc.) will delineate the thresholds beyond which Nrf2 enhancement becomes problematic for the heart.
Ostensibly, boosting Nrf2 will increase the total antioxidant capacity within the heart and neutralize ROS that perpetuate necrotic and fibrotic pathways, leading to the concept of “the more antioxidant capacity, the better”. In spite of this theory, results from well-controlled clinical trials of supplemental antioxidants (selenium, vitamin E, beta-carotene, etc.) have returned disappointing results where risk was either unchanged or even enhanced [56]. Results from previously reviewed meta-studies with 156,663 and 188,209 total participants found no significant effects of antioxidant/vitamin supplements on cardiovascular risk [56]). However, a recent meta-study of selenium and other antioxidants only found significant risk reduction for selenium across 43 studies (possibly because such minerals, similarly to zinc, are important constituents of antioxidant enzymes and not activators of Nrf2) [57]). Consequently, the Selenium and Vitamin E Cancer Prevention trial (N = 35,533) found that supplementation increased diabetes and prostate cancer risks, while a beta-carotene study did find inverse relationships with lower cardiovascular risk but could not completely rule out the effects of confounding variables (i.e., accidents and injuries) [57,58]. In general, antioxidants have proven to be poor substitutes for generally healthy lifestyle habits (e.g., no tobacco use, moderate diet, moderate exercise, stress reduction, good sleep habits) and excessive antioxidant use is associated with increased all-cause mortality (vitamin E), oxidative stress (ascorbic acid), and cancer risk (vitamin A) [59].
In similar fashion, Nrf2 exogenous enhancers have not shown promise in either preventing or treating cardiovascular diseases and several trials have ended early because of deleterious heart effects after treatment (see Section 4.2 and Section 4.3). For this reason, external and sustained enhancement of the antioxidant response out of context with other regulatory factors (e.g., autophagy) could counterintuitively damage the myocardium through pathways not yet fully elucidated (Figure 1). More antioxidant capacity is, in light of these studies, definitely not better.
3.2. Nrf2 in the Failing Heart: Autophagy as a Keystone Mechanism
Aging and failing hearts experience stiffening from fibrosis caused by immune responses to myocardial necrosis, increased ROS from aging and senescent mitochondria, lipofuscin accumulation from lysosomal degradation, deficiencies in calmodulin signaling/calcium flux (RYR2, SERCA2a) and increased maladaptive remodeling due to high blood pressure that stems from glucose dysregulation and hyperkalemia [60,61,62]. Additionally, autophagic capacity drops as suppression factors like mTOR are overexpressed by chronically high AKT levels while chronic IGF-1 expression, long touted as a youth-sustaining factor, paradoxically ages the heart rapidly as it has been shown to downregulate autophagy by suppression of autophagosome formation and increases in AKT/mTOR [63,64].
Hyperglycemia has been shown to modulate autophagy via AMPK and ROS induction of the ERK/JNK-p53 mechanism [65,66,67]. Additionally, fasting is a potent activator of autophagy even under increased peroxide generation by mitochondria in animals [66,68]. In type 2 diabetics, while initially protective, mitophagy (i.e., autophagy of damaged mitochondria) may eventually drive cells towards reduced energy as mitochondria are damaged by increased metabolic activity and are recycled faster than replacement [69]. However, the loss of autophagic capacity, especially in pancreatic β cells and diabetic hearts, may also be important in progression to end-stage disease [69,70]). Thus, patients who do not possess a fully intact autophagy capacity (e.g., heart failure or type 2 diabetics) may be harmed by artificial Nrf2 enhancement.
Wu et al. recently reported a putative mechanism for this effect in pressure-overloaded hearts that involves dysfunctional autophagy, restricting phosphorylated Fyn and ERK from translocating to the nucleus and downregulating Nrf2 activity that would otherwise restrict angiotensin expression [71]. In such cases, subsequent activation of angiotensin II (Ang-II) receptors by Ang-II production would increase blood pressure and eventual hypertrophy [71]. Additionally, interactions between autophagic control factor p62 and Keap1 mean that reduction in upstream p62/AKT/mTOR result in increased Nrf2 activation and further exacerbation of Ang-II-induced maladaptive remodeling (Figure 1) [72].

Figure 1.
The Vicious Cycle of Nrf2 in Cardiac Hypertrophy. Aged and failing hearts have dysfunctional autophagy (bottom), which cannot downregulate Nrf2 transcription of Angiotensinogen and AngII, increasing blood pressure and mechanical induction of hypertrophy, pro-hypertrophic miRNA, necrosis, and fibroblast activation (right). Hypertrophic cells increase ROS output and decompensation within the heart occurs, increasing the ischemic microenvironment and generating even more ROS in a vicious cycle (top) [71,72]. Created in BioRender.com.
Figure 1.
The Vicious Cycle of Nrf2 in Cardiac Hypertrophy. Aged and failing hearts have dysfunctional autophagy (bottom), which cannot downregulate Nrf2 transcription of Angiotensinogen and AngII, increasing blood pressure and mechanical induction of hypertrophy, pro-hypertrophic miRNA, necrosis, and fibroblast activation (right). Hypertrophic cells increase ROS output and decompensation within the heart occurs, increasing the ischemic microenvironment and generating even more ROS in a vicious cycle (top) [71,72]. Created in BioRender.com.

3.3. Nrf2 in the Kidneys
Because of their role in blood filtering and dependence on over ¼ of cardiac output to function, the kidneys are inextricably linked to the heart [73]. As in the heart, Nrf2 plays an important role in defense against bloodborne sources of ROS (e.g., hyperglycemia, nitrosamines, xenobiotics) and the dense, fine capillary network within kidneys is easily damaged. The primary basal ROS within the kidney are produced by epithelial cells that use mitochondrial respiration for ATP generation that drives glomerular filtration [74]. Such ROS are also important messengers in secondary pathways, including hormone secretion and vascular reactivity [73]. Transient ischemia from heart failure, atherosclerosis, or chronic kidney diseases happens from occluded blood flow and creates excessive ROS from reperfusion injury that can easily damage delicate epithelial cells within the glomerular network and release inflammatory factors that locally propagate ROS production [73]. To compensate, Nrf2, in addition to its suite of ARE-mediated antioxidant enzymes, also produces pentose phosphate that generates NADPH which serves as a local and direct antioxidant [73]. Nrf2 is also protective against heavy metal insult from cadmium or arsenic and glutathione production by Nrf2 may attenuate damage from hyperglycemia in addition to reduction in inflammation through cytokine and NLRP3 inflammasome suppression [75]). Once transcription has been activated, Nrf2 can then be degraded in its canonical manner (proteasome via β-TrCP or Hrd1) and is thus prevented from overaccumulation [75,76]).
Unfortunately, as in the heart, Nrf2 has the potential to inflict harm as a report by Rush et al. (as reviewed by Nezu and Suzuki) revealed that sustained increases of Nrf2 in injured kidneys from treatment with bardoloxone-methyl (CDDO-Me) results in proteinuria and malformed podocyte feet [76,77]. This was thought to be due to inactivation of Keap1 suppression of Nrf2 by electrophilic effect [76]. Thus, in light of the links between deficient autophagy, Nrf2, and myocardial maladaptation, similarly suppressed autophagy in aged or damaged kidneys may also modulate chronic status and permanent damage. However, as sustained autophagy after acute kidney injury has been shown to promote fibrosis, further kidney-specific studies on the relationship between autophagy, Nrf2, and kidney fibrosis with regard to acute and chronic kidney injury microenvironments are required [78].
4. Clinical Nrf2 Modulators
Upregulation or downregulation of Nrf2 activity by exogenous modulators can be separated into 3 groups, namely those that increase Nrf2 levels, those that facilitate Nrf2 transcription, or those that affect translocation/stability. Most electrophilic modulators derived from triterpinoids, organosulfur compounds, and stilbenes act directly on Keap1 Cys151, 273, 288 or combinations of these or other cysteines to reduce Keap1 binding affinity to cytosolic Nrf2 [79]. However, inorganic compounds, such as LiCl, may alternately activate Nrf2 through GSK-3 suppression and DHA may activate the p65/MAPK/IKK-mediated upregulation of Nrf2 [80]. By suppression of Keap1 expression, chlorogenic acid compounds (such as CGA) may also increase cytosolic free Nrf2 [81]. Other compounds, such as the fumaric acid derivatives (Tecfidera and others), increase Nrf2 activity by increasing the export of the Bach1 competitive transcription factor [82]). These compounds have been extensively reviewed [83,84]. Table 4 contains a list of currently known and reported regulators.
Table 4.
Exogenous Activators/Suppressors of Nrf2 [79].
4.1. Exercise versus Exogenous Regulation
Exercise is universally accepted as heart-healthy, countering cardiomyopathy and resulting in a dramatic upregulation of Nrf2 and associated downstream elements [100]. In this fashion, Nrf2 is closely tied to muscular endurance against ROS and other associated oxidative byproducts of myocyte mitochondrial respiration. Within skeletal muscle itself, Nrf2 is part of the antioxidant response to ROS generated by aerobic respiration within striated myocytes and helps to reduce citrate synthase and COX-mediated inflammation [101]. In cases of exercise, muscle contractions (100 and 50 Hz) and long periods of aerobic exertion have been found to stimulate Nrf2 response [101]. So why does exercise-mediated Nrf2 elevation prevent cardiomyopathy instead of possibly promoting it as seen with exogenous compounds? The answer may lie in both autophagy competence and miRNA-mediated control of myocyte cell size and growth.
In the heart, murine models of exercise have reported that moderate exercise stabilizes the Nrf2 promoter in myocardial cells while it also increases AMPK phosphatase activity on mTOR to suppress its negative regulation of autophagic induction [100,102]. Additionally, it upregulates pro-autophagic factors FOXO3 and HIF-1 while also upregulating mitochondrial biogenesis factor PGC-1α through increased AMPK activity [102,103]. The simultaneous increase of autophagy with induction of short-term, shear-stress mediated pumping action, activates both SIRT1 transcription pathways as well as production of numerous anti-hypertrophic miRNAs (miR-1, -133, -26 and many others) that can, in the case of miR-1, inhibit PP2A and regulate heart rhythm or, in the case of miR-133, control hypertrophy by suppressing RHOA, NELF-A/WHSC2, and CDC42 [104,105,106,107]. Counterintuitively, pro-hypertrophic miRNAs are also expressed simultaneously (miR-143, -103, 130a, and others) that function to regulate both differentiation and cardiomyocyte morphology [108]. Furthermore, miR-29 has been specifically noted to downregulate collagen formation (reducing fibrosis) and miR-27a/b and -143 control blood pressure by action on angiotensin even as miR-27a regulates myosin heavy chain gene β-MHC (Figure 2) [105,109]. These miRNAs then act in opposing concert to mediate a controlled growth that results in myocytes that grow stronger but not larger; in effect, these cells become better adapted and more efficient. A partial list of such miRNAs affected by exercise are summarized in Table 5. In contrast, exogenous regulators of Nrf2 target only Nrf2 and do not seem to engage systemic regulatory machinery that provides anti-hypertrophic signaling in addition to the antioxidant response (Figure 2). In murine models, CDDO treatment has been shown to cause large changes in miRNA expression but this effect has not yet been studied in clinical trials of Nrf2 exogenous modulators [110]. Thus, simple administration of Nrf2 enhancers may not maintain the same benefit as exercise and clinical trials of such compounds would do well to include exercise/non-exercise groups whenever possible to determine the effect of this systemic machinery on cardiac hypertrophy. Future studies on human pan-miRNA expression profiles, especially miRNAs that regulate hypertrophy, will be useful in determining the molecular impact of artificial Nrf2 enhancement on the potential pathogenesis of cardiomyopathy.
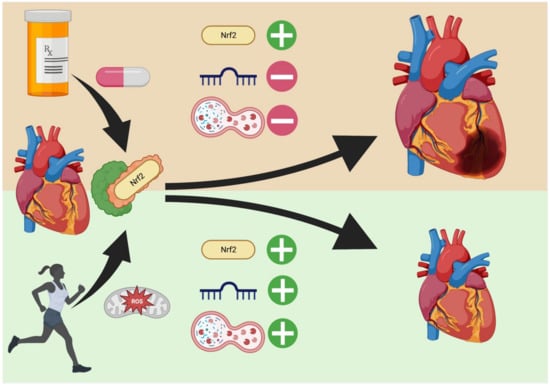
Figure 2.
Exercise vs. Endogenous Nrf2 Regulators in the Heart. Exogenous regulators (top) increase Nrf2 levels through interaction with Keap1 but the effect on cardiac-related miRNA and autophagy is poorly studied. Conversely, exercise (bottom) upregulates not only Nrf2, but also pro- and anti-hypertrophic miRNA generation that allow for controlled remodeling. Additionally, autophagic enhancement removes the danger of necrosis from damaged organelles and reduces fibrosis from protein aggregates. Created in BioRender.com.
Table 5.
Hypertrophy-Modulating Factors Upregulated by Exercise [105,111].
4.2. Clinical Trials with Exogenous Nrf2 Modulators
To explore the potential of developing cardiac pathologies after exogenous Nrf2 modulation, ClinicalTrials.gov was searched for each compound in Table 3 and results were filtered as follows: ALL interventional trials (randomized clinical trials), any phase (Early Phase 1, Phase 1, Phase 2, Phase 3, Phase 4), with results. In cases where studies exceeded 35 (e.g., ascorbic acid), 10 of the topmost results were used. Notable cardiac-related side effects were tallied and are displayed in Table 6.
Table 6.
Selected clinical trial updates and dispositions for Nrf2 Exogenous Regulators (data from ClinicalTrials.gov).
4.3. Clinical Trial Commentary: Reata Bardoloxone Trials
As seen in Table 4, the incidence of reported cardiac-related adverse events has remained quite low (usually less than 10%) but the number of completed trials with no results outweighs, in the cases of CDDO-Me and sulforaphane, completed trials containing reported adverse events. Even if positive results are not reported, the lack of adverse event reporting contributes to the issue of whether Nrf2 exogenous modulation has any negative effects on the heart as reported in the CDDO-Me Reata clinical trials (2007–2014; ClinicalTrials.gov: NCT01549769, NCT01351675, NCT01500798, NCT01551446, NCT01655186, NCT01576887, NCT00550849) [8]. If Nrf2 upregulation by other Keap1-Cys151-acting compounds, such as dimethyl fumarate or ursodiol (a gallstone dissolver), activated Nrf2 at the same level as CDDO, more trials could be expected to end in termination for patient safety/adverse event reasons. However, it seems as if only the Reata trials were affected because several other CDDO-Me trials were successfully completed, albeit without reported results. This raises several important questions with regard to Nrf2 regulation in chronic diseases. First, what miRNA does CDDO-Me regulate and are transcription profiles in sufferers of chronic pulmonary or metabolic diseases different from healthy volunteers? Second, since functional autophagy is indicated to play an important role in Nrf2-mediated pathogenesis, molecular evaluation of autophagy should be mandated in patients before such Nrf2-modulating compounds are tested, especially in diabetics or those with cardiac/pulmonary diseases [136]. Finally, other strong Nrf2 activators (such as organosulfur compounds or regular exercise) should be tested alongside CDDO-Me and any other compound suspected of causing Nrf2-mediated cardiomyopathy. These three precautions would give invaluable data as to the true cause of any cardiac maladaptation due to Nrf2-mediated hypertrophy and also verify miRNA-related silencing with regard to Nrf2 expression and downstream elements.
4.4. Clinical Perspective: Usefulness of Nrf2 Modulation in Heart Pathologies
Despite the potential to ablate ROS-mediated cardiomyocyte damage, curative applications of Nrf2 modulators for cardiac pathologies have yet to be reported. With regard to the heart, only animal models of heart failure have shown promise, with Nrf2 activators such as curcumin and CDDO-Me increasing exercise capacity, stroke volume, and cardiac output [137,138,139]). Conversely, diet and exercise carry extensive evidence for cardiac benefit. Furthermore, the possibility of exacerbating cardiomyopathy with Nrf2 exogenous modulation and concerns over chemotherapy resistance from Nrf2-mediated antioxidant enzymes upregulated in cancer cells make the use of such compounds questionable for clinical applications [47]. However, some studies have shown promise in wound healing, particularly within diabetic or hyperglycemic milieus, through the activation of Nrf2 targets HO-1 and NQO1 via hyperbaric oxygen therapy [140].
While clinical applications of Nrf2 activators have centered around cancers, kidney diseases, multiple sclerosis, and other inflammation-mediated diseases, topical Nrf2 activation in wound healing remains an underexplored topic and localized upregulation of Nrf2-mediated antioxidants might be of some value in the surgical suite [140]. Additionally, dental inflammation, primarily initiated upon microbial challenge by the NLRP3-mediated, pattern recognizing inflammasome, has been reported to be ablated by Nrf2 through NF-κB downregulation, countering ROS-mediated activation [141]. Nrf2 is also apparently capable of upregulating NLRP3 upon challenge with solid irritants (e.g., alum, silica crystals) [141]. Finally, the success of Nrf2 activators in animal models naturally points towards the potential of Nrf2 manipulation in animal and veterinary medicine, especially in wound healing. Thus, the exploration of Nrf2 manipulation for human clinical purposes must orient towards localized and isolated systems (i.e., oral, digestive, neurological) instead of systemic increase through oral or intravenous administration of compounds.
5. Conclusions
Nrf2, once regarded as a potential key to unlock novel therapies in the cardiovascular and cancer fields, has now assumed the role of a double-edged sword: when properly regulated, it can reduce ROS and increase wound healing but carries the possibility of chemotherapy resistance and cardiomyopathy when overexpressed by exogenous manipulation through natural and synthetic compounds. Clinical trials have not reported significant effects in diverse human disease systems while only animal trials seem to hold promise for veterinary therapeutic development. However, numerous reports indicate that exercise, as a natural Nrf2 upregulator, simultaneously increases miRNA that prevent cardiomyopathy during remodeling and exercise response. Furthermore, functional autophagy prevents pathological effects of Nrf2 activation and autophagic activators, such as fasting, may also be important in controlling unwanted Nrf2 effects [66]. Therefore, detailed studies on the regulatory microenvironment of the heart during exercise, fasting, and exogenous Nrf2 stimulation may provide insight into the complex regulatory system that controls cardiac remodeling.
Author Contributions
Conceptualization, B.J.M. and Y.H.; writing—original draft preparation, B.J.M. and H.K.; writing—review and editing, B.J.M., H.K. and Y.H.; supervision, Y.H.; project administration, B.J.M. and Y.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sykiotis, G.P.; Bohmann, D. Stress-Activated Cap’n’collar Transcription Factors in Aging and Human Disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Mathis, B.J.; Cui, T. The Role of Nrf2 in the Cardiovascular System and Atherosclerosis. In Nrf2 and its Modulation in Inflammation; Progress in Inflammation Research; Springer: Berlin/Heidelberg, Germany, 2020; pp. 97–127. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated Turnover of Nrf2 Is Determined by at Least Two Separate Protein Domains, the Redox-sensitive Neh2 Degron and the Redox-insensitive Neh6 Degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef]
- Hushpulian, D.M.; Kaidery, N.A.; Ahuja, M.; Poloznikov, A.A.; Sharma, S.M.; Gazaryan, I.G.; Thomas, B. Challenges and Limitations of Targeting the Keap1-Nrf2 Pathway for Neurotherapeutics: Bach1 De-Repression to the Rescue. Front. Aging Neurosci. 2021, 13, 673205. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The universal protein knowledgebase in Nucleic acids research. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Mathis, B.J.; Cui, T. CDDO and Its Role in Chronic Diseases. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2016; Volume 929, pp. 291–314. Available online: http://www.ncbi.nlm.nih.gov/pubmed/27771930 (accessed on 2 November 2022).
- Poh, J.; Ponsford, A.H.; Boyd, J.; Woodsmith, J.; Stelzl, U.; Wanker, E.; Harper, N.; MacEwan, D.; Sanderson, C.M. A functionally defined high-density NRF2 interactome reveals new conditional regulators of ARE transactivation. Redox Biol. 2020, 37, 101686. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Qin, J.-J.; Cheng, X.-D.; Zhang, J.; Zhang, W.-D. Dual roles and therapeutic potential of Keap1-Nrf2 pathway in pancreatic cancer: A systematic review. Cell Commun. Signal. 2019, 17, 121. [Google Scholar] [CrossRef]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The Carboxy-Terminal Neh3 Domain of Nrf2 Is Required for Transcriptional Activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kong, A.-N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. 2008, 48, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Ortet, P.C.; Muellers, S.N.; Viarengo-Baker, L.A.; Streu, K.; Szymczyna, B.R.; Beeler, A.B.; Allen, K.N.; Whitty, A. Recapitulating the Binding Affinity of Nrf2 for KEAP1 in a Cyclic Heptapeptide, Guided by NMR, X-ray Crystallography, and Machine Learning. J. Am. Chem. Soc. 2021, 143, 3779–3793. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef]
- Naidu, S.D.; Muramatsu, A.; Saito, R.; Asami, S.; Honda, T.; Hosoya, T.; Itoh, K.; Yamamoto, M.; Suzuki, T.; Dinkova-Kostova, A.T. C151 in KEAP1 is the main cysteine sensor for the cyanoenone class of NRF2 activators, irrespective of molecular size or shape. Sci. Rep. 2018, 23, 8037. [Google Scholar] [CrossRef]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef]
- Park, J.-Y.; Kim, S.; Sohn, H.Y.; Koh, Y.H.; Jo, C. TFEB activates Nrf2 by repressing its E3 ubiquitin ligase DCAF11 and promoting phosphorylation of p62. Sci. Rep. 2019, 9, 14354. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef]
- Brewer, A.C.; Murray, T.V.; Arno, M.; Zhang, M.; Anilkumar, N.P.; Mann, G.E.; Shah, A.M. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic. Biol. Med. 2011, 51, 205–215. [Google Scholar] [CrossRef]
- Theodore, M.; Kawai, Y.; Yang, J.; Kleshchenko, Y.; Reddy, S.P.; Villalta, F.; Arinze, I.J. Multiple Nuclear Localization Signals Function in the Nuclear Import of the Transcription Factor Nrf2. J. Biol. Chem. 2008, 283, 8984–8994. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell. Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [PubMed]
- De la Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar]
- Zang, H.; Mathew, R.O.; Cui, T. The Dark Side of Nrf2 in the Heart. Front. Physiol. 2020, 11, 722. [Google Scholar] [CrossRef]
- Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anti-cancer Therapies. Antioxidants 2020, 9, 1151. [Google Scholar] [CrossRef]
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002, 21, 5216–5224. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, J.; Wei, X.; Niu, C.; Jia, M.; Li, Q.; Meng, D. Bach1: Function, Regulation, and Involvement in Disease. Oxidative Med. Cell. Longev. 2018, 2018, 1347969. [Google Scholar] [CrossRef]
- Nishizawa, H.; Matsumoto, M.; Shindo, T.; Saigusa, D.; Kato, H.; Suzuki, K.; Sato, M.; Ishii, Y.; Shimokawa, H.; Igarashi, K. Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor BACH1. J. Biol. Chem. 2020, 295, 69–82. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Chen, P.-H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef]
- Wang, W.; Li, S.; Wang, H.; Li, B.; Shao, L.; Lai, Y.; Horvath, G.; Wang, Q.; Yamamoto, M.; Janicki, J.S.; et al. Nrf2 enhances myocardial clearance of toxic ubiquitinated proteins. J. Mol. Cell. Cardiol. 2014, 72, 305–315. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Öhlund, D.; et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Thimmulappa, R.K.; Kumar, V.; Cui, W.; Kumar, S.; Kombairaju, P.; Zhang, H.; Margolick, J.; Matsui, W.; Macvittie, T.; et al. NRF2-mediated Notch pathway activation en-hances hematopoietic reconstitution following myelosuppressive radiation. J. Clin. Investig. 2014, 124, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Chartoumpekis, D.V.; Kensler, T.W. Crosstalk between Nrf2 and Notch signaling. Free Radic. Biol. Med. 2015, 88, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Chai, D.; Zhang, L.; Xi, S.; Cheng, Y.; Jiang, H.; Hu, R. Nrf2 Activation Induced by Sirt1 Ameliorates Acute Lung Injury After Intestinal Ischemia/Reperfusion Through NOX4-Mediated Gene Regulation. Cell. Physiol. Biochem. 2018, 46, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, K.; Papagiannakopoulos, T.; Motohashi, H. Metabolic features of cancer cells in NRF2 addiction status. Biophys. Rev. 2020, 12, 435–441. [Google Scholar] [CrossRef]
- Dodson, M.; Anandhan, A.; Zhang, D.D.; Madhavan, L. An NRF2 Perspective on Stem Cells and Ageing. Front. Aging 2021, 2, 690686. [Google Scholar] [CrossRef]
- Safran, M.; Rosen, N.; Twik, M.; BarShir, R.; Stein, T.I.; Dahary, D.; Fishilevich, S.; Lancet, D. The GeneCards Suite; Springer: Berlin/Heidelberg, Germany, 2021; pp. 27–56. [Google Scholar] [CrossRef]
- Namani, A.; Rahaman, M.; Chen, M.; Tang, X. Gene-expression signature regulated by the KEAP1-NRF2-CUL3 axis is associated with a poor prognosis in head and neck squamous cell cancer. BMC Cancer 2018, 18, 46. [Google Scholar] [CrossRef]
- De Freitas Alves, A.; de Moura, A.C.; Andreolla, H.F.; Gorini da Veiga, A.B.; Fiegenbaum, M.; Giovenardi, M.; Almeida, A. Gene expression evaluation of antioxidant enzymes in patients with hepatocellular carcinoma: RT-qPCR and bioinformatic analyses. Genet. Mol. Biol. 2021, 44, e20190373. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef]
- Li, W.; Jain, M.R.; Chen, C.; Yue, X.; Hebbar, V.; Zhou, R.; Kong, A.-N.T. Nrf2 Possesses a Redox-insensitive Nuclear Export Signal Overlapping with the Leucine Zipper Motif. J. Biol. Chem. 2005, 280, 28430–28438. [Google Scholar] [CrossRef]
- Burroughs, A.F.; Eluhu, S.; Whalen, D.; Goodwin, J.S.; Sakwe, A.M.; Arinze, I.J. PML-Nuclear Bodies Regulate the Stability of the Fusion Protein Dendra2-Nrf2 in the Nucleus. Cell. Physiol. Biochem. 2018, 47, 800–816. [Google Scholar] [CrossRef] [PubMed]
- Walters, T.S.; McIntosh, D.J.; Ingram, S.M.; Tillery, L.; Motley, E.D.; Arinze, I.J.; Misra, S. SUMO-Modification of Human Nrf2 at K110 and K533 Regulates Its Nucleocytoplasmic Localization, Stability and Transcriptional Activity. Cell. Physiol. Biochem. 2021, 55, 141–159. [Google Scholar] [PubMed]
- Qin, Q.; Qu, C.; Niu, T.; Zang, H.; Qi, L.; Lyu, L.; Wang, X.; Nagarkatti, M.; Nagarkatti, P.; Janicki, J.S.; et al. Nrf2-Mediated Cardiac Maladaptive Remodeling and Dysfunction in a Setting of Autophagy Insufficiency. Hypertension 2016, 67, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.I.E.; Buckberg, G.D. The Myocardial Oxygen Supply:Demand Index Revisited. J. Am. Heart Assoc. 2014, 3, e000285. [Google Scholar] [CrossRef] [PubMed]
- Moris, D.; Spartalis, M.; Tzatzaki, E.; Spartalis, E.; Karachaliou, G.-S.; Triantafyllis, A.S.; Karaolanis, G.I.; Tsilimigras, D.I.; Theocharis, S. The role of reactive oxygen species in myocardial redox signaling and regulation. Ann. Transl. Med. 2017, 5, 324. [Google Scholar] [CrossRef] [PubMed]
- Noseda, M.; Abreu-Paiva, M.; Schneider, M.D. The Quest for the Adult Cardiac Stem Cell. Circ. J. 2015, 79, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Li, Z.; Bao, P.; Chen, M.; Zhang, M.; Yan, F.; Xu, Y.; Ji, C.; Hu, X.; Sanchis, D. Nrf2 deficiency aggravates Angiotensin II-induced cardiac injury by in-creasing hypertrophy and enhancing IL-6/STAT3-dependent inflammation. Biochim. Biophys. Acta 2019, 1865, 1253–1264. [Google Scholar] [CrossRef]
- Syed, A.M.; Kundu, S.; Ram, C.; Kulhari, U.; Kumar, A.; Mugale, M.N.; Mohapatra, P.; Murty, U.S.; Sahu, B.D. Up-regulation of Nrf2/HO-1 and inhibition of TGF-β1/Smad2/3 signaling axis by daphnetin alleviates transverse aortic constriction-induced cardiac remodeling in mice. Free Radic. Biol. Med. 2022, 186, 17–30. [Google Scholar] [CrossRef]
- Gutiérrez-Cuevas, J.; Galicia-Moreno, M.; Monroy-Ramírez, H.C.; Sandoval-Rodriguez, A.; García-Bañuelos, J.; Santos, A.; Armendariz-Borunda, J. The Role of NRF2 in Obesity-Associated Cardiovascular Risk Factors. Antioxidants 2022, 11, 235. [Google Scholar] [CrossRef]
- Zhou, S.; Sun, W.; Zhang, Z.; Zheng, Y. The Role of Nrf2-Mediated Pathway in Cardiac Remodeling and Heart Failure. Oxidative Med. Cell. Longev. 2014, 2014, 260429. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; An, L.; Taylor, M.R.G.; Chen, Q.M. Nrf2 signaling in heart failure: Expression of Nrf2, Keap1, antioxidant, and detoxification genes in dilated or ischemic cardiomyopathy. Physiol. Genom. 2022, 54, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.A. Antioxidants and coronary artery disease: From pathophysiology to preventive therapy. Coron. Artery Dis. 2015, 26, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, A.D.J.; Kitts, D.; Giovannucci, E.L.; Sahye-Pudaruth, S.; Paquette, M.; Mejia, S.B.; Patel, D.; Kavanagh, M.; Tsirakis, T.; Kendall, C.W.C.; et al. Selenium, antioxidants, cardiovascular disease, and all-cause mortality: A systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2020, 112, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Weinstein, S.J.; Yu, K.; Männistö, S.; Albanes, D. Serum Beta Carotene and Overall and Cause-Specific Mortality. Circ. Res. 2018, 123, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Henkel, R.; Sandhu, I.S.; Agarwal, A. The excessive use of antioxidant therapy: A possible cause of male infertility? Andrologia 2019, 51, e13162. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.P.; Butler, J.; Rossignol, P.; Pitt, B.; Anker, S.D.; Kosiborod, M.; Lund, L.H.; Bakris, G.L.; Weir, M.R.; Zannad, F. Abnormalities of Potassium in Heart Failure: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 2836–2850. [Google Scholar] [CrossRef]
- Abdellatif, M.; Sedej, S.; Carmona-Gutierrez, D.; Madeo, F.; Kroemer, G. Autophagy in Cardiovascular Aging. Circ. Res. 2018, 123, 803–824. [Google Scholar] [CrossRef]
- Marks, A.R. Calcium cycling proteins and heart failure: Mechanisms and therapeutics. J. Clin. Investig. 2013, 123, 46–52. [Google Scholar] [CrossRef]
- Bitto, A.; Lerner, C.; Torres, C.; Roell, M.; Malaguti, M.; Perez, V.; Lorenzini, A.; Hrelia, S.; Ikeno, Y.; Matzko, M.E.; et al. Long-Term IGF-I Exposure Decreases Autophagy and Cell Viability. PLoS ONE 2010, 5, e12592. [Google Scholar] [CrossRef]
- Yang, M.; Wen, T.; Chen, H.; Deng, J.; Yang, C.; Zhang, Z. Knockdown of insulin-like growth factor 1 exerts a protective effect on hypoxic injury of aged BM-MSCs: Role of autophagy. Stem Cell Res. Ther. 2018, 9, 284. [Google Scholar] [CrossRef] [PubMed]
- Ben Wang, B.; Shi, Y.; Chen, J.; Shao, Z.; Ni, L.; Lin, Y.; Wu, Y.; Tian, N.; Zhou, Y.; Sun, L.; et al. High glucose suppresses autophagy through the AMPK pathway while it induces autophagy via oxidative stress in chondrocytes. Cell Death Dis. 2021, 12, 506. [Google Scholar] [CrossRef] [PubMed]
- Bagherniya, M.; Butler, A.E.; Barreto, G.E.; Sahebkar, A. The effect of fasting or calorie restriction on autophagy induction: A review of the literature. Ageing Res. Rev. 2018, 47, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Long, J.; Liu, J. Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Elsevier: Amsterdam, The Netherlands, 2014; pp. 105–115. [Google Scholar]
- Salin, K.; Villasevil, E.M.; Anderson, G.J.; Auer, S.K.; Selman, C.; Hartley, R.C.; Mullen, W.; Chinopoulos, C.; Metcalfe, N.B. Decreased mitochondrial metabolic requirements in fasting animals carry an oxidative cost. Funct. Ecol. 2018, 32, 2149–2157. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Lim, Y.-M.; Lee, M.-S. Role of autophagy in diabetes and endoplasmic reticulum stress of pancreatic β-cells. Exp. Mol. Med. 2012, 44, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Takemura, G.; Goto, K.; Tsujimoto, A.; Mikami, A.; Ogino, A.; Watanabe, T.; Morishita, K.; Okada, H.; Kawasaki, M.; et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy 2015, 11, 1146–1160. [Google Scholar] [CrossRef]
- Wu, W.; Qin, Q.; Ding, Y.; Zang, H.; Li, D.-S.; Nagarkatti, M.; Nagarkatti, P.; Wang, W.; Wang, X.; Cui, T. Autophagy Controls Nrf2-Mediated Dichotomy in Pressure Over-loaded Hearts. Front. Physiol. 2021, 13, 12. [Google Scholar]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88 Pt B, 199–204. [Google Scholar] [CrossRef]
- Nezu, M.; Suzuki, N. Roles of Nrf2 in Protecting the Kidney from Oxidative Damage. Int. J. Mol. Sci. 2020, 21, 2951. [Google Scholar] [CrossRef]
- Kriz, W. Adenosine and ATP: Traffic regulators in the kidney. J. Clin. Investig. 2004, 114, 611–613. [Google Scholar] [CrossRef]
- Guerrero-Hue, M.; Rayego-Mateos, S.; Vázquez-Carballo, C.; Palomino-Antolín, A.; García-Caballero, C.; Opazo-Rios, L.; Morgado-Pascual, J.L.; Herencia, C.; Mas, S.; Ortiz, A. Protective Role of Nrf2 in Renal Disease. Antioxidants 2020, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Nezu, M.; Suzuki, N. Nrf2 activation for kidney disease treatment—A mixed blessing? Kidney Int. 2021, 99, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Rush, B.M.; Bondi, C.D.; Stocker, S.D.; Barry, K.M.; Small, S.A.; Ong, J.; Jobbagy, S.; Stolz, D.B.; Bastacky, S.I.; Chartoumpekis, D.V. Genetic or pharmacologic Nrf2 activation increases proteinuria in chronic kidney disease in mice. Kidney Int. 2021, 99, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxidative Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Quan, J.I.; Li, L.; Kinghorn, K.J.; Ivanov, D.K.; Tain, L.S.; Slack, C.; Kerr, F.; Nespital, T.; Thornton, J.; Hardy, J.; et al. Lithium Promotes Longevity through GSK3/NRF2-Dependent Hormesis. Cell Rep. 2016, 15, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Gu, X.; Gao, J.; Wang, Z.; Liu, G.; Barkema, H.W.; Han, B. Chlorogenic acid promotes the Nrf2/HO-1 anti-oxidative pathway by activating p21Waf1/Cip1 to resist dexamethasone-induced apoptosis in osteoblastic cells. Free Radic. Biol. Med. 2019, 137, 1–12. [Google Scholar] [CrossRef]
- Ahuja, M.; Kaidery, N.A.; Yang, L.; Calingasan, N.; Smirnova, N.; Gaisin, A.; Gaisina, I.N.; Gazaryan, I.; Hushpulian, D.M.; Kaddour-Djebbar, I.; et al. Distinct Nrf2 Signaling Mechanisms of Fumaric Acid Esters and Their Role in Neuroprotection against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Experimental Parkinson’s-Like Disease. J. Neurosci. 2016, 36, 6332–6351. [Google Scholar] [CrossRef]
- Unoki, T.; Akiyama, M.; Kumagai, Y. Nrf2 Activation and Its Coordination with the Protective Defense Systems in Response to Electrophilic Stress. Int. J. Mol. Sci. 2020, 21, 545. [Google Scholar] [CrossRef]
- Egbujor, M.C.; Petrosino, M.; Zuhra, K.; Saso, L. The Role of Organosulfur Compounds as Nrf2 Activators and Their Antioxidant Effects. Antioxidants 2022, 11, 1255. [Google Scholar] [CrossRef]
- Shin, J.W.; Chun, K.-S.; Kim, D.-H.; Kim, S.-J.; Kim, S.H.; Cho, N.-C.; Na, H.-K.; Surh, Y.-J. Curcumin induces stabilization of Nrf2 protein through Keap1 cysteine modification. Biochem. Pharmacol. 2020, 173, 113820. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Eggler, A.L.; Liu, D.; Liu, G.; Mesecar, A.D.; van Breemen, R.B. Sites of alkylation of human Keap1 by natural chemoprevention agents. J. Am. Soc. Mass Spectrom. 2007, 18, 2226–2232. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Weng, X.; Bao, X.; Bai, X.; Lv, Y.; Zhang, S.; Chen, Y.; Zhao, C.; Zeng, M.; Huang, J.; et al. A novel anti-atherosclerotic mechanism of quercetin: Competitive binding to KEAP1 via Arg483 to inhibit macrophage pyroptosis. Redox Biol. 2022, 57, 102511. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Buendia, I.; Parada, E.; Navarro, E.; Rada, P.; Cuadrado, A.; López, M.G.; García, A.G.; León, R. Melatonin-sulforaphane hybrid ITH12674 induces neuro-protection in oxidative stress conditions by a “drug-prodrug” mechanism of action. Br. J. Pharmacol. 2015, 172, 1807–1821. [Google Scholar] [CrossRef]
- Liang, N.; Dupuis, J.H.; Yada, R.Y.; Kitts, D.D. Chlorogenic acid isomers directly interact with Keap 1-Nrf2 signaling in Caco-2 cells. Mol. Cell. Biochem. 2019, 457, 105–118. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, S.; Fu, X.; Zhou, W.; Hong, W.; Zou, D.; Li, X.; Liu, J.; Ran, P.; Li, B. tert-Butylhydroquinone mobilizes intracellular-bound zinc to stabilize Nrf2 through inhibiting phosphatase activity. Am. J. Physiol. Physiol. 2015, 309, C148–C158. [Google Scholar] [CrossRef]
- Mostafavi-Pour, Z.; Ramezani, F.; Keshavarzi, F.; Samadi, N. The role of quercetin and vitamin C in Nrf2-dependent oxidative stress production in breast cancer cells. Oncol. Lett. 2017, 13, 1965–1973. [Google Scholar] [CrossRef]
- Gao, L.; Wang, J.; Sekhar, K.R.; Yin, H.; Yared, N.F.; Schneider, S.N.; Sasi, S.; Dalton, T.P.; Anderson, M.E.; Chan, J.Y.; et al. Novel n-3 Fatty Acid Oxidation Products Activate Nrf2 by Destabilizing the Association between Keap1 and Cullin3. J. Biol. Chem. 2007, 282, 2529–2537. [Google Scholar] [CrossRef]
- Ahmadi, Z.; Ashrafizadeh, M. Melatonin as a potential modulator of Nrf2. Fundam. Clin. Pharmacol. 2019, 34, 11–19. [Google Scholar] [CrossRef]
- Fuda, H.; Miyanaga, S.; Furukawa, T.; Umetsu, S.; Joko, S.; Roan, Y.; Suzuki, H.; Hui, S.-P.; Watanabe, M.; Chiba, H. Flazin as a Promising Nrf2 Pathway Activator. J. Agric. Food Chem. 2019, 67, 12844–12853. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Sun, K.; Wang, X.; Pan, H.; Zhu, J.; Ji, X.; Li, X. Chrysin suppresses proliferation, migration, and invasion in glioblastoma cell lines via mediating the ERK/Nrf2 signaling pathway. Drug Des. Dev. Ther. 2018, 12, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Sebens, S.; Krebs, S.; Geismann, C.; Grossmann, M.; Kruse, M.-L.; Schreiber, S.; Schäfer, H. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene 2012, 32, 4825–4835. [Google Scholar] [CrossRef] [PubMed]
- Gęgotek, A.; Skrzydlewska, E. Antioxidative and Anti-Inflammatory Activity of Ascorbic Acid. Antioxidants 2022, 11, 1993. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef]
- Chian, S.; Thapa, R.; Chi, Z.; Wang, X.J.; Tang, X. Luteolin inhibits the Nrf2 signaling pathway and tumor growth in vivo. Biochem. Biophys. Res. Commun. 2014, 447, 602–608. [Google Scholar] [CrossRef]
- Shanmugam, G.; Challa, A.K.; Devarajan, A.; Athmanathan, B.; Litovsky, S.H.; Krishnamurthy, P.; Davidson, C.J.; Rajasekaran, N.S. Exercise Mediated Nrf2 Signaling Protects the Myocardium from Isoproterenol-Induced Pathological Remodeling. Front. Cardiovasc. Med. 2019, 6, 68. [Google Scholar] [CrossRef]
- Kitaoka, Y. The Role of Nrf2 in Skeletal Muscle on Exercise Capacity. Antioxidants 2021, 10, 1712. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Cretoiu, D.; Li, G.; Xiao, J. Exercise-mediated regulation of autophagy in the cardiovascular system. J. Sport Health Sci. 2019, 9, 203–210. [Google Scholar] [CrossRef]
- Sorriento, D.; Di Vaia, E.; Iaccarino, G. Physical Exercise: A Novel Tool to Protect Mitochondrial Health. Front. Physiol. 2021, 12, 660068. [Google Scholar] [CrossRef]
- Wen, D.-T.; Zheng, L.; Li, J.-X.; Lu, K.; Hou, W.-Q. The activation of cardiac dSir2-related pathways mediates physical exercise resistance to heart aging in old Drosophila. Aging 2019, 11, 7274–7293. [Google Scholar] [CrossRef]
- Fernandes, T.; Baraúna, V.G.; Negrão, C.E.; Phillips, M.I.; Oliveira, E.M. Aerobic exercise training promotes physiological cardiac remodeling involving a set of microRNAs. Am. J. Physiol. Circ. Physiol. 2015, 309, H543–H552. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dong, X.; Wang, Z.; Wu, J. MicroRNA-1 in Cardiac Diseases and Cancers. Korean J. Physiol. Pharmacol. 2014, 18, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Carè, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.-L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhao, S.-P.; Zhao, Y.-H. MicroRNA-143/-145 in Cardiovascular Diseases. BioMed Res. Int. 2015, 2015, 531740. [Google Scholar] [CrossRef]
- Nishi, H.; Ono, K.; Horie, T.; Nagao, K.; Kinoshita, M.; Kuwabara, Y.; Watanabe, S.; Takaya, T.; Tamaki, Y.; Takanabe-Mori, R.; et al. MicroRNA-27a Regulates Beta Cardiac Myosin Heavy Chain Gene Expression by Targeting Thyroid Hormone Receptor β1 in Neonatal Rat Ventricular Myocytes. Mol. Cell. Biol. 2011, 31, 744–755. [Google Scholar] [CrossRef]
- Livingstone, M.C.; Johnson, N.M.; Roebuck, B.D.; Kensler, T.W.; Groopman, J.D. Profound changes in miRNA expression during cancer initiation by aflatoxin B1 and their abrogation by the chemopreventive triterpenoid CDDO-Im. Mol. Carcinog. 2017, 56, 2382–2390. [Google Scholar] [CrossRef]
- Brown, G.R.; Hem, V.; Katz, K.S.; Ovetsky, M.; Wallin, C.; Ermolaeva, O.; Tolstoy, I.; Tatusova, T.; Pruitt, K.; Maglott, D.R.; et al. Gene: A gene-centered information resource at NCBI. Nucleic Acids Res. 2014, 43, D36–D42. [Google Scholar] [CrossRef]
- Chen, J.-F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, E.T.; Hammond, S.M.; Conlon, F.L.; Wang, D.-Z. The Role of MicroRNA-1 and MicroRNA-133 in Skeletal Muscle Proliferation and Differentiation. Nat. Genet. 2005, 38, 228–233. [Google Scholar] [CrossRef]
- Jeyabal, P.; Thandavarayan, R.A.; Joladarashi, D.; Babu, S.S.; Krishnamurthy, S.; Bhimaraj, A.; Youker, K.A.; Kishore, R.; Krishnamurthy, P. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem. Biophys. Res. Commun. 2016, 471, 423–429. [Google Scholar] [CrossRef]
- Wu, M.; Liu, X.; Li, Z.; Huang, X.; Guo, H.; Guo, X.; Yang, X.; Li, B.; Xuan, K.; Jin, Y. SHED aggregate exosomes shuttled miR-26a promote angiogenesis in pulp regeneration via TGF-β/SMAD2/3 signalling. Cell Prolif. 2021, 54, e13074. [Google Scholar] [CrossRef]
- Castaigne, A.D.; Duval, A.M.; Dubois-Rande, J.L.; Herve, C.; Jan, F.; Louvard, Y. Prehospital Administration of Anisoylated Plasminogen Streptokinase Activator Complex in Acute Myocardial Infarction. Drugs 1987, 33, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Liu, H.; Guo, J.; Yu, Y.; Yang, D.; He, F.; Du, Z. MicroRNA-98 negatively regulates myocardial infarction-induced apoptosis by down-regulating Fas and caspase-3. Sci. Rep. 2017, 7, 7460. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lu, Y.; Li, Z.; Wang, Q. microRNA-133: Expression, Function and Therapeutic Potential in Muscle Diseases and Cancer. Curr. Drug Targets 2014, 15, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Shen, Z.; Zhou, S. Function of microRNA-145 and mechanisms underlying its role in malignant tumor diagnosis and treatment. Cancer Manag. Res. 2019, 11, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, J.; Ramanujam, D.P.; Sassi, Y.; Ahles, A.; Jentzsch, C.; Werfel, S.; Leierseder, S.; Loyer, X.; Giacca, M.; Zentilin, L.; et al. MiR-378 Controls Cardiac Hypertrophy by Combined Repression of Mitogen-Activated Protein Kinase Pathway Factors. Circulation 2013, 127, 2097–2106. [Google Scholar] [CrossRef]
- Hullinger, T.G.; Montgomery, R.L.; Seto, A.G.; Dickinson, B.A.; Semus, H.M.; Lynch, J.M.; Dalby, C.M.; Robinson, K.; Stack, C.; Latimer, P.A.; et al. Inhibition of miR-15 Protects Against Cardiac Ischemic Injury. Circ. Res. 2012, 110, 71–81. [Google Scholar] [CrossRef]
- Zheng, M.; Wang, M. A narrative review of the roles of the miR-15/107 family in heart disease: Lessons and prospects for heart disease. Ann. Transl. Med. 2021, 9, 66. [Google Scholar] [CrossRef]
- Jenike, A.E.; Halushka, M.K. miR-21: A non-specific biomarker of all maladies. Biomark. Res. 2021, 9, 18. [Google Scholar] [CrossRef]
- Li, S.; Ren, J.; Sun, Q. The expression of microRNA-23a regulates acute myocardial infarction in patients and in vitro through targeting PTEN. Mol. Med. Rep. 2018, 17, 6866–6872. [Google Scholar] [CrossRef]
- Li, D.; Shen, M.; Deng, X.; Bai, Y. MicroRNA miR-27a-3p accelerates cardiac hypertrophy by targeting neuro-oncological ventral antigen 1. Bioengineered 2022, 13, 8982–8993. [Google Scholar] [CrossRef]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Tréguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Rech, M.; Kuhn, A.R.; Lumens, J.; Carai, P.; van Leeuwen, R.; Verhesen, W.; Verjans, R.; Lecomte, J.; Liu, Y.; Luiken, J.J.; et al. AntagomiR-103 and -107 Treatment Affects Cardiac Function and Metabolism. Mol. Ther. Nucleic Acids 2018, 14, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Tao, T.; Chen, S.; Liang, C.; Qiu, Y.; Zhou, Y.; Zhang, R. MicroRNA-143 promotes cardiac ischemia-mediated mitochondrial impairment by the inhibition of protein kinase Cepsilon. Basic Res. Cardiol. 2017, 112, 60. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Li, C.; Zhang, H.; Qiu, S.; Fu, T.; Xu, Y. Downregulation of miR-146a Contributes to Cardiac Dysfunction Induced by the Tyrosine Kinase Inhibitor Sunitinib. Front. Pharmacol. 2019, 10, 914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ji, R.; Liao, X.; Castillero, E.; Kennel, P.J.; Brunjes, D.L.; Franz, M.; Möbius-Winkler, S.; Drosatos, K.; George, I.; et al. MicroRNA-195 Regulates Metabolism in Failing Myocardium Via Alterations in Sirtuin 3 Expression and Mitochondrial Protein Acetylation. Circulation 2018, 137, 2052–2067. [Google Scholar] [CrossRef] [PubMed]
- Gabisonia, K.; Prosdocimo, G.; Aquaro, G.D.; Carlucci, L.; Zentilin, L.; Secco, I.; Ali, H.; Braga, L.; Gorgodze, N.; Bernini, F.; et al. MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature 2019, 569, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, Y.; Sun, X. The functions of microRNA-208 in the heart. Diabetes Res. Clin. Pract. 2020, 160, 108004. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Huang, M.; Li, Z.; Jia, F.; Ghosh, Z.; Lijkwan, M.A.; Fasanaro, P.; Sun, N.; Wang, X.; Martelli, F.; et al. MicroRNA-210 as a Novel Therapy for Treatment of Ischemic Heart Disease. Circulation 2010, 122, S124–S131. [Google Scholar] [CrossRef]
- Verjans, R.; Peters, T.; Beaumont, F.J.; van Leeuwen, R.; van Herwaarden, T.; van Verhesen, W.; Munts, C.; Bijnen, M.; Henkens, M.; Diez, J.; et al. MicroRNA-221/222 Family Coun-teracts Myocardial Fibrosis in Pressure Overload–Induced Heart Failure. Hypertension 2018, 71, 280–288. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, X.; Su, T.; Li, H.; Huang, Q.; Wu, D.; Yang, C.; Han, Z. Circulating miR-499 are novel and sensitive biomarker of acute myocardial infarction. J. Thorac. Dis. 2015, 7, 303–308. [Google Scholar] [CrossRef]
- Wan, Q.; Xu, T.; Ding, W.; Zhang, X.; Ji, X.; Yu, T.; Yu, W.; Lin, Z.; Wang, J. miR-499-5p Attenuates Mitochondrial Fission and Cell Apoptosis via p21 in Doxorubicin Cardiotoxicity. Front. Genet. 2019, 9, 734. [Google Scholar] [CrossRef] [PubMed]
- Zang, H.; Wu, W.; Qi, L.; Tan, W.; Nagarkatti, P.; Nagarkatti, M.; Wang, X.; Cui, T. Autophagy Inhibition Enables Nrf2 to Exaggerate the Progression of Diabetic Cardiomyopathy in Mice. Diabetes 2020, 69, 2720–2734. [Google Scholar] [CrossRef] [PubMed]
- Wafi, A.M.; Hong, J.; Rudebush, T.L.; Yu, L.; Hackfort, B.; Wang, H.; Schultz, H.D.; Zucker, I.H.; Gao, L. Curcumin improves exercise performance of mice with coronary artery ligation-induced HFrEF: Nrf2 and antioxidant mechanisms in skeletal muscle. J. Appl. Physiol. 2019, 126, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Gao, L.; Zucker, I.H. Regulation of Nrf2 signaling pathway in heart failure: Role of extracellular vesicles and non-coding RNAs. Free Radic. Biol. Med. 2021, 167, 218–231. [Google Scholar] [CrossRef]
- Tian, C.; Gao, L.; Zhang, A.; Hackfort, B.T.; Zucker, I.H. Therapeutic Effects of Nrf2 Activation by Bardoxolone Methyl in Chronic Heart Failure. J. Pharmacol. Exp. Ther. 2019, 371, 642–651. [Google Scholar] [CrossRef]
- Victor, P.; Sarada, D.; Ramkumar, K.M. Pharmacological activation of Nrf2 promotes wound healing. Eur. J. Pharmacol. 2020, 886, 173395. [Google Scholar] [CrossRef]
- Schieffer, L.; Manzl, C.; Schatz, C.; Haybaeck, J.; Crismani, A. Nrf2 in the Field of Dentistry with Special Attention to NLRP3. Antioxidants 2022, 11, 149. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).