1. Introduction
Pancreatic cancer is currently one of the cancers with the highest mortality and lowest survival rate of all cancers. Pancreatic cancer has been reported to have an overall 5-year survival rate of less than 7%, accounting for the fourth in tumor related lethality, and it is expected to reach the second position by 2030 [
1]. Although radical surgery increases the 5-year survival rate of pancreatic cancer to 15–20%, its long-term survival rate has not improved significantly [
2]. Therefore, novel therapeutic strategies for pancreatic cancer need to be developed urgently.
The biological characteristics of tumor cells are modulated by the microenvironment in which they reside, in which s are important mediators of “communication” between cells, and can transport molecules such as proteins, mRNAs, or miRNAs to recipient cells, thereby modulating metastasis and therapeutic resistance. It has been confirmed that stromal cells weaving complex communication networks with breast cancer cells via exosomes enhances therapeutic resistance [
3]. In addition, miR-208a reached lung cancer cells to target p21 via exosomal trafficking, thus affecting lung cancer proliferation and radiotherapy sensitivity [
4]. In a related study of pancreatic cancer, Wang et al. showed that plasma derived exosomal miR-19b expression levels in pancreatic cancer patients were significantly lower than those in other pancreatic tumor patients, chronic pancreatitis patients, and healthy volunteers, suggesting that plasma derived exosomal miR-19b may be a promising diagnostic marker for pancreatic cancer [
5]. Another study found increased expression of miR-155 in PDAC cells chronically exposed to gemcitabine, and miR-155 was able to promote exosome secretion, which in turn resulted in the resistance of tumor cells to gemcitabine [
6].
For decades, an increasing number of studies have demonstrated that cellular metabolism is closely related to malignant tumorigenesis. However, unlike normal cells, cancer cells reprogram metabolism to meet the substantial material and energy demands of their rapid proliferation. Many metabolic pathways appear reprogrammed in cancer, including glycolysis, the TCA cycle, glutaminolysis, the electron transport chain, and the pentose phosphate pathway [
7,
8]. Since the discovery of the Warburg effect, more and more studies have proved that the metabolism of cancer cells plays a crucial role in cancer survival and growth, and glutamine plays a more important role in cancer metabolism than previously thought. It is well documented that several human pancreatic ductal adenocarcinoma (PDAC) cell lines rely on the Kirsten ratsarcoma viral oncogene homolog regulated non canonical glutamine (Gln) metabolic pathway to promote tumor cell proliferation and growth [
9]. In PDAC cells, KRAS specific dependent downregulation of glutamate dehydrogenase (GLUD1) and the upregulation of aspartate aminotransferase 1 (GOT1) promotes the derivation of glutamine into aspartate, which is converted into oxaloacetate (OAA) in the cytoplasm by GOT1, followed by a series of transformations into pyruvate, ultimately promoting the production of NADPH for maintaining redox balance [
9]. In addition, the proliferation of PDAC cells is also inhibited upon selective inhibition of GOT1 expression in tumor cells, and these phenomena suggest that targeting GOT1 may be a novel approach for the treatment of pancreatic ductal carcinoma [
10]. A recent publication showed that aspartate aminotransferases (GOTS) were identified as key metabolic enzymes for human PDAC. Yang et al. found that acetylation of GOT2 could promote ATP production and stimulate NADPH generation to inhibit ROS generation [
11]. These studies collectively establish that GOTS play an important role in redox regulation in human pancreatic cancer, laying the foundation for the treatment of cancer by targeting GOTS.
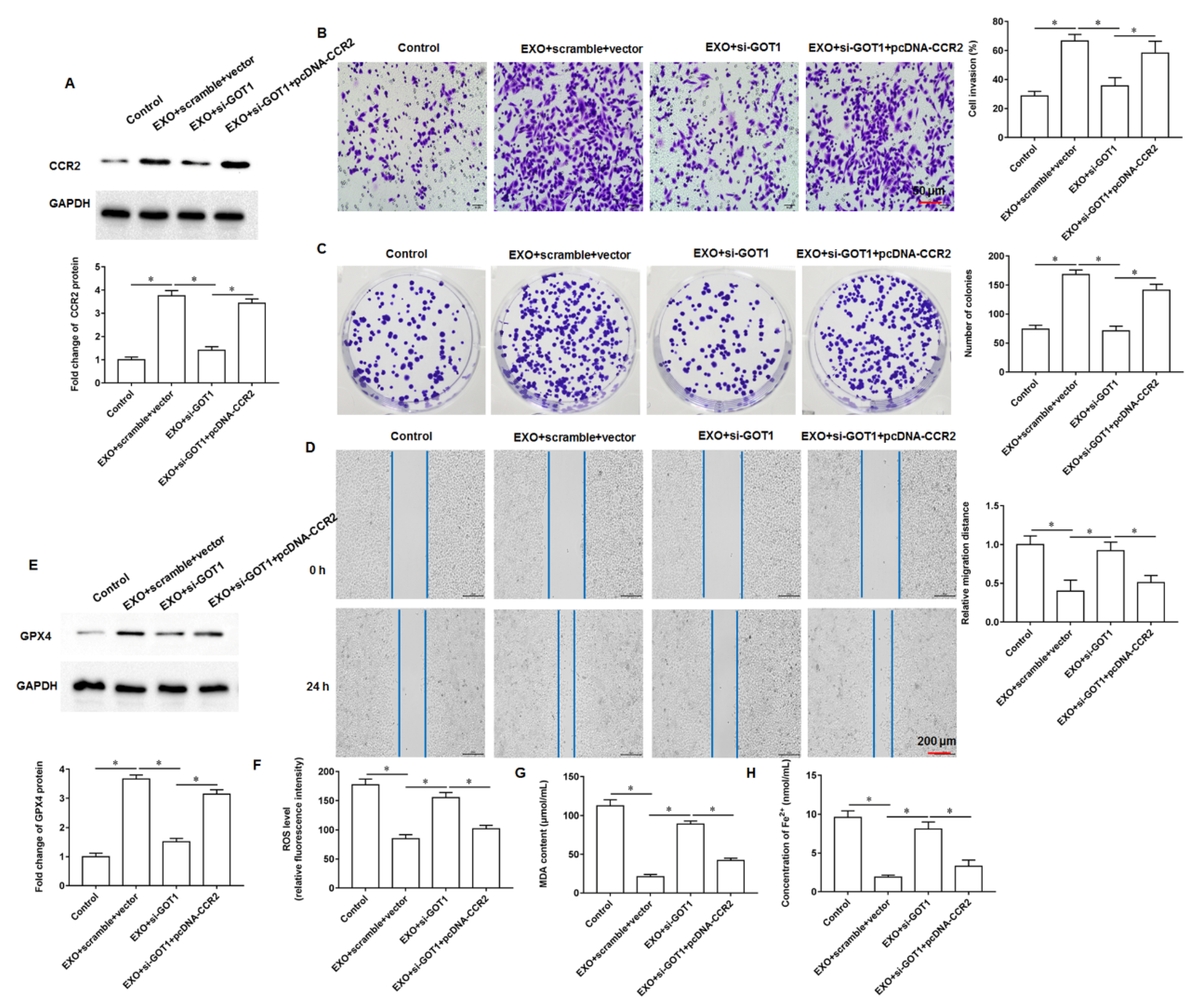
The tumor microenvironment is a key factor influencing tumor growth, spread, and metastasis. As an important and widely studied chemokine in the tumor microenvironment, monocytechemoattractant protein-1 (MCP-1/CCL2) is one of the important members of the CC subfamily of chemokines with two forms, autocrine or paracrine. CCL2 exerts its biological effects mainly through binding to its receptor C-C motif chemokine receptor-2 (CCR2), and numerous studies have shown that the chemokine CCL2 and its receptor CCR2 are highly expressed in a variety of malignancies. Zhuang et al. found that CCL2 could activate the hedgehog signaling pathway of HCC cells through the CCL2/CCR2 molecular axis, and up regulate the expression of snail and vimentin, as well as down regulate E-cadherin expression to promote HCC cell invasion and epithelial mesenchymal transition [
12]. Li et al. showed that CCR2 expression was abnormally elevated in HCC tissues and significantly correlated with tumor volume, metastasis and clinical stage, and that the high expression of CCR2 in HCC tissues was significantly associated with the poor prognosis of patients [
13]. Independently, CCL2 was reported to recruit monocytes and reduce CD8
+ T cell infiltration in pancreatic tumors, and CCL2 inhibition and monocyte neutralisation increased the sensitivity of PDAC to immune checkpoint blockade [
14]. Currently, although numerous studies have confirmed the cancer promoting role of CCR2 in multiple tumors, its role in pancreatic cancer progression is not well understood. Therefore, investigating the role and possible mechanisms of CCR2 in pancreatic cancer progression will be of great importance to unearth new therapies against pancreatic cancer.
2. Materials and Methods
2.1. Cell Culture
The human pancreatic cancer cell lines AsPC-1 (RRID: CVCL_0152), BxPC-3 (RRID: CVCL_0186), PANC-1 (RRID: CVCL_0480) and SW1990 (RRID: CVCL_1723), and the normal human pancreatic ductal epithelial cell line HPDE were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were incubated in medium containing 10% FBS and 100 U/mL penicillin and 100 μg/mL streptomycin (Sigma, St. Louis, MO, USA) in RPMI 1640 medium (Gibco, Rockville, MD, USA) at 5% CO2 at 37 °C. The pcDNA-GOT1, pcDNA-CCR2 and their negative control (vector) were purchased from RiboBio Co., Ltd. (Guangzhou, China) and transfected using RiboBio Transfection Kit (RiboBio Co., Ltd.). Small interfering RNA against GOT1 (si-GOT1), CCR2 (si-CCR2) and their negative control (scramble) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All transfection reagent transfected into cells using Lipofectamine 3000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The cell lines used in this study have been STR authenticated and determined to be contamination free. The absence of mycoplasma was checked every 6 months (MycoAlert™ mycoplasma detection kit, Lonza, Ozyme, Saint-Cyr L’Ecole, France).
2.2. Animals
Adult nude mice (weight 20–22 g, Wuhan Experimental Animal Center, China) were housed in a specific pathogen-free environment under the condition of 12-h light/12-h dark cycle, free access to food and water, and acclimatized to their surroundings for three days. These nude mice were randomly divided into two groups (n = 8 per group) including scramble group (exosomes were secreted by PANC-1 cells that transfected with scramble) and si-GOT1 group (exosomes were secreted by PANC-1 cells that transfected with si-GOT1). The exosomes (250 μL) secreted by PANC-1 cells transfected with scramble or si-GOT1, were subcutaneously injected into the armpits of nude mice. The tumor volume was closely observed and measured on days 8, 11, 14, 17, 20 and 23 after injection. On day 23, nude mice were euthanized, and tumor tissues were collected subjected to subsequent studies. All experiments were conducted in accordance with the Animal Ethics Committee of Union Hospital, Tongji Medical College, Huazhong University of Science and Technology.
2.3. Exosome Extraction and Identification
The supernatant of cell culture medium was collected, and the cell components and dead cells were removed by low-speed centrifugation (300× g × 10 min, 2000× g × 10 min) at 4 °C. The supernatant containing exosomes was retained and the cell debris was removed by high-speed centrifugation (10,000× g × 70 min). The supernatant containing extracellular vesicles was retained and the exosomes were precipitated by ultracentrifugation (100,000× g × 70 min). Appropriate amount of PBS was taken to resuspend the extracellular vesicle precipitation, and then ultracentrifuged again (100,000× g × 70 min) to eliminate contaminated proteins. The precipitation was collected and divided, and stored at −80 °C for future use. The size and concentration of the exosomes were analyzed by a nanoparticle tracking analysis (NTA) instrument (ZetaView, Particle Metrix, Meerbusch, Germany). The morphology of exosomes was observed using a transmission electron microscopy (TEM, StarJoy, Japan; JSM-7800F).
2.4. RT-qPCR
Total RNA was isolated from tumor tissues by using TRIzol (Invitrogen, Carlsbad, CA, USA). Single-stranded cDNA was synthesized with the PrimeScript Reagent Kit (Promega, USA). Real-time qPCR was conducted by using SYBR Premix Ex TaqTM Kit (Applied Biosystems, Foster City, CA, USA). The reaction was run in ABI7500 Real-time PCR system (Applied Biosystems, Carlsbad, CA, USA). GAPDH was used as an endogenous control. The PCR cycling conditions consisted of: 95 °C for 3 min; then, 35 cycle amplification for 20 s at 95 °C, 30 s at 55 °C, 15 s at 72 °C; followed by 1 min at 72 °C. The primers used in this study were synthesized by Sangon Biotech (Shanghai, China). The expression level was normalized by using the 2−ΔΔCt method.
2.5. Western Blotting
Briefly, the cells were lysed for 20 min on ice in ice-cold lysis buffer (Roche). The lysates were centrifuged at 12,000× g for 20 min at 4 °C to obtain a clear lysate. The protein content of each sample was determined using the BCA Protein Assay Kit (Thermo Scientific). Then, equal amounts of proteins(15 μg/lane) were separated on a 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidenedifluoride (PVDF) membranes (Bio-Rad, Hercules, CA, USA).The membranes were blocked in 5% (w/v) nonfat dry milk in TBST (Tris-buffered saline-0.1% Tween) at 25 °C for 3 h and then incubated with the following primary antibodies: GAPDH antibody (1:1000, Abcam, ab8245), CD63 antibody (1:1000, Abcam, ab217345), TSG101 antibody (1:2000, Abcam, ab125011), Alix antibody (1:8000, Abcam, ab88388), GOT1 antibody (1:500, Abcam, ab85857), GXP4 antibody (1:800, Abcam, ab75810), CCR2 antibody (1:900, Abcam, ab203128), Nrf2 antibody (1:1000, Abcam, ab92946) and HO-1 antibody (1:2000, Abcam, ab52947). Then, the membranes were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1:10,000, Abcam, ab205718) for 1 h. The protein bands were visualized by using the Enhanced chemiluminescence reagents (Millipore, MA, USA). The expression of relative protein was obtained by the gray value ratio of the target protein to the internal reference GAPDH and analyzed with the ImageJ software (National Institutes of Health, Bethesda, MA, USA).
2.6. Immunohistochemical Assay
Briefly, tumor tissue sections were pretreated with trypsin (0.05%) for 10 min and then treated with 3% (v/v) H2O2. Sections were then blocked with 10% goat serum for 1 h at room temperature. After washing with PBS, anti-GOT1 antibody, anti-CCR2, anti-Nrf2 or anti-HO-1 (1:50 dilution) was applied to the sections, and the sections were incubated overnight at 4 °C. Sections were then washed with PBS for 15 min and incubated with biotinylated secondary antibodies by using the Histostain Plus Kit (Invitrogen, Carlsbad, CA, USA). Sections were washed and incubated with 3, 30-diaminobenzidine (DAB) substrate for 2 min.
2.7. MTT Assay
Cell viability was determined by MTT kits (Dojindo, Kumamoto, Japan). The PANC-1 cells were seeded in a 96-well plate at 1.5 × 104 cells/well. At 24, 48 and 72 h after transfection, the supernatant was discarded and 20 μL of MTT solution was added to each well. After incubating at 37 °C for 4 h, the absorbance at 490 nm was measured and recorded. Each experimental procedure was processed at least three times.
2.8. Transwell Cell Invasion Assay
Cells were seeded into the upper chamber of Transwell chambers (8.0 μm pore size; Millipore Corporation, Boston, MA, USA) coated with Matrigel (BD Bioscience, Franklin lakes, NJ, USA). The complete medium was added into the lower chamber. After incubation at 37 °C for 48 h, cells on the upper chamber were removed with cotton swabs, while cells on the lower champer were fixed with 70% ethanol and stained with 0.1% crystal violet. The invasive cells were counted under a light microscope (Olympus, Tokyo, Japan).
2.9. Cell Colony Formation Assay
PANC-1 and SW1990 cells were seeded in 6-well plates. Media were renewed every 3 days during culture. Two weeks later, cell supernatant was removed, and proliferative colonies were incubated with paraformaldehyde (Sigma) and crystal violet (Sigma), respectively. Cell colony-forming ability was illustrated via counting cell numbers. A colony was deemed when cell numbers >50.
2.10. Wound-Healing Assay
PANC-1 and SW1990 cells were cultured in 6-well plates until their confluence reached about 100%. Then, cell wounds were created and cells were washed using phosphate buffer solution (PBS; Thermo Fisher, Waltham, MA, USA). Cells were continued to be cultured in serum-free media for 24 h. Finally, cell migratory ability was determined via calculating the width of wounds under microscope (Nikon) with a 100(×) magnification.
2.11. Co-IP Assay
After 48 h of transfection, cells were washed twice with cold PBS, and then RIPA lysis buffer was added for lysis. The supernatant collected by centrifugation was incubated with corresponding antibodies at 4 °C overnight. Then, we added 100 μL of Protein A agarose beads to capture the antigen-antibody complex, and slowly shook the mixture at 4 °C overnight. The agarose beads-antigen-antibody complex was collected by instantaneous centrifugation, and washed with ice-cold PBS. Then, the complex was boiled with protein loading buffer to free the antigen, antibody and beads. After centrifugation, the supernatant was taken for electrophoresis to detect the expression of the interaction protein.
2.12. ROS Content Detection
Cells were plated at 1 × 104 density seeded in culture flasks. ROS kit was used to measure ROS levels according to the manufacturer’s instructions. Dichloro-dihydro-fluorescein diacetate (DCFH-DA; 10 μM) was added to the cells and incubated for 20 min at 37 °C. The cells were then digested and suspended. The cell suspensions were centrifuged at 1000× g for 10 min and washed twice with phosphate-buffered saline (PBS). The cells were collected after centrifugation for fluorescence detection. Flow cytometry was used to measure fluorescence intensity. The positive area of DCFH-DA was ROS fluorescence intensity.
2.13. Determination of MDA and Fe2+ Contents
The secretion of MDA in PANC-1 cell supernatant was detected by ELISA kits (Sigma) following the manufacturer’s instructions. Similarly, the levels of Fe2+ were detected by Iron Colorimetric Assay Kits (Sigma).
2.14. Statistical Analysis
All statistical analyses were performed by using the SPSS software (ver. 21.0; SPSS, Chicago, IL, USA). The quantitative data derived from three independent experiments were expressed as mean ± SD. Comparisons between two groups were made by the Student’s t-test. Data between multiple groups were performed with one-way analysis of variance (ANOVA) followed by post hoc analysis with LSD test. p < 0.05 was considered statistically significant.
4. Discussion
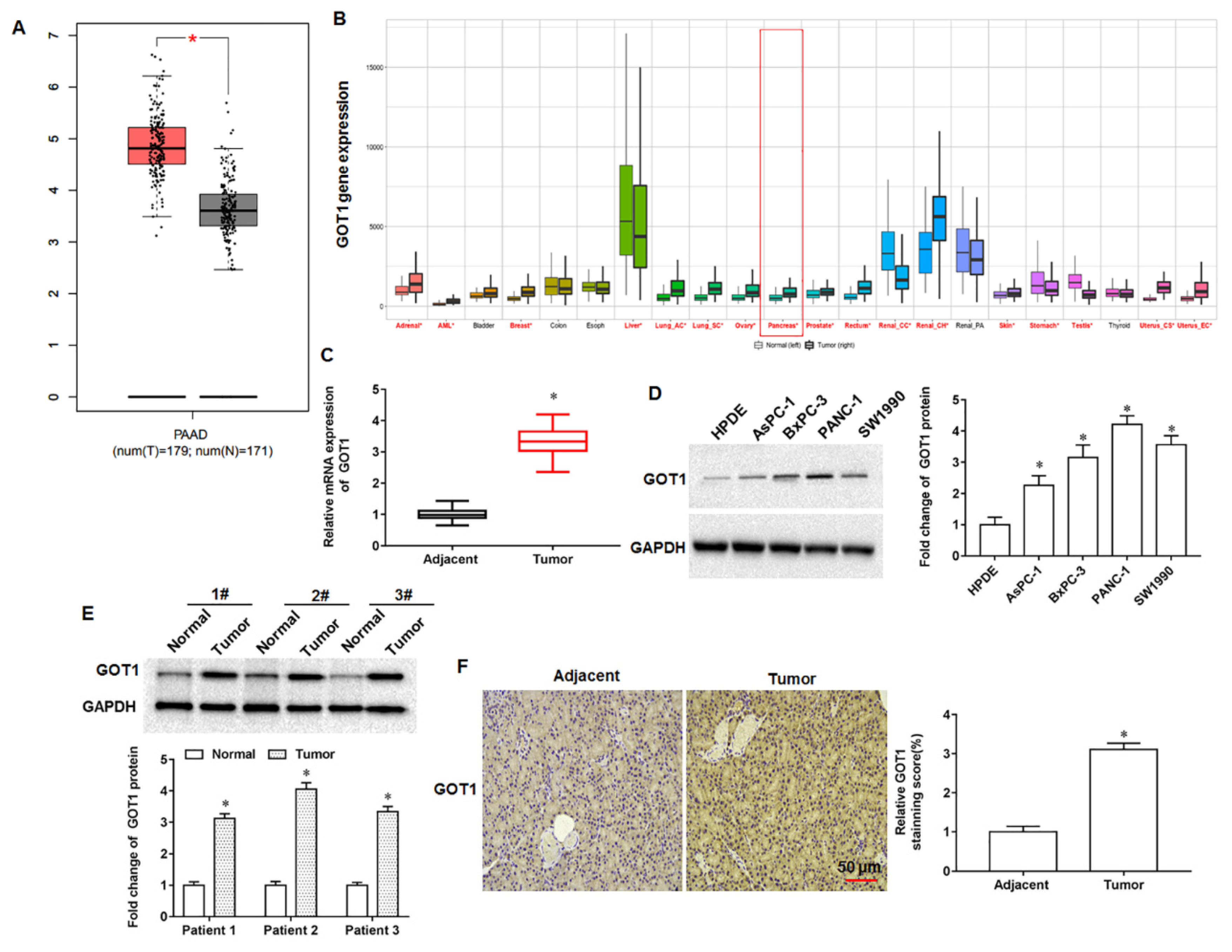
The GOT1 gene is located on human chromosome 10 and is approximately 34,955 bp in length. Tumor cell proliferation requires not only the maintenance of intracellular redox homeostasis but also the production of large amounts of RNA, DNA and proteins, and therefore the presence of aspartate must be sufficient. Studies have found that aspartate can be produced not only by Gln in the mitochondrial matrix but also catalytically by GOT1 in the cytoplasmic matrix, so the expression of GOT1 in cells is tightly associated with cell proliferation and the synthesis of aspartate [
17]. Oxidative stress is the excessive production of highly reactive molecules such as RNS and ROS in the body, and the degree of oxidation exceeds the scavenging degree of oxides, thus leading to tissue damage. Relative to normal cells, the characteristics of tumor cells are manifested in the increased production rate of reactive oxygen species and the altered redox reaction environment, so that the redox balance inside tumor cells is critical to maintain the survival and proliferation of tumor cells. Tumor cells not only undergo glycolysis to produce NADPH to maintain cellular redox balance, but can also generate NADPH through a non-canonical glutamine pathway mediated by GOT1 [
18]. Wiley et al. experimentally demonstrated that aminooxyacetic acid can decrease the NAD+/NADH ratio by inhibiting GOT1, leading to cellular senescence [
19]. Another scholar found that ziprasidone could induce glutamine metabolism disorder and redox state imbalance of PDAC cells by targeting GOT1, thereby inhibiting tumor cell proliferation, migration and inducing cell apoptosis [
20].
Ferroptosis, a newly discovered mode of programmed death, is a non-apoptotic cell death modality that depends on lipid peroxidation driven processes that require intracellular enrichment of available iron. When cells undergo iron death, a shrunken volume of mitochondria, an increased density of bilayer membranes, and a decrease or disappearance of mitochondrial cristae are clearly observed. Moreover, when iron death occurs, glutathione (GSH) in the cell is depleted, and glutathione peroxidase 4 (GPX4) activity declines, and lipid oxides cannot be metabolized by the GPX4 catalyzed glutathione reduction reaction, followed by the oxidation of lipids by ferrous ions in a manner similar to the Fenton reaction to produce large amounts of reactive oxygen species (ROS), prompting the cell to undergo iron death. Currently, it is found that iron death is polygenically regulated and mainly involves genetic alterations in aspects of iron homeostasis and lipid peroxidative metabolism. Currently, studies have confirmed that GOT1 inhibition promotes pancreatic cancer cell death by triggering ferroptosis [
21]. Wang et al. found that miR-9-5p inhibited pancreatic cancer cell proliferation, invasion, glutamine metabolism and redox homeostasis by downregulating GOT1 expression, suggesting that miR-9-5p may serve as a prognostic or therapeutic target in pancreatic cancer [
22]. Tumor cells have a high activity of antioxidant system, so tumor cells can have a strong ability to tolerate oxidative stress.
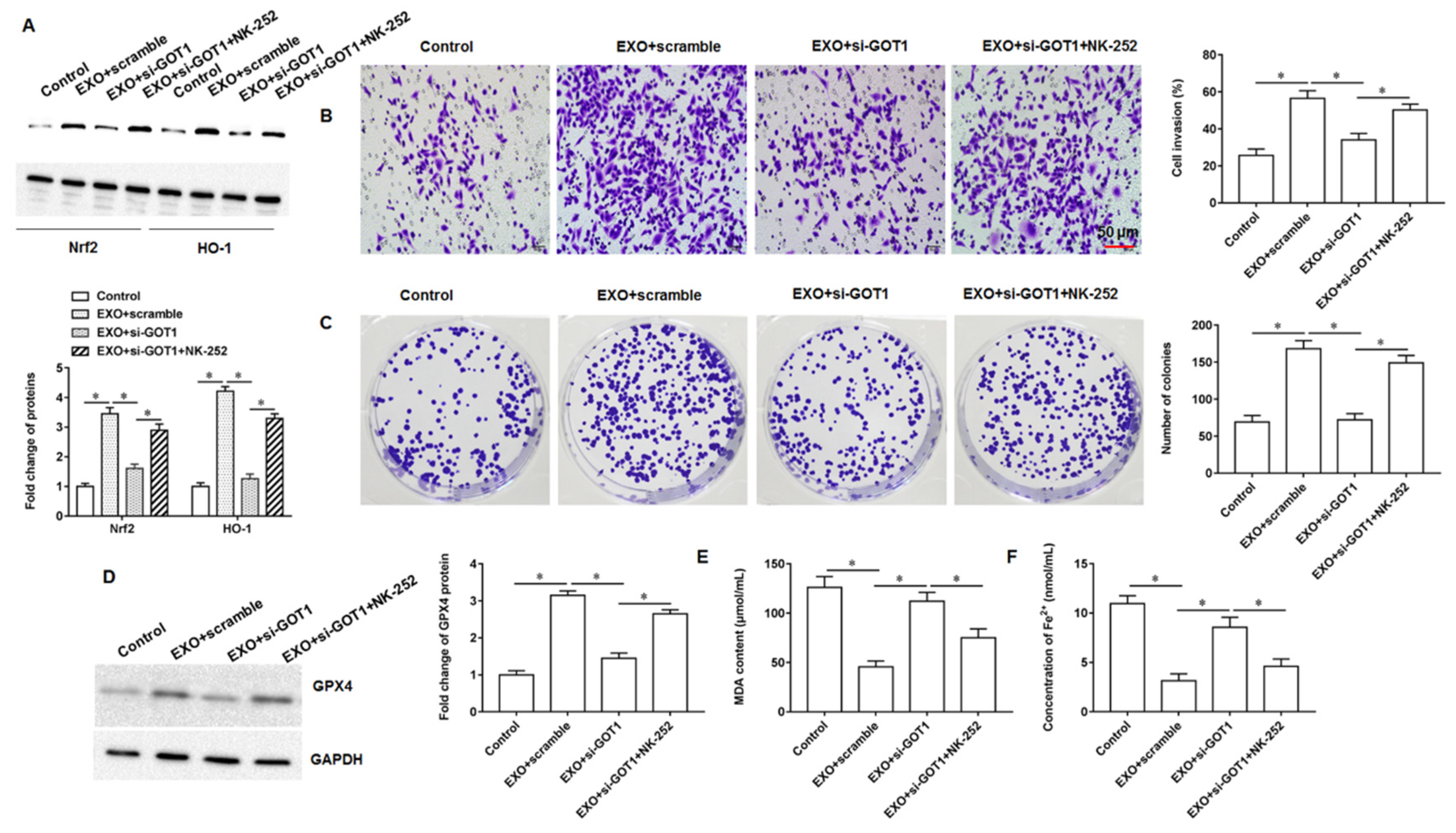
Nuclear factor E2 related factor 2 (Nrf2) is a core transcription factor regulating cellular oxidative stress, which can exert powerful antioxidant/antiapoptotic effects and is an important mechanism of drug resistance in tumor cells. When cells or organisms are exposed to ROS, it can cause the modification of the cytoskeleton associated inhibitory protein Keap1, promoting the dissociation of the keap1-Nrf2 complex, resulting in the translocation of Nrf2 from the cytoplasm into the nucleus. Next, Nrf2 that enters the nucleus can bind to the antioxidant response element, thereby activating the expression of downstream genes that regulate iron and ROS metabolism, including heme oxygenase 1 (HO-1), γ-Glutamylcysteine synthetase and quinone oxidoreductase 1. Yang et al. demonstrated that cetuximab was able to promote RSL3 induced ferroptosis by inhibiting the Nrf2/HO-1 signaling pathway in KRAS mutant colorectal cancer [
23]. In bladder cancer progression, erianin is able to promote bladder cancer cell death and cell cycle arrest, and mechanistic studies have shown that this is mediated by accelerating ferroptosis via inducing Nrf2 inactivation [
24]. Kuang et al. found that ferroptosis activators promoted mgst1 expression in PDAC cell lines in a Nrf2 dependent manner, and overexpressing mgst1 restored the resistance to ferroptosis in cells with Nrf2 expression knockdown [
25].
In this study, we found that GOT1 protein enriched in exosomes secreted by pancreatic cancer cells promoted tumor cell proliferation, invasion and migration and inhibited cell iron death. Furthermore, mechanistic studies revealed that exosomal GOT1 suppressed pancreatic cancer cell iron death and accelerated pancreatic cancer progression by activating the Nrf2/HO-1 axis via upregulation of CCR2 expression, and our study may provide a new reference for pancreatic cancer treatment.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







