
Targeting of the ELR+CXCL/CXCR1/2 Pathway Is a Relevant Strategy for the Treatment of Paediatric Medulloblastomas
 , , , and
, , , and
Abstract
:1. Research in Context
1.1. Evidence before This Study
1.2. Added Value of This Study
1.3. Implications of All the Available Evidence
2. Introduction
3. Materials and Methods
3.1. Chemistry
3.2. Cell Culture
Human Medulloblastoma Cells
3.3. Flow Cytometry
CXCR1/2 Labelling
3.4. In Vitro Assays of Cell Behaviour
3.4.1. Cerebellar Organotypic Model
3.4.2. Quantification of C29 in Mouse Organs Postmortem
3.4.3. In Silico Analysis
3.4.4. Statistical Analysis
4. Results
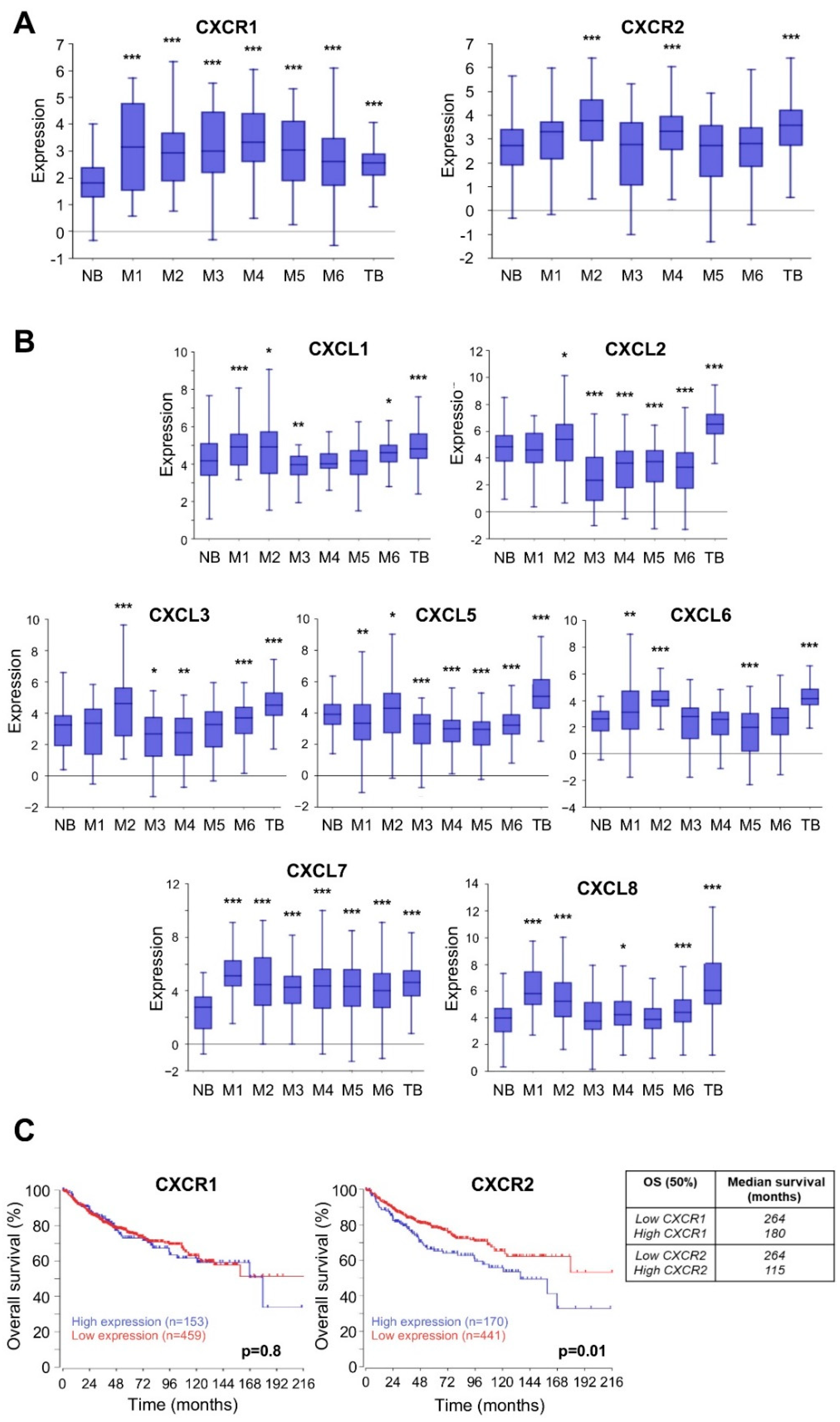
4.1. Members of ELR+CXCL/CXCR Axis Are Overexpressed and Associated with Poor Prognosis in MB Patients
4.2. Expression of ELR+CXCL-CXCR1/2 Family Members in MB Cells
4.3. Inhibition of Cell Metabolism by the Novel Pharmacological CXCR1/2 Inhibitor C29 in MB Cells
4.4. C29 Reduces Proliferation, Migration, and Invasion of Naïve MB Cells
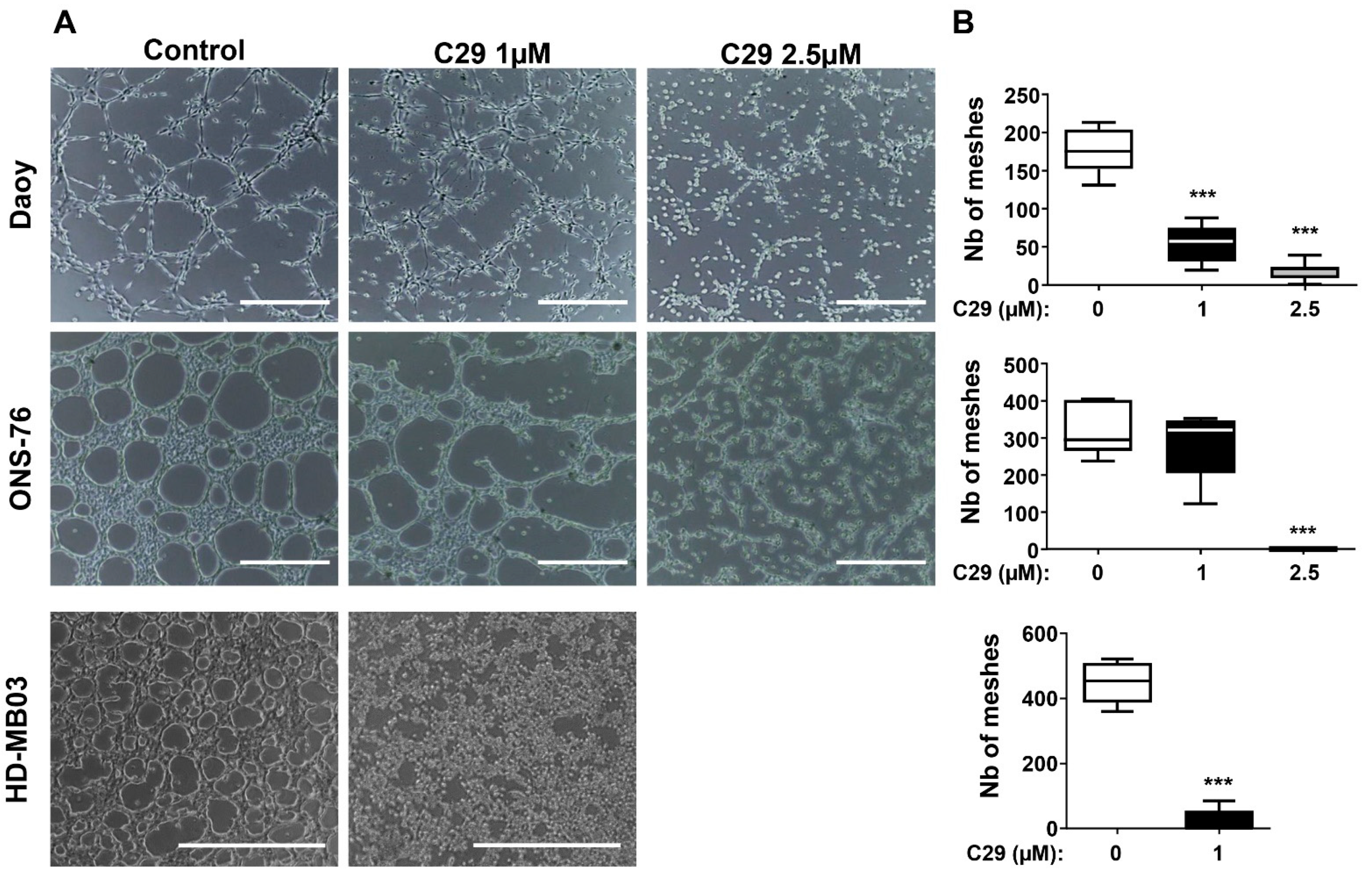
4.5. C29 Inhibits the Formation of Pseudo-Vascular Structures
4.6. C29 Is Active in the Ex Vivo Organotypic Model of the Cerebellum and Crosses the Blood–Brain Barrier In Vivo
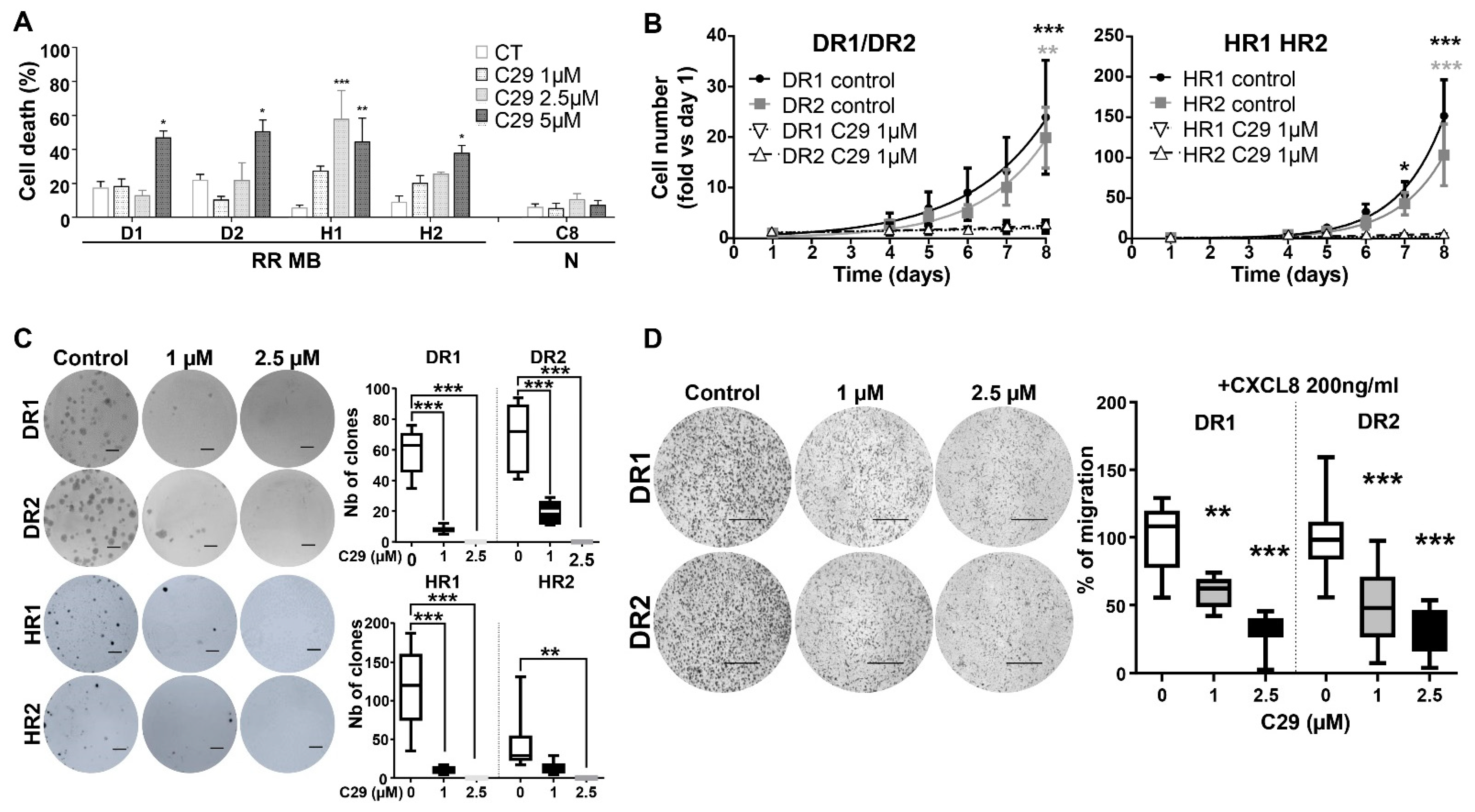
4.7. C29 Is Effective in Radiation-Resistant MB Cell Lines
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gajjar, A.; Chintagumpala, M.; Ashley, D.; Kellie, S.; Kun, L.E.; Merchant, T.E.; Woo, S.; Wheeler, G.; Ahern, V.; Krasin, M.J.; et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): Long-term results from a prospective, multicentre trial. Lancet Oncol. 2006, 7, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Packer, R.J.; Gajjar, A.; Vezina, G.; Rorke-Adams, L.; Burger, P.C.; Robertson, P.L.; Bayer, L.; LaFond, D.; Donahue, B.R.; Marymont, M.H.; et al. Phase III Study of Craniospinal Radiation Therapy Followed by Adjuvant Chemotherapy for Newly Diagnosed Average-Risk Medulloblastoma. J. Clin. Oncol. 2006, 24, 4202–4208. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.C.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma Comprises Four Distinct Molecular Variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef]
- Shih, D.J.; Northcott, P.A.; Remke, M.; Korshunov, A.; Ramaswamy, V.; Kool, M.; Luu, B.; Yao, Y.; Wang, X.; Dubuc, A.M.; et al. Cytogenetic Prognostication Within Medulloblastoma Subgroups. J. Clin. Oncol. 2014, 32, 886–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strieter, R.M.; Polverini, P.J.; Kunkel, S.L.; Arenberg, D.A.; Burdick, M.D.; Kasper, J.; Dzuiba, J.; Van Damme, J.; Walz, A.; Marriott, D.; et al. The Functional Role of the ELR Motif in CXC Chemokine-mediated Angiogenesis. J. Biol. Chem. 1995, 270, 27348–27357. [Google Scholar] [CrossRef] [Green Version]
- Vandercappellen, J.; Van Damme, J.; Struyf, S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008, 267, 226–244. [Google Scholar] [CrossRef] [PubMed]
- Dufies, M.; Grytsai, O.; Ronco, C.; Camara, O.; Ambrosetti, D.; Hagege, A.; Parola, J.; Mateo, L.; Ayrault, M.; Giuliano, S.; et al. New CXCR1/CXCR2 inhibitors represent an effective treatment for kidney or head and neck cancers sensitive or refractory to reference treatments. Theranostics 2019, 9, 5332–5346. [Google Scholar] [CrossRef]
- Bihannic, L.; Ayrault, O. Insights into cerebellar development and medulloblastoma. Bull. Cancer 2016, 103, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, T.N.; Patmore, D.M.; Boop, S.; Boulos, N.; Jacus, M.O.; Patel, Y.T.; Roussel, M.F.; Finkelstein, D.; Goumnerova, L.; Perreault, S.; et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 2016, 29, 508–522. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.H.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stütz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajjar, A.; Bowers, D.; Karajannis, M.; Leary, S.; Witt, H.; Gottardo, N. Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape. J. Clin. Oncol. 2015, 33, 2986–2998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajjar, A.; Robinson, G.W.; Smith, K.S.; Lin, T.; Merchant, T.E.; Chintagumpala, M.; Mahajan, A.; Su, J.; Bouffet, E.; Bartels, U.; et al. Outcomes by Clinical and Molecular Features in Children with Medulloblastoma Treated with Risk-Adapted Therapy: Results of an International Phase III Trial (SJMB03). J. Clin. Oncol. 2021, 39, 822–835. [Google Scholar] [CrossRef]
- Cavalli, F.M.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, J.C.; Hill, R.M.; Megahed, H.; Lusher, M.E.; Schwalbe, E.C.; Cole, M.; Hogg, T.L.; Gilbertson, R.J.; Ellison, D.W.; Bailey, S.; et al. TP53 Mutations in Favorable-Risk Wnt/Wingless-Subtype Medulloblastomas. J. Clin. Oncol. 2011, 29, e344–e346. [Google Scholar] [CrossRef]
- Goschzik, T.; Mynarek, M.; Doerner, E.; Schenk, A.; Spier, I.; Warmuth-Metz, M.; Bison, B.; Obrecht, D.; Struve, N.; Kortmann, R.-D.; et al. Genetic alterations of TP53 and OTX2 indicate increased risk of relapse in WNT medulloblastomas. Acta Neuropathol. 2022, 144, 1143–1156. [Google Scholar] [CrossRef]
- Northcott, P.A.; Robinson, G.W.; Kratz, C.P.; Mabbott, D.J.; Pomeroy, S.L.; Clifford, S.C.; Rutkowski, S.; Ellison, D.W.; Malkin, D.; Taylor, M.D.; et al. Medulloblastoma. Nat. Rev. Dis. Prim. 2019, 5, 11. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [Green Version]
- Asuthkar, S.; Velpula, K.K.; Nalla, A.K.; Gogineni, V.R.; Gondi, C.S.; Rao, J.S. Irradiation-induced angiogenesis is associated with an MMP-9-miR-494-syndecan-1 regulatory loop in medulloblastoma cells. Oncogene 2014, 33, 1922–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, H.; Eggert, A.; Janss, A.; Wiewrodt, R.; Zhao, H.; Sutton, L.; Rorke, L.; Phillips, P.; Grotzer, M. Angiogenic profile of childhood primitive neuroectodermal brain tumours/medulloblastomas. Eur. J. Cancer 2001, 37, 2064–2072. [Google Scholar] [CrossRef]
- Yang, F.; Jove, V.; Xin, H.; Hedvat, M.; Van Meter, T.E.; Yu, H. Sunitinib Induces Apoptosis and Growth Arrest of Medulloblastoma Tumor Cells by Inhibiting STAT3 and AKT Signaling Pathways. Mol. Cancer Res. 2010, 8, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Grizzi, F.; Weber, C.; Di Ieva, A. Antiangiogenic Strategies in Medulloblastoma: Reality or Mystery. Pediatr. Res. 2008, 63, 584–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, A.S.; Krailo, M.; Chi, S.; Villaluna, D.; Springer, L.; Williams-Hughes, C.; Fouladi, M.; Gajjar, A. Temozolomide with irinotecan versus temozolomide, irinotecan plus bevacizumab for recurrent medulloblastoma of childhood: Report of a COG randomized Phase II screening trial. Pediatr. Blood Cancer 2021, 68, e29031. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotondi, M.; Coperchini, F.; Latrofa, F.; Chiovato, L. Role of Chemokines in Thyroid Cancer Microenvironment: Is CXCL8 the Main Player? Front. Endocrinol. 2018, 9, 314. [Google Scholar] [CrossRef]
- Sparmann, A.; Bar-Sagi, D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004, 6, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, K. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev. 2001, 12, 375–391. [Google Scholar] [CrossRef]
- Mestas, J.; Burdick, M.D.; Reckamp, K.; Pantuck, A.; Figlin, R.A.; Strieter, R.M. The role of CXCR2/CXCR2 ligand biological axis in renal cell carcinoma. J. Immunol. 2005, 175, 5351–5357. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.J.; Gallagher, R.; Seaton, A.; Wilson, C.; Scullin, P.; Pettigrew, J.; Stratford, I.J.; Williams, K.J.; Johnston, P.G.; Waugh, D.J. HIF-1 and NF-kappaB-mediated upregulation of CXCR1 and CXCR2 expression promotes cell survival in hypoxic prostate cancer cells. Oncogene 2007, 26, 7333–7345. [Google Scholar] [CrossRef]
- Merritt, W.M.; Lin, Y.G.; Han, L.Y.; Kamat, A.A.; Spannuth, W.A.; Schmandt, R.; Urbauer, D.; Pennacchio, L.A.; Cheng, J.-F.; Nick, A.M.; et al. Dicer, Drosha, and Outcomes in Patients with Ovarian Cancer. N. Engl. J. Med. 2008, 359, 2641–2650. [Google Scholar] [CrossRef] [Green Version]
- Brat, D.J.; Bellail, A.C.; Van Meir, E.G. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro-Oncology 2005, 7, 122–133. [Google Scholar] [CrossRef]
- Wu, S.; Singh, S.; Varney, M.L.; Kindle, S.; Singh, R.K. Modulation of CXCL-8 expression in human melanoma cells regulates tumor growth, angiogenesis, invasion, and metastasis. Cancer Med. 2012, 1, 306–317. [Google Scholar] [CrossRef]
- Wang, B.; Hendricks, D.T.; Wamunyokoli, F.; Parker, M.I. A Growth-Related Oncogene/CXC Chemokine Receptor 2 Autocrine Loop Contributes to Cellular Proliferation in Esophageal Cancer. Cancer Res. 2006, 66, 3071–3077. [Google Scholar] [CrossRef] [Green Version]
- Botton, T.; Puissant, A.; Cheli, Y.; Tomic, T.; Giuliano, S.; Fajas, L.; Deckert, M.; Ortonne, J.-P.; Bertolotto, C.; Tartare-Deckert, S.; et al. Ciglitazone negatively regulates CXCL1 signaling through MITF to suppress melanoma growth. Cell Death Differ. 2011, 18, 109–121. [Google Scholar] [CrossRef]
- Miyake, M.; Hori, S.; Morizawa, Y.; Tatsumi, Y.; Nakai, Y.; Anai, S.; Torimoto, K.; Aoki, K.; Tanaka, N.; Shimada, K.; et al. CXCL1-Mediated Interaction of Cancer Cells with Tumor-Associated Macrophages and Cancer-Associated Fibroblasts Promotes Tumor Progression in Human Bladder Cancer. Neoplasia 2016, 18, 636–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echizen, K.; Hirose, O.; Maeda, Y.; Oshima, M. Inflammation in gastric cancer: Interplay of the COX-2/prostaglandin E2 and Toll-like receptor/MyD88 pathways. Cancer Sci. 2016, 107, 391–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Sun, H.; Wei, J.; Cen, B.; DuBois, R.N. CXCL1 Is Critical for Premetastatic Niche Formation and Metastasis in Colorectal Cancer. Cancer Res. 2017, 77, 3655–3665. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Jian, Z.; Jia, B.; Chang, L. CXCL7 promotes proliferation and invasion of cholangiocarcinoma cells. Oncol. Rep. 2017, 37, 1114–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; Li, E.; Liu, Y.; Xie, W.; Huang, C.; Song, J.; Zhang, W.; Zheng, Y.; Wang, H.; Wang, Q. CTAPIII/CXCL7: A novel biomarker for early diagnosis of lung cancer. Cancer Med. 2018, 7, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Dufies, M.; Giuliano, S.; Viotti, J.; Borchiellini, D.; Cooley, L.S.; Ambrosetti, D.; Guyot, M.; Ndiaye, P.D.; Parola, J.; Claren, A.; et al. CXCL7 is a predictive marker of sunitinib efficacy in clear cell renal cell carcinomas. Br. J. Cancer 2017, 117, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Desurmont, T.; Skrypek, N.; Duhamel, A.; Jonckheere, N.; Millet, G.; Leteurtre, E.; Gosset, P.; Duchene, B.; Ramdane, N.; Hebbar, M.; et al. Overexpression of chemokine receptor CXCR2 and ligand CXCL7 in liver metastases from colon cancer is correlated to shorter disease-free and overall survival. Cancer Sci. 2015, 106, 262–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Berk, R.; Kosir, M.A. CXCL7-Mediated Stimulation of Lymphangiogenic Factors VEGF-C, VEGF-D in Human Breast Cancer Cells. J. Oncol. 2010, 2010, 939407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, H.; Debnath, B.; Neamati, N. Role of the CXCL8-CXCR1/2 Axis in Cancer and Inflammatory Diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef]
- Waugh, D.J.; Wilson, C. The Interleukin-8 Pathway in Cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Dubey, S.; Varney, M.L.; Dave, B.J.; Singh, R.K. IL-8 Directly Enhanced Endothelial Cell Survival, Proliferation, and Matrix Metalloproteinases Production and Regulated Angiogenesis. J. Immunol. 2003, 170, 3369–3376. [Google Scholar] [CrossRef] [Green Version]
- Reiland, J.; Furcht, L.T.; McCarthy, J.B. CXC-chemokines stimulate invasion and chemotaxis in prostate carcinoma cells through the CXCR2 receptor. Prostate 1999, 41, 78–88. [Google Scholar] [CrossRef]
- Grepin, R.; Guyot, M.; Giuliano, S.; Boncompagni, M.; Ambrosetti, D.; Chamorey, E.; Scoazec, J.Y.; Negrier, S.; Simonnet, H.; Pages, G. The CXCL7/CXCR1/2 axis is a key driver in the growth of clear cell renal cell carcinoma. Cancer Res. 2014, 74, 873–883. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Bondarenko, M.; Le Grand, M.; Shaked, Y.; Raviv, Z.; Chapuisat, G.; Carrère, C.; Montero, M.-P.; Rossi, M.; Pasquier, E.; Carré, M.; et al. Metronomic Chemotherapy Modulates Clonal Interactions to Prevent Drug Resistance in Non-Small Cell Lung Cancer. Cancers 2021, 13, 2239. [Google Scholar] [CrossRef]
- Rossi, M.; Talbot, J.; Piris, P.; Le Grand, M.; Montero, M.-P.; Matteudi, M.; Agavnian-Couquiaud, E.; Appay, R.; Keime, C.; Williamson, D.; et al. Beta-blockers disrupt mitochondrial bioenergetics and increase radiotherapy efficacy independently of beta-adrenergic receptors in medulloblastoma. eBioMedicine 2022, 82, 104149. [Google Scholar] [CrossRef] [PubMed]
- Badisa, R.B.; Darling-Reed, S.F.; Joseph, P.; Cooperwood, J.S.; Latinwo, L.M.; Goodman, C.B. Selective cytotoxic activities of two novel synthetic drugs on human breast carcinoma MCF-7 cells. Anticancer. Res. 2009, 29, 2993–2996. [Google Scholar] [PubMed]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’Er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular Channel Formation by Human Melanoma Cells in Vivo and in Vitro: Vasculogenic Mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, L.; Liang, N.; Zhang, J.; Xie, J.; Liu, F.; Xu, D.; Yu, X.; Tian, Y. Advanced research on vasculogenic mimicry in cancer. J. Cell. Mol. Med. 2015, 19, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Penco-Campillo, M.; Comoglio, Y.; Feliz Morel, A.J.; Hanna, R.; Durivault, J.; Leloire, M.; Mejias, B.; Pagnuzzi, M.; Morot, A.; Burel-Vandenbos, F.; et al. VEGFC negatively regulates the growth and aggressiveness of medulloblastoma cells. Commun. Biol. 2020, 3, 579. [Google Scholar] [CrossRef] [PubMed]
- Grépin, R.; Ambrosetti, D.; Marsaud, A.; Gastaud, L.; Amiel, J.; Pedeutour, F.; Pagès, G. The Relevance of Testing the Efficacy of Anti-Angiogenesis Treatments on Cells Derived from Primary Tumors: A New Method for the Personalized Treatment of Renal Cell Carcinoma. PLoS ONE 2014, 9, e89449. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Gil, J. A role for CXCR2 in senescence, but what about in cancer? Cancer Res. 2009, 69, 2167–2170. [Google Scholar] [CrossRef] [Green Version]
- Ginestier, C.; Liu, S.; Diebel, M.E.; Korkaya, H.; Luo, M.; Brown, M.; Wicinski, J.; Cabaud, O.; Charafe-Jauffret, E.; Birnbaum, D.; et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Investig. 2010, 120, 485–497. [Google Scholar] [CrossRef]
- Mhaidat, N.M.; Thorne, R.F.; Zhang, X.D.; Hersey, P. Regulation of Docetaxel-Induced Apoptosis of Human Melanoma Cells by Different Isoforms of Protein Kinase C. Mol. Cancer Res. 2007, 5, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Eisemann, T.; Wechsler-Reya, R.J. Coming in from the cold: Overcoming the hostile immune microenvironment of medulloblastoma. Genes Dev. 2022, 36, 514–532. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Filipponi, D.; Pagnuzzi-Boncompagni, M.; Pages, G. Inhibiting ALK2/ALK3 Signaling to Differentiate and Chemo-Sensitize Medulloblastoma. Cancers 2022, 14, 2095. [Google Scholar] [CrossRef] [PubMed]
- Bilton, R.; Mazure, N.; Trottier, E.; Hattab, M.; Déry, M.A.; Richard, D.E.; Pouysségur, J.; Brahimi-Horn, M.C. Arrest-defective-1 protein, an acetyltransferase, does not alter stability of hypoxia-inducible factor (HIF)-1alpha and is not induced by hypoxia or HIF. J. Biol. Chem. 2005, 280, 31132–31140. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Capillary Temp (°C) | 275 |
| Sheath Gas Flow (u.a.): | 50 |
| Aux/Sweep Gas Flow (u.a.) | 10 |
| Negative Mode | |
| Source Voltage (kV) | 4.50 |
| Source Current (uA) | 80.00 |
| Capillary Voltage (V) | −10.00 |
| Tube Lens Offset (V) | −50.00 |
| All Groups (n = 612) | WNT (n = 63) | SHH (n = 172) | Group 4 (n = 264) | Group 3 (n = 113) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Last Quartile | OS (%) 5y | p-Value | OS (%) 5y | p-Value | OS (%) 5y | p-Value | OS (%) 5y | p-Value | OS (%) 5y | p-Value | |||||
| CXCR1 | Low | 76% | 0.799 | Low | 98% | 0.917 | Low | 80% | 0.904 | Low | 78% | 0.159 | Low | 55% | 0.294 |
| High | 72% | High | 100% | High | 80% | High | 68% | High | 68% | ||||||
| CXCR2 | Low | 79% | 0.001 | Low | 98% | 0.724 | Low | 79% | 0.203 | Low | 79% | 0.157 | Low | 55% | 0.583 |
| High | 68% | High | 100% | High | 82% | High | 65% | High | 60% | ||||||
| CXCL1 | Low | 78% | 0.283 | Low | 98% | 0.153 | Low | 84% | 0.93 | Low | 82% | 0.0072 | Low | 54% | 0.528 |
| High | 70% | High | 100% | High | 74% | High | 58% | High | 67% | ||||||
| CXCL2 | Low | 78% | 0.305 | Low | 96% | 0.148 | Low | 84% | 0.168 | Low | 76% | 0.854 | Low | 58% | 0.582 |
| High | 69% | High | 100% | High | 72% | High | 76% | High | 52% | ||||||
| CXCL3 | Low | 78% | 0.122 | Low | 98% | 0.313 | Low | 87% | 0.022 | Low | 77% | 0.206 | Low | 55% | 0.621 |
| High | 70% | High | 100% | High | 62% | High | 68% | High | 60% | ||||||
| CXCL5 | Low | 78% | 0.381 | Low | 98% | 0.844 | Low | 83% | 0.53 | Low | 78% | 0.531 | Low | 56% | 0.629 |
| High | 72% | High | 100% | High | 78% | High | 68% | High | 62% | ||||||
| CXCL6 | Low | 78% | 0.32 | Low | 77% | 0.32 | Low | 78% | 0.739 | Low | 73% | 0.256 | Low | 61% | 0.365 |
| High | 72% | High | 73% | High | 82% | High | 82% | High | 47% | ||||||
| CXCL7 | Low | 77% | 0.828 | Low | 100% | 0.054 | Low | 82% | 0.274 | Low | 70% | 0.748 | Low | 76% | 0.045 |
| High | 72% | High | 92% | High | 68% | High | 77% | High | 51% | ||||||
| CXCL8 | Low | 78% | 0.13 | Low | 98% | 0.785 | Low | 82% | 0.135 | Low | 78% | 0.238 | Low | 59% | 0.813 |
| High | 68% | High | 100% | High | 68% | High | 66% | High | 52% | ||||||
| SCORE | −2 | −1 | −5 | −5 | 0 | ||||||||||
| C29 Gavage | Acute (3 h) | Chronic (5 days) |
|---|---|---|
| 100 mg/kg | ng C29/mg cerebellum | |
| Cerebellum 1 | 0 | 0 |
| Cerebellum 2 | 0 | 0 |
| Cerebellum 3 | 0 | 0 |
| Cerebellum 4 | 0 | 0 |
| Cerebellum 5 | 5.394 | 0 |
| 200 mg/kg | ng C29/mg cerebellum | |
| Cerebellum 1 | 13.1 | 0 |
| Cerebellum 2 | 9.3 | 0 |
| Cerebellum 3 | 7.4 | 0 |
| Cerebellum 4 | 4.7 | 0 |
| Cerebellum 5 | 6.4 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penco-Campillo, M.; Molina, C.; Piris, P.; Soufi, N.; Carré, M.; Pagnuzzi-Boncompagni, M.; Picco, V.; Dufies, M.; Ronco, C.; Benhida, R.; et al. Targeting of the ELR+CXCL/CXCR1/2 Pathway Is a Relevant Strategy for the Treatment of Paediatric Medulloblastomas. Cells 2022, 11, 3933. https://doi.org/10.3390/cells11233933
Penco-Campillo M, Molina C, Piris P, Soufi N, Carré M, Pagnuzzi-Boncompagni M, Picco V, Dufies M, Ronco C, Benhida R, et al. Targeting of the ELR+CXCL/CXCR1/2 Pathway Is a Relevant Strategy for the Treatment of Paediatric Medulloblastomas. Cells. 2022; 11(23):3933. https://doi.org/10.3390/cells11233933
Chicago/Turabian StylePenco-Campillo, Manon, Clément Molina, Patricia Piris, Nouha Soufi, Manon Carré, Marina Pagnuzzi-Boncompagni, Vincent Picco, Maeva Dufies, Cyril Ronco, Rachid Benhida, and et al. 2022. "Targeting of the ELR+CXCL/CXCR1/2 Pathway Is a Relevant Strategy for the Treatment of Paediatric Medulloblastomas" Cells 11, no. 23: 3933. https://doi.org/10.3390/cells11233933