

HMGB1 Promotes In Vitro and In Vivo Skeletal Muscle Atrophy through an IL-18-Dependent Mechanism

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.3. Western Blot Analysis
2.4. Enzyme-Linked Immunosorbent Assay (ELISA)
2.5. Immunofluorescence Staining
2.6. Hematoxylin and Eosin Staining
2.7. HMGB1 Knockdown in C2C12 Myoblast Cell Lines
2.8. Transfection and Luciferase Reporter Assay
2.9. Chromatin Immunoprecipitation-Quantitative Polymerase Chain Reaction (ChIP-qPCR) Assay
2.10. Glycerol-Induced Muscle Injury Mouse Model
2.11. Micro-Computed Tomography Analysis
2.12. Immunohistochemistry and Dystrophin Staining
2.13. Reagent and Resources
2.14. Statistical Analysis
3. Results
3.1. HMGB1 Induces Skeletal Muscle Atrophy, and Attenuates Myogenic Marker Expression in Myoblasts
3.2. HMGB1 Inhibits Myoblast Differentiation via an IL-18-Dependent Mechanism
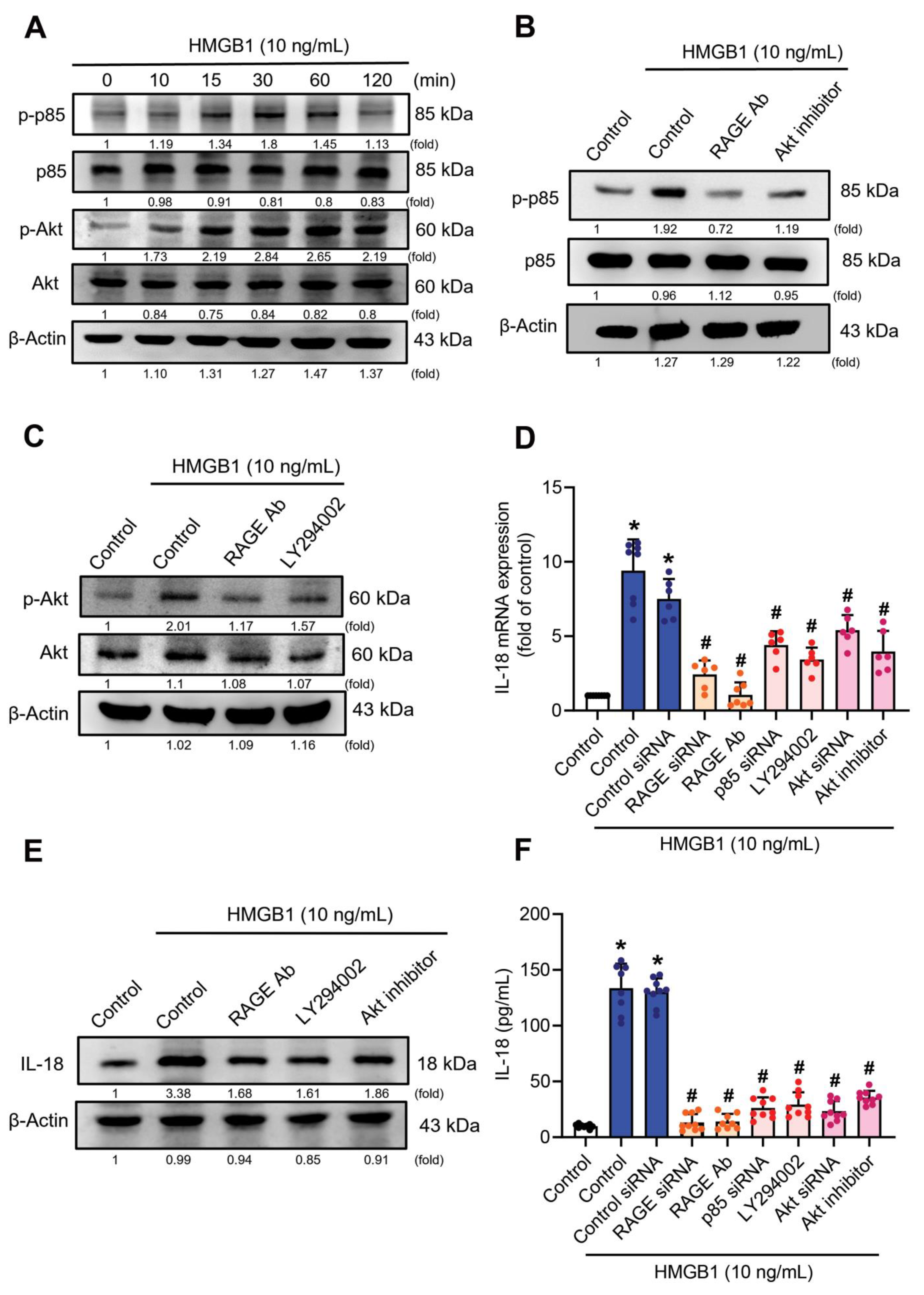
3.3. RAGE/p85/Akt Signaling Is Involved in HMGB1-Induced Increases of IL-18 Expression
3.4. mTOR Signaling Is Required for the HMGB1-Promotes IL-18 Expression
3.5. HMGB1-Induced Increases of IL-18 Expression in C2C12 Myoblasts Requires c-Jun Phosphorylation
3.6. Inhibition of HMGB1 Rescues In Vivo Skeletal Muscle Injury
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP-1 | activation protein-1 |
| ChIP | chromatin immunoprecipitation |
| BSA | bovine serum albumin |
| DAB | 3,3ʹ-diaminobenzidine tetrahydrochloride |
| DAPI | 4,6-diamidino-2-phenylindole |
| DMEM | Dulbecco’s modified Eagle’s medium |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GIMI | glycerol-induced muscle injury |
| GPU | graphics processing unit |
| H&E | hematoxylin and eosin |
| HMGB1 | high-mobility group box-1 |
| IGF-1 | insulin-like growth factor 1 |
| IHC | immunohistochemistry |
| IL-18 | interleukin-18 |
| mAb | monoclonal antibody |
| MuRF-1 | muscle RING-finger-1 |
| MyHC | myosin heavy chain |
| MyoD | myogenic differentiation |
| NF-κB | nuclear factor-κB |
| Pax-7 | paired box 7 |
| pfu | plaque-forming units |
| PBS | phosphate-buffered saline |
| PTA | phosphotungstic acid |
| qRT-PCR | quantitative real-time polymerase chain reaction |
| RAGE | receptor for advanced glycation end-products |
| ROI | region of interest |
| SD | standard deviation |
| shRNA | short hairpin RNA |
| siRNA | small interfering RNA |
| TA | tibialis anterior |
| TLR4 | toll-like receptor 4 |
References
- Fanzani, A.; Conraads, V.M.; Penna, F.; Martinet, W. Molecular and cellular mechanisms of skeletal muscle atrophy: An update. J. Cachex. Sarcopenia Muscle 2012, 3, 163–179. [Google Scholar] [CrossRef]
- Thomas, D.R. Loss of skeletal muscle mass in aging: Examining the relationship of starvation, sarcopenia and cachexia. Clin. Nutr. 2007, 26, 389–399. [Google Scholar] [CrossRef]
- Powers, S.K.; Lynch, G.S.; Murphy, K.; Reid, M.B.; Zijdewind, I. Disease-Induced Skeletal Muscle Atrophy and Fatigue. Med. Sci. Sports Exerc. 2016, 48, 2307–2319. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef]
- Ebert, S.M.; Al-Zougbi, A.; Bodine, S.C.; Adams, C.M. Skeletal Muscle Atrophy: Discovery of Mechanisms and Potential Therapies. Physiology 2019, 34, 232–239. [Google Scholar] [CrossRef]
- Suetta, C.; Hvid, L.G.; Justesen, L.; Christensen, U.; Neergaard, K.; Simonsen, L.; Ortenblad, N.; Magnusson, S.P.; Kjaer, M.; Aagaard, P. Effects of aging on human skeletal muscle after immobilization and retraining. J. Appl. Physiol. 2009, 107, 1172–1180. [Google Scholar] [CrossRef]
- Jameson, T.S.O.; Kilroe, S.P.; Fulford, J.; Abdelrahman, D.R.; Murton, A.J.; Dirks, M.L.; Stephens, F.B.; Wall, B.T. Muscle damaging eccentric exercise attenuates disuse-induced declines in daily myofibrillar protein synthesis and transiently prevents muscle atrophy in healthy men. Am. J. Physiol. Metab. 2021, 321, E674–E688. [Google Scholar] [CrossRef]
- Cheung, W.W.; Hao, S.; Wang, Z.; Ding, W.; Zheng, R.; Gonzalez, A.; Zhan, J.; Zhou, P.; Li, S.; Esparza, M.C.; et al. Vitamin D repletion ameliorates adipose tissue browning and muscle wasting in infantile nephropathic cystinosis-associated cachexia. J. Cachex. Sarcopenia Muscle 2019, 11, 120–134. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo Transcription Factors Induce the Atrophy-Related Ubiquitin Ligase Atrogin-1 and Cause Skeletal Muscle Atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Baczek, J.; Silkiewicz, M.; Wojszel, Z.B. Myostatin as a Biomarker of Muscle Wasting and other Pathologies-State of the Art and Knowledge Gaps. Nutrients 2020, 12, 2401. [Google Scholar] [CrossRef]
- Hawke, T.J.; Garry, D.J. Myogenic satellite cells: Physiology to molecular biology. J. Appl. Physiol. 2001, 91, 534–551. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, F.; Barberi, L.; Dobrowolny, G.; Villa Nova Bacurau, A.; Nicoletti, C.; Rizzuto, E.; Rosenthal, N.; Scicchitano, B.M.; Musarò, A. Effects of IGF-1 isoforms on muscle growth and sarcopenia. Aging Cell 2019, 18, e12954. [Google Scholar] [CrossRef]
- Howard, E.E.; Pasiakos, S.M.; A. Fussell, M.; Rodriguez, N.R. Skeletal Muscle Disuse Atrophy and the Rehabilitative Role of Protein in Recovery from Musculoskeletal Injury. Adv. Nutr. Int. Rev. J. 2020, 11, 989–1001. [Google Scholar] [CrossRef]
- Deger, S.M.; Hung, A.M.; Gamboa, J.; Siew, E.D.; Ellis, C.D.; Booker, C.; Sha, F.; Li, H.; Bian, A.; Stewart, T.G.; et al. Systemic inflammation is associated with exaggerated skeletal muscle protein catabolism in maintenance hemodialysis patients. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Gutiérrez, J.M.; Escalante, T.; Hernández, R.; Gastaldello, S.; Saravia-Otten, P.; Rucavado, A. Why is Skeletal Muscle Regeneration Impaired after Myonecrosis Induced by Viperid Snake Venoms? Toxins 2018, 10, 182. [Google Scholar] [CrossRef]
- Yanay, N.; Rabie, M.; Nevo, Y. Impaired Regeneration in Dystrophic Muscle-New Target for Therapy. Front. Mol. Neurosci. 2020, 13, 69. [Google Scholar]
- Li, S.; Zhou, X.; Zeng, R.; Lin, L.; Zou, X.; Yan, Y.; Lu, Z.; Xia, J.; Zhang, L.; Ni, S.; et al. YAP1 silencing attenuated lung injury/fibrosis but worsened diaphragmatic function by regulating oxidative stress and inflammation response in mice. Free Radic. Biol. Med. 2022, 193, 485–498. [Google Scholar] [CrossRef]
- Nguyen, T.T.N.; Choi, H.; Jun, H.-S. Preventive Effects of Dulaglutide on Disuse Muscle Atrophy Through Inhibition of Inflammation and Apoptosis by Induction of Hsp72 Expression. Front. Pharmacol. 2020, 11, 90. [Google Scholar] [CrossRef]
- Helmers, S.B.; Bruton, M.; Loell, I.; Ulfgren, A.-K.; Gracie, A.J.; McInnes, I.B.; Lundberg, I.E. Expression of interleukin-18 in muscle tissue of patients with polymyositis or dermatomyositis and effects of conventional immunosuppressive treatment. Rheumatology 2018, 57, 2149–2157. [Google Scholar] [CrossRef]
- Wang, R.; Wu, W.; Li, W.; Huang, S.; Li, Z.; Liu, R.; Shan, Z.; Zhang, C.; Wang, S. Activation of NLRP3 Inflammasome Promotes Foam Cell Formation in Vascular Smooth Muscle Cells and Atherogenesis Via HMGB. J. Am. Hear. Assoc. 2018, 7, e008596. [Google Scholar] [CrossRef]
- Kashiwamura, S.-I.; Ueda, H.; Okamura, H. Roles of Interleukin-18 in Tissue Destruction and Compensatory Reactions. J. Immunother. 2002, 25, S4–S11. [Google Scholar] [CrossRef]
- Li, C.-W.; Yu, K.; Shyh-Chang, N.; Li, G.-X.; Jiang, L.-J.; Yu, S.-L.; Xu, L.-Y.; Liu, R.-J.; Guo, Z.-J.; Xie, H.-Y.; et al. Circulating factors associated with sarcopenia during ageing and after intensive lifestyle intervention. J. Cachexia. Sarcopenia Muscle 2019, 10, 586–600. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, W.; Gao, R.; Su, Y.; Umehara, H.; Dong, L.; Gong, F. The role of high mobility group box chromosomal protein 1 in rheumatoid arthritis. Rheumatology 2013, 52, 1739–1747. [Google Scholar] [CrossRef][Green Version]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell. Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef]
- Day, J.; Otto, S.; Cash, K.; Eldi, P.; Hissaria, P.; Proudman, S.; Limaye, V.; Hayball, J. Aberrant Expression of High Mobility Group Box Protein 1 in the Idiopathic Inflammatory Myopathies. Front. Cell Dev. Biol. 2020, 8, 226. [Google Scholar] [CrossRef]
- Harris, H.E.; Andersson, U.; Pisetsky, D.S. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012, 8, 195–202. [Google Scholar] [CrossRef]
- Zong, M.; Bruton, J.; Grundtman, C.; Yang, H.; Li, J.H.; Alexanderson, H.; Palmblad, K.; Andersson, U.; Harris, H.E.; Lundberg, I.; et al. TLR4 as receptor for HMGB1 induced muscle dysfunction in myositis. Ann. Rheum. Dis. 2012, 72, 1390–1399. [Google Scholar] [CrossRef]
- Chiappalupi, S.; Sorci, G.; Vukasinovic, A.; Salvadori, L.; Sagheddu, R.; Coletti, D.; Renga, G.; Romani, L.; Donato, R.; Riuzzi, F. Targeting RAGE prevents muscle wasting and prolongs survival in cancer cachexia. J. Cachex. Sarcopenia Muscle 2020, 11, 929–946. [Google Scholar] [CrossRef]
- He, Q.; You, H.; Li, X.-M.; Liu, T.-H.; Wang, P.; Wang, B.-E. HMGB1 Promotes the Synthesis of Pro-IL-1β and Pro-IL-18 by Activation of p38 MAPK and NF-κB Through Receptors for Advanced Glycation End-products in Macrophages. Asian Pac. J. Cancer Prev. 2012, 13, 1365–1370. [Google Scholar] [CrossRef]
- Su, C.-M.; Wang, I.-C.; Liu, S.-C.; Sun, Y.; Jin, L.; Wang, S.-W.; Lee, H.-P.; Tseng, W.-P.; Tang, C.-H. Hypoxia induced mitogenic factor (HIMF) triggers angiogenesis by increasing interleukin-18 production in myoblasts. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef][Green Version]
- Wu, M.-H.; Lee, T.-H.; Lee, H.-P.; Li, T.-M.; Lee, I.-T.; Shieh, P.-C.; Tang, C.-H. Kuei-Lu-Er-Xian-Jiao extract enhances BMP-2 production in osteoblasts. Biomedicine 2017, 7, 2. [Google Scholar] [CrossRef]
- Lu, C.-C.; Tsai, H.-C.; Yang, D.-Y.; Wang, S.-W.; Tsai, M.-H.; Hua, C.-H.; Chen, K.-J.; Chen, M.Y.-C.; Lien, M.-Y.; Tang, C.-H. The Chemokine CCL4 Stimulates Angiopoietin-2 Expression and Angiogenesis via the MEK/ERK/STAT3 Pathway in Oral Squamous Cell Carcinoma. Biomedicines 2022, 10, 1612. [Google Scholar] [CrossRef]
- Achudhan, D.; Liu, S.; Lin, Y.; Lee, H.; Wang, S.; Huang, W.; Wu, Y.; Kuo, Y.; Tang, C. Antcin K inhibits VEGF-dependent angiogenesis in human rheumatoid arthritis synovial fibroblasts. J. Food Biochem. 2021, 46, e14022. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Suzuki, T.; Arai, H.; Tanabe, S.; Atomi, Y. Continuous mild heat stress induces differentiation of mammalian myoblasts, shifting fiber type from fast to slow. Am. J. Physiol. Physiol. 2010, 298, C140–C148. [Google Scholar] [CrossRef]
- Kasprzycka, P.; Archacka, K.; Kowalski, K.; Mierzejewski, B.; Zimowska, M.; Grabowska, I.; Piotrowski, M.; Rafałko, M.; Ryżko, A.; Irhashava, A.; et al. The factors present in regenerating muscles impact bone marrow-derived mesenchymal stromal/stem cell fusion with myoblasts. Stem Cell Res. Ther. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Liu, C.-M.; Hsieh, C.-L.; He, Y.-C.; Lo, S.-J.; Liang, J.-A.; Hsieh, T.-F.; Josson, S.; Chung, L.W.K.; Hung, M.-C.; Sung, S.-Y. In Vivo Targeting of ADAM9 Gene Expression Using Lentivirus-Delivered shRNA Suppresses Prostate Cancer Growth by Regulating REG4 Dependent Cell Cycle Progression. PLoS ONE 2013, 8, e53795. [Google Scholar] [CrossRef]
- Su, C.-M.; Chiang, Y.-C.; Huang, C.-Y.; Hsu, C.-J.; Fong, Y.-C.; Tang, C.-H. Osteopontin Promotes Oncostatin M Production in Human Osteoblasts: Implication of Rheumatoid Arthritis Therapy. J. Immunol. 2015, 195, 3355–3364. [Google Scholar] [CrossRef]
- Zhong, H.; May, M.J.; Jimi, E.; Ghosh, S. The Phosphorylation Status of Nuclear NF-ΚB Determines Its Association with CBP/p300 or HDAC-1. Mol. Cell 2002, 9, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Kawai, H.; Nishino, H.; Kusaka, K.; Naruo, T.; Tamaki, Y.; Iwasa, M. Experimental glycerol myopathy: A histological study. Acta Neuropathol. 1990, 80, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Londhe, P.; Guttridge, D.C. Inflammation induced loss of skeletal muscle. Bone 2015, 80, 131–142. [Google Scholar] [CrossRef]
- Schierbeck, H.; Lundbäck, P.; Palmblad, K.; Klevenvall, L.; Erlandsson-Harris, H.; Andersson, U.; Ottosson, L. Monoclonal Anti-HMGB1 (High Mobility Group Box Chromosomal Protein 1) Antibody Protection in Two Experimental Arthritis Models. Mol. Med. 2011, 17, 1039–1044. [Google Scholar] [CrossRef]
- Tieland, M.; Franssen, R.; Dullemeijer, C.; van Dronkelaar, C.; Kim, H.K.; Ispoglou, T.; Zhu, K.; Prince, R.L.; van Loon, L.J.; De Groot, L.C.P.G.M. The impact of dietary protein or amino acid supplementation on muscle mass and strength in elderly people: Individual participant data and meta-analysis of RCT’s. J. Nutr. Health Aging 2017, 21, 994–1001. [Google Scholar] [CrossRef]
- Li, R.; Zou, X.; Huang, H.; Yu, Y.; Zhang, H.; Liu, P.; Pan, S.; Ouyang, Y.; Shang, Y. HMGB1/PI3K/Akt/mTOR Signaling Participates in the Pathological Process of Acute Lung Injury by Regulating the Maturation and Function of Dendritic Cells. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Uluçkan, O.; Guinea-Viniegra, J.; Jimenez, M.; Wagner, E.F. Signalling in inflammatory skin disease by AP-1 (Fos/Jun). Clin. Exp. Rheumatol. 2015, 33, S44–S49. [Google Scholar]
- Howard, E.E.; Pasiakos, S.M.; Blesso, C.N.; Fussell, M.A.; Rodriguez, N.R. Divergent Roles of Inflammation in Skeletal Muscle Recovery From Injury. Front. Physiol. 2020, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, O.; Reid, M.B.; Berghe, G.V.D.; Vanhorebeek, I.; Hermans, G.; Rich, M.M.; Larsson, L. The Sick and the Weak: Neuropathies/Myopathies in the Critically Ill. Physiol. Rev. 2015, 95, 1025–1109. [Google Scholar] [CrossRef]
- Wang, M.; Gauthier, A.; Daley, L.; Dial, K.; Wu, J.; Woo, J.; Lin, M.; Ashby, C.; Mantell, L.L. The Role of HMGB1, a Nuclear Damage-Associated Molecular Pattern Molecule, in the Pathogenesis of Lung Diseases. Antioxidants Redox Signal. 2019, 31, 954–993. [Google Scholar] [CrossRef]
- Li, L.; Liu, H.; Tao, W.; Wen, S.; Fu, X.; Yu, S. Pharmacological Inhibition of HMGB1 Prevents Muscle Wasting. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Kamiya, M.; Mizoguchi, F.; Yasuda, S. Amelioration of inflammatory myopathies by glucagon-like peptide-1 receptor agonist via suppressing muscle fibre necroptosis. J. Cachex- Sarcopenia Muscle 2022, 13, 2118–2131. [Google Scholar] [CrossRef]
- Kamiya, M.; Mizoguchi, F.; Kawahata, K.; Wang, D.; Nishibori, M.; Day, J.; Louis, C.; Wicks, I.P.; Kohsaka, H.; Yasuda, S. Targeting necroptosis in muscle fibers ameliorates inflammatory myopathies. Nat. Commun. 2022, 13, 166. [Google Scholar] [CrossRef]
- Yang, X.; Xue, P.; Liu, X.; Xu, X.; Chen, Z. HMGB1/autophagy pathway mediates the atrophic effect of TGF-β1 in denervated skeletal muscle. Cell Commun. Signal. 2018, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.W.; Kim, J.-Y.; Yoon, J.P.; Suh, D.W.; Yeo, W.J.; Lee, Y.-S. Atrogin1-induced loss of aquaporin 4 in myocytes leads to skeletal muscle atrophy. Sci. Rep. 2020, 10, 14189. [Google Scholar] [CrossRef]
- Careccia, G.; Saclier, M.; Tirone, M.; Ruggieri, E.; Principi, E.; Raffaghello, L.; Torchio, S.; Recchia, D.; Canepari, M.; Gorzanelli, A.; et al. Rebalancing expression of HMGB1 redox isoforms to counteract muscular dystrophy. Sci. Transl. Med. 2021, 13, eaay8416. [Google Scholar] [CrossRef]
- Tirone, M.; Tran, N.L.; Ceriotti, C.; Gorzanelli, A.; Canepari, M.; Bottinelli, R.; Raucci, A.; Di Maggio, S.; Santiago, C.; Mellado, M.; et al. High mobility group box 1 orchestrates tissue regeneration via CXCR4. J. Exp. Med. 2017, 215, 303–318. [Google Scholar] [CrossRef]
- Vezzoli, M.; Castellani, P.; Corna, G.; Castiglioni, A.; Bosurgi, L.; Monno, A.; Brunelli, S.; Manfredi, A.A.; Rubartelli, A.; Rovere-Querini, P. High-Mobility Group Box 1 Release and Redox Regulation Accompany Regeneration and Remodeling of Skeletal Muscle. Antioxidants Redox Signal. 2011, 15, 2161–2174. [Google Scholar] [CrossRef]
- Witt, S.H.; Granzier, H.; Witt, C.C.; Labeit, S. MURF-1 and MURF-2 Target a Specific Subset of Myofibrillar Proteins Redundantly: Towards Understanding MURF-dependent Muscle Ubiquitination. J. Mol. Biol. 2005, 350, 713–722. [Google Scholar] [CrossRef]
- Vilchinskaya, N.; Altaeva, E.; Lomonosova, Y. Gaining insight into the role of FoxO1 in the progression of disuse-induced skeletal muscle atrophy. Adv. Biol. Regul. 2022, 85, 100903. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Parr, T.; Brameld, J.M. Myosin heavy chain mRNA isoforms are expressed in two distinct cohorts during C2C12 myogenesis. J. Muscle Res. Cell Motil. 2011, 32, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wei, G.; Liu, B.; Zhou, X.; Xiao, H.; Dong, N.; Li, F. The Role of High Mobility Group Box 1 Protein in Interleukin-18-Induced Myofibroblastic Transition of Valvular Interstitial Cells. Cardiology 2016, 135, 168–178. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef]
- Muth, I.E.; Zschüntzsch, J.; Kleinschnitz, K.; Wrede, A.; Gerhardt, E.; Balcarek, P.; Schreiber-Katz, O.; Zierz, S.; Dalakas, M.C.; Voll, R.; et al. HMGB1 and RAGE in skeletal muscle inflammation: Implications for protein accumulation in inclusion body myositis. Exp. Neurol. 2015, 271, 189–197. [Google Scholar] [CrossRef]
- Riuzzi, F.; Sorci, G.; Donato, R. The Amphoterin (HMGB1)/Receptor for Advanced Glycation End Products (RAGE) Pair Modulates Myoblast Proliferation, Apoptosis, Adhesiveness, Migration, and Invasiveness. J. Biol. Chem. 2006, 281, 8242–8253. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): From Risk Factors to Therapeutic Targeting. Cells 2020, 9, 383. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Zhang, X.; Peng, A.; He, M.; Lei, Z.; Wang, Y. TLR4-HMGB1 signaling pathway affects the inflammatory reaction of autoimmune myositis by regulating MHC-I. Int. Immunopharmacol. 2016, 41, 74–81. [Google Scholar] [CrossRef]
- Mahdy, M.A.A. Glycerol-induced injury as a new model of muscle regeneration. Cell Tissue Res. 2018, 374, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Choi, S.; Liu, X.; Zhang, M.; Schageman, J.J.; Lee, S.Y.; Hart, R.; Lin, L.; Thurmond, F.A.; Williams, R.S. Highly Coordinated Gene Regulation in Mouse Skeletal Muscle Regeneration. J. Biol. Chem. 2003, 278, 8826–8836. [Google Scholar] [CrossRef] [PubMed]
- Mahdy, M.A.A.; Warita, K.; Hosaka, Y.Z. Glycerol induces early fibrosis in regenerating rat skeletal muscle. J. Veter. Med. Sci. 2018, 80, 1646–1649. [Google Scholar] [CrossRef]
- Rigon, M.; Hörner, S.J.; Straka, T.; Bieback, K.; Gretz, N.; Hafner, M.; Rudolf, R. Effects of ASC Application on Endplate Regeneration Upon Glycerol-Induced Muscle Damage. Front. Mol. Neurosci. 2020, 13, 107. [Google Scholar] [CrossRef]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a Late Mediator of Endotoxin Lethality in Mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Fortin, C.F.; Ear, T.; McDonald, P.P. utocrine role of endogenous interleukin-18 on inflammatory cytokine generation by human neutrophils. FASEB J. 2008, 23, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 Binding Protein. Front. Immunol. 2013, 4, 289. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Jonuleit, H.; Schmitt, E.; Müller, G.; Yamauchi, H.; Kurimoto, M.; Knop, J.; Enk, A.H. Production of functional IL-18 by different subtypes of murine and human dendritic cells (DC): DC-derived IL-18 enhances IL-12-dependent Th1 development. Eur. J. Immunol. 1998, 28, 3231–3239. [Google Scholar] [CrossRef]
- Robertson, S.E.; Young, J.D.; Kitson, S.; Pitt, A.; Evans, J.; Roes, J.; Karaoglu, D.; Santora, L.; Ghayur, T.; Liew, F.Y.; et al. Expression and alternative processing of IL-18 inhuman neutrophils. Eur. J. Immunol. 2006, 36, 722–731. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, T.-L.; Tang, C.-H.; Chang, S.L.-Y.; Tsai, C.-H.; Chen, H.-T.; Su, C.-M. HMGB1 Promotes In Vitro and In Vivo Skeletal Muscle Atrophy through an IL-18-Dependent Mechanism. Cells 2022, 11, 3936. https://doi.org/10.3390/cells11233936
Ho T-L, Tang C-H, Chang SL-Y, Tsai C-H, Chen H-T, Su C-M. HMGB1 Promotes In Vitro and In Vivo Skeletal Muscle Atrophy through an IL-18-Dependent Mechanism. Cells. 2022; 11(23):3936. https://doi.org/10.3390/cells11233936
Chicago/Turabian StyleHo, Trung-Loc, Chih-Hsin Tang, Sunny Li-Yun Chang, Chun-Hao Tsai, Hsien-Te Chen, and Chen-Ming Su. 2022. "HMGB1 Promotes In Vitro and In Vivo Skeletal Muscle Atrophy through an IL-18-Dependent Mechanism" Cells 11, no. 23: 3936. https://doi.org/10.3390/cells11233936
APA StyleHo, T.-L., Tang, C.-H., Chang, S. L.-Y., Tsai, C.-H., Chen, H.-T., & Su, C.-M. (2022). HMGB1 Promotes In Vitro and In Vivo Skeletal Muscle Atrophy through an IL-18-Dependent Mechanism. Cells, 11(23), 3936. https://doi.org/10.3390/cells11233936