

Bioinformatic Analysis Revealed the Essential Regulatory Genes and Pathways of Early and Advanced Atherosclerotic Plaque in Humans

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microarray Data Collection
2.2. Gene Set Enrichment and Differentially Expressed Genes Analysis
2.3. Gene Expression Patterns Analysis
2.4. Constructing Weighted Co-expression Gene Networks
2.5. GO and KEGG Enrichment Analyses
2.6. Protein-Protein Interaction Network Analysis
2.7. Logistic Regression Models with the ROC Curve
3. Results
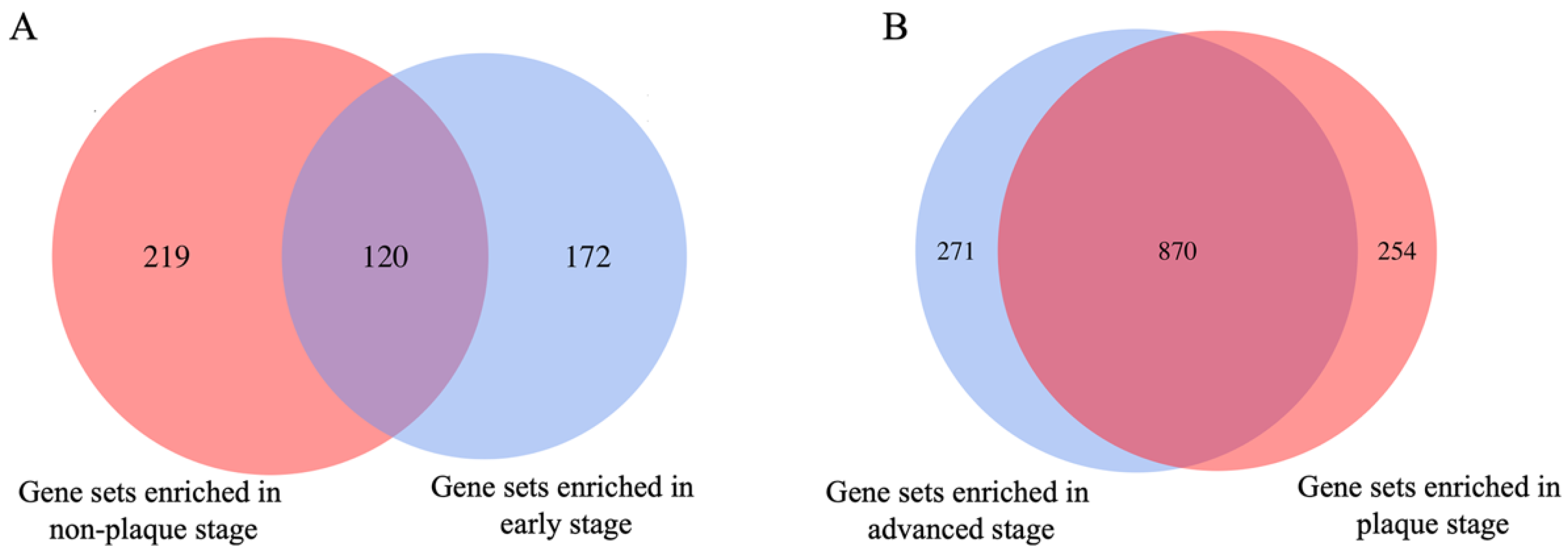
3.1. Identification of Key Gene Sets in Different Stages of Atherosclerotic Plaque
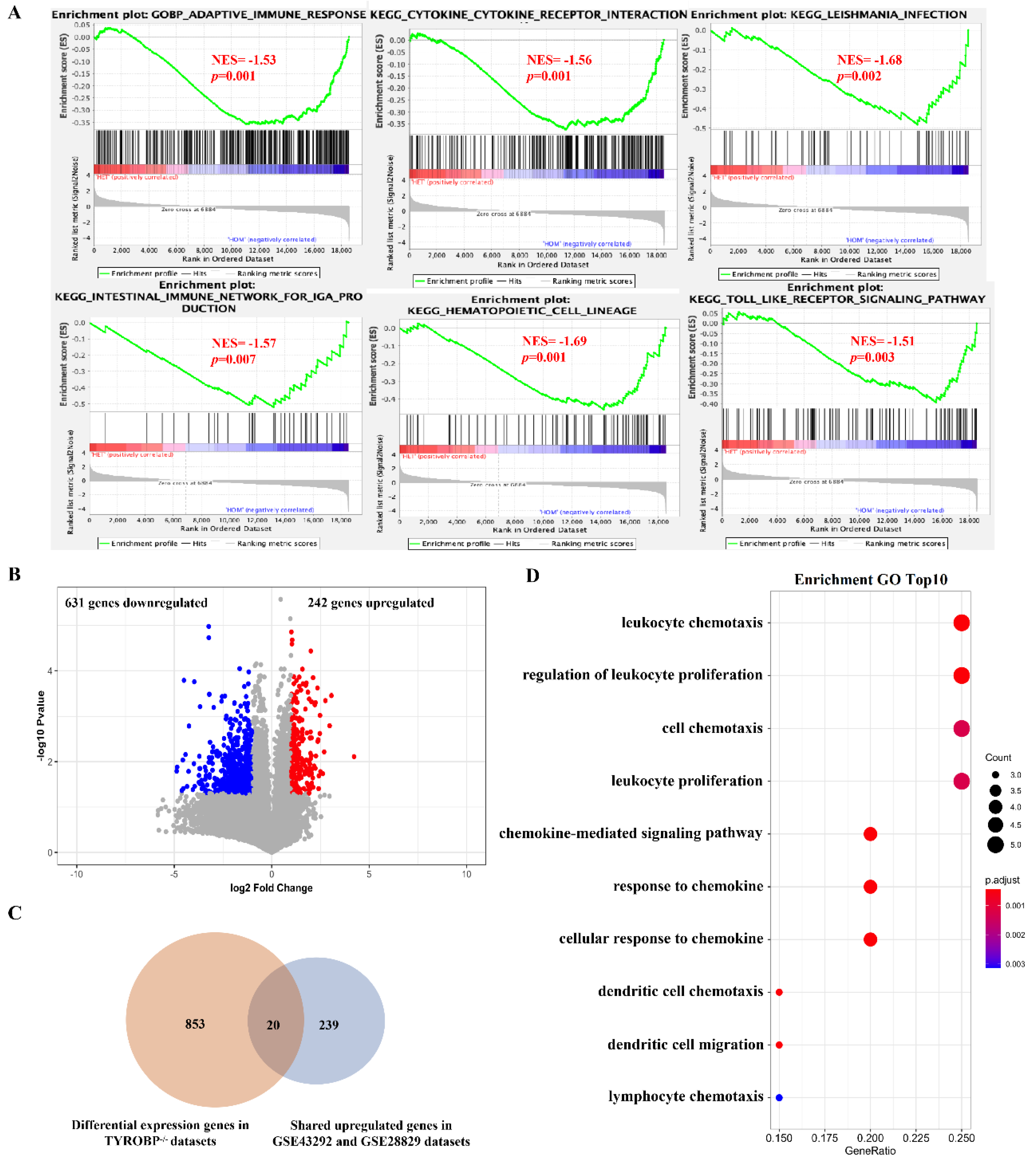
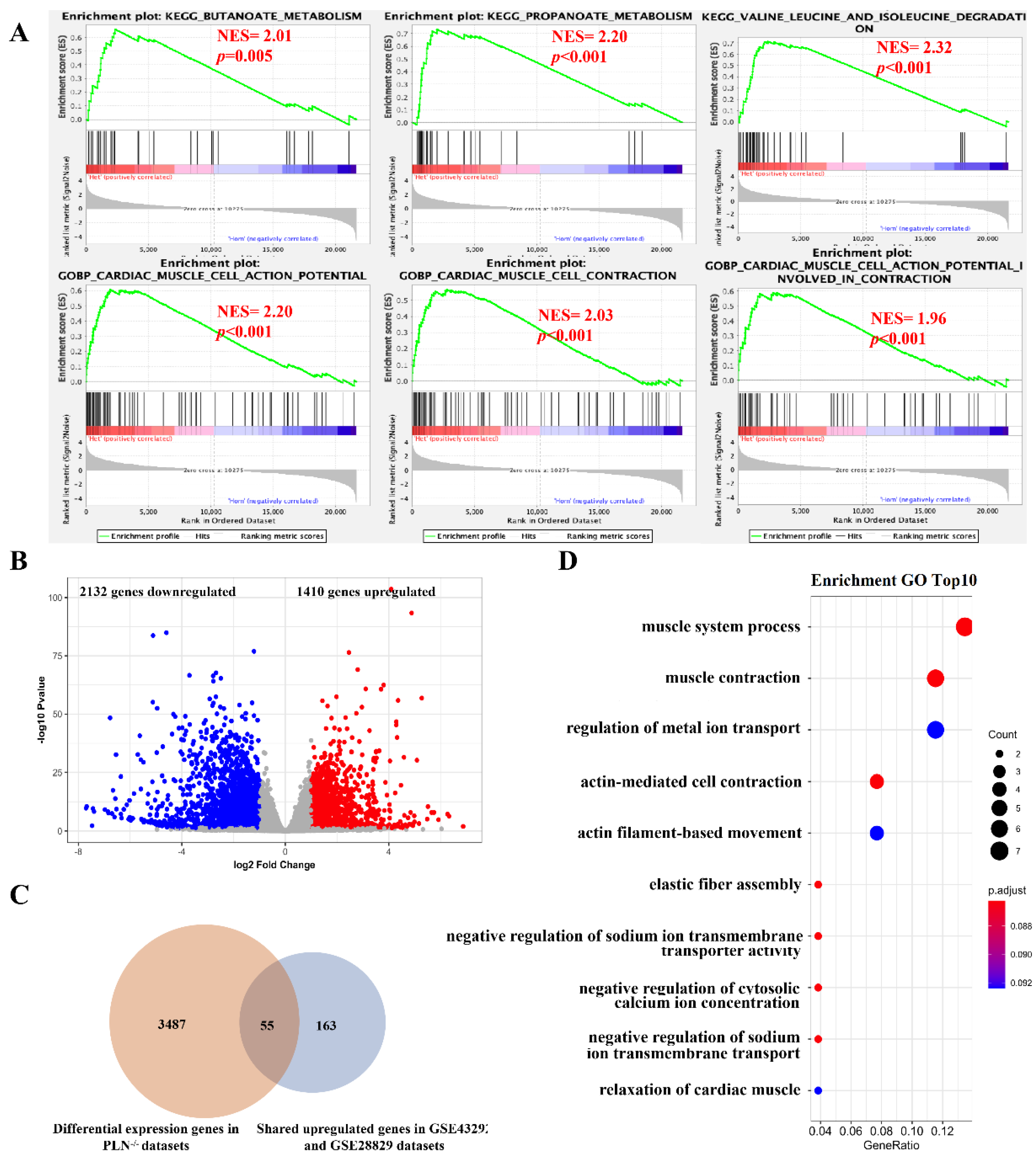
3.2. The Mainly Regulated Pathways in Different Stages of Atherosclerotic Plaque
3.3. Remodeling of the Molecular Interaction Structure in Atherosclerosis
3.4. Identification of Key Genes As Potential Therapeutic targets for Atherosclerosis
3.5. Validation of Potential Therapeutic Target Genes Using Gene Deficiency Mouse Expression Pro-Files
4. Discussion
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aziz, M.; Yadav, K. Pathogenesis of atherosclerosis a review. Med. Clin. Rev. 2016, 2, 1–6. [Google Scholar]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research progress on the relationship between atherosclerosis and inflammation. Biomole 2018, 8, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Nakamura, T.; Enomoto, S.; Kawahito, H.; Kurata, H.; Nakahara, Y.; Ijichi, T. Prevalence of coronary artery disease in Japanese patients with cerebral infarction. Circ. J. 2008, 72, 404–408. [Google Scholar] [CrossRef] [Green Version]
- Hiwasa, T.; Zhang, X.-M.; Kimura, R.; Ohno, M.; Chen, P.-M.; Nishi, E.; Ono, K.; Kimura, T.; Kamitsukasa, I.; Wada, T. Elevated adiponectin antibody levels in sera of patients with atherosclerosis-related coronary artery disease, cerebral infarction and diabetes mellitus. J. Circ. Biomark 2016, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, C.; Wu, X.; Xie, Q.; Li, L.; Chen, Y.; Yan, H.; Ren, P.; Huang, X. Identification of genes and pathways related to atherosclerosis comorbidity and depressive behavior via RNA-seq and bioinformation analysis in ApoE−/− mice. Ann. Transl. Med. 2019, 7, 733. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [Green Version]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Guella, I.; Rimoldi, V.; Asselta, R.; Francolini, M.; Martinelli, N.; Girelli, D.; Peyvandi, F.; Tubaro, M.; Merlini, P.A.; Mannucci, P.M.; et al. Association and functional analyses of MEF2A as a susceptibility gene for premature myocardial infarction and coronary artery disease. Circ.-Cardiovasc. Gene 2009, 2, 165–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivapalaratnam, S.; Motazacker, M.M.; Maiwald, S.; Hovingh, G.K.; Kastelein, J.; Levi, M.; Trip, M.; Dallinga-Thie, G.M. Genome-Wide Association Studies in Atherosclerosis. Curr. Atheroscler. Rep. 2011, 13, 225–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perumal, V.; Mahalingam, V. Insights from the clustering of microarray data associated with the heart disease. Bioinformation 2013, 9, 759–765. [Google Scholar] [CrossRef]
- Boyce, K.; Kriete, A.; Nagatomi, S.; Kelder, B.; Coschigano, K.; Kopchick, J.J. Phenotypical enrichment strategies for microarray data analysis applied in a type II diabetes study. Omics 2005, 9, 251. [Google Scholar] [CrossRef]
- Hou, C.; Feng, W.; Wei, S.; Wang, Y.; Xu, X.; Wei, J.; Ma, Z.; Du, Y.; Guo, J.; He, Y. Bioinformatics Analysis of Key Differentially Expressed Genes in Nonalcoholic Fatty Liver Disease Mice Models. Gene Expr. 2018, 19, 25–35. [Google Scholar] [CrossRef]
- Wang, J.; Wei, B.; Cao, S.; Xu, F.; Chen, W.; Lin, H.; Du, C.; Sun, Z. Identification by microarray technology of key genes involved in the progression of carotid atherosclerotic plaque. Genes Genet Syst. 2014, 89, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Zhang, X.; Pan, L.; Tian, X.; Dong, P. Identification of Key Pathways and Genes in Advanced Coronary Atherosclerosis Using Bioinformatics Analysis. Biomed. Res. Int. 2017, 2017, 4323496. [Google Scholar] [CrossRef] [Green Version]
- Perisic, L.; Aldi, S.; Sun, Y.; Folkersen, L.; Razuvaev, A.; Roy, J.; Lengquist, M.; Åkesson, S.; Wheelock, C.; Maegdefessel, L. Gene expression signatures, pathways and networks in carotid atherosclerosis. J. Intern. Med. 2016, 279, 293–308. [Google Scholar] [CrossRef] [Green Version]
- Perisic, L.M.; Rykaczewska, U.; Razuvaev, A.; Sabater-Lleal, M.; Lengquist, M.; Miller, C.L.; Ericsson, I.; Rhl, S.; Kronqvist, M.; Aldi, S. Phenotypic Modulation of Smooth Muscle Cells in Atherosclerosis Is Associated With Downregulation of LMOD1, SYNPO2, PDLIM7, PLN, and SYNM. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1947–1961. [Google Scholar] [CrossRef] [Green Version]
- Ayari, H.; Bricca, G. Identification of two genes potentially associated in iron-heme homeostasis in human carotid plaque using microarray analysis. J. Biosci. 2013, 38, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Manthey, H.D.; Drechsler, M.; Lievens, D.; Megens, R.T.; Soehnlein, O.; Busch, M.; Manca, M.; Koenen, R.R.; Pelisek, J. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 2012, 125, 1673–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charakida, M.; O'Neil, F.; Masi, S.; Papageorgiou, N.; Tousoulis, D. Inflammatory disorders and atherosclerosis: New therapeutic approaches. Curr. Pharm Des. 2011, 17, 4111–4120. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic. Acids. Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic. Acids. Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Barabasi, A.-L.; Oltvai, Z.N. Network biology: Understanding the cell's functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Zhao, L.; Zhao, W.; Weng, J. Dissecting the mechanism of carotid atherosclerosis from the perspective of regulation. Int. J. Mol. Med. 2014, 34, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Kuai, D.; Yang, Y.; Yang, G.; Wei, Z.; Zhao, W. Screening of potential gene markers for predicting carotid atheroma plaque formation using bioinformatics approaches. Mol. Med. Rep. 2017, 15, 2039–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Qin, X.; Zhou, X.; Zhou, J.; Wen, P.; Chen, S.; Wu, M.; Wu, Y.; Zhuang, J. Analysis of genes and underlying mechanisms involved in foam cells formation and atherosclerosis development. PeerJ 2020, 8, e10336. [Google Scholar] [CrossRef]
- Yang, R.; Yao, L.; Du, C.; Wu, Y. Identification of key pathways and core genes involved in atherosclerotic plaque progression. Ann. Transl. Med. 2021, 9, 267. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, D.; Liu, Y.; Zhang, X.; Wan, Z.; Wang, J.; Su, T.; Duan, L.; Wang, Y.; Zhang, Y. Six-gene signature associated with immune cells in the progression of atherosclerosis discovered by comprehensive bioinformatics analyses. Cardiovasc. Ther. 2020, 2020, 1230513. [Google Scholar] [CrossRef]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Mussbacher, M.; Galkina, E.V. Functional Role of B Cells in Atherosclerosis. Cells 2021, 10, 270. [Google Scholar] [CrossRef]
- Chen, M.; Chen, S.; Yang, D.; Zhou, J.; Liu, B.; Chen, Y.; Ye, W.; Zhang, H.; Ji, L.; Zheng, Y. Weighted Gene Co-expression Network Analysis Identifies Crucial Genes Mediating Progression of Carotid Plaque. Front. Physiol. 2021, 12, 601952. [Google Scholar] [CrossRef]
- Demigné, C.; Yacoub, C.; Morand, C.; RéMésy, C. Interactions between propionate and amino acid metabolism in isolated sheep hepatocytes. Br. J. Nutr. 1991, 65, 301–317. [Google Scholar] [CrossRef] [Green Version]
- Layman, D.K.; Walker, D.A. Potential importance of leucine in treatment of obesity and the metabolic syndrome. J. Nutr. 2006, 136, 319S–323S. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Jiang, H.; Li, L.; Chen, F.; Liu, Y.; Zhou, M.; Wang, J.; Jiang, J.; Li, X.; Fan, X. Branched-chain amino acid catabolism promotes thrombosis risk by enhancing tropomodulin-3 propionylation in platelets. Circulation 2020, 142, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Granger, C.B.; Craig, D.; Haynes, C.; Bain, J.; Stevens, R.D.; Hauser, E.R.; Newgard, C.B.; Kraus, W.E.; Newby, L.K. Validation of the association between a branched chain amino acid metabolite profile and extremes of coronary artery disease in patients referred for cardiac catheterization. Atherosclerosis 2014, 232, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, K.; Page, S.; Walli, A.K.; Neumeier, D.; Baeuerle, P.A. Role of nuclear factor-kappa B in atherogenesis. Exp. Physiol. 1997, 82, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Pan, H.; Xue, C.; Auerbach, B.J.; Fan, J.; Reilly, M.P. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 2020, 142, 2060–2075. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Kovanen, P.T.; Rezaee, M.; Banach, M.; Sahebkar, A. Ca2+ Flux: Searching for a role in efferocytosis of apoptotic cells in atherosclerosis. J. Clin. Med. 2019, 8, 2047. [Google Scholar] [CrossRef] [Green Version]
- Martinet, W.; De Meyer, G.R. Autophagy in atherosclerosis. Curr. Atheroscler. Rep. 2008, 10, 216–223. [Google Scholar] [CrossRef]
- Somers, K.D.; Murphey, M.M. Multinucleation in response to cytochalasin B: A common feature in several human tumor cell lines. Cancer Res. 1982, 42, 2575–2578. [Google Scholar]
- Tsiantoulas, D.; Sage, A.P.; Mallat, Z.; Binder, C.J. Targeting B cells in atherosclerosis: Closing the gap from bench to bedside. TArter. Throm. Vasc. 2015, 35, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Litvinova, L.S.; Mazunin, I.O.; Gutsol, A.A.; Sokhonevich, N. Dose-response effect of steroid hormones on Gfi1 and U2af1l4 gene expression in T lymphocytes at different stages of differentiation. Mol. Biol. 2013, 47, 572–580. [Google Scholar] [CrossRef]
- Al Barashdi, M.A.; Ali, A.; McMullin, M.F.; Mills, K. Protein tyrosine phosphatase receptor type C (PTPRC or CD45). J. Clin. Pathol. 2021, 74, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Takai, T.; Ono, M.; Hiki Da, M.; Ohmori, H.; Ravetch, J.V. Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature 1996, 379, 346–349. [Google Scholar] [CrossRef]
- Chen, L.; Yuan, L.; Wang, Y.; Wang, G.; Zhu, Y.; Cao, R.; Qian, G.; Xie, C.; Liu, X.; Xiao, Y. Co-expression network analysis identified FCER1G in association with progression and prognosis in human clear cell renal cell carcinoma. Int. J. Biol. Sci. 2017, 13, 1361–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Zhang, Q.; Zheng, H.; Hong, C.; Yi, L. Novel Mutations in the β2 Integrin Gene (ITGB2) in a Moderate Leukocyte Adhesion Defect type 1 Patient. Arch. Iran. Med. 2018, 21, 296–301. [Google Scholar]
- Gratuze, M.; Leyns, C.E.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulland, T.K.; Colonna, M. TREM2—a key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, H.; Chen, Y.; Wang, S.; Chen, Z.; Liu, Z.; Wang, J. Identifying RBM47, HCK, CD53, TYROBP, and HAVCR2 as hub genes in advanced atherosclerotic plaques by network-based analysis and validation. Front Genet. 2021, 11, 1770. [Google Scholar] [CrossRef]
- Nanda, V.; Wang, T.; Pjanic, M.; Liu, B.; Nguyen, T.; Matic, L.P.; Hedin, U.; Koplev, S.; Ma, L.; Franzén, O. Functional regulatory mechanism of smooth muscle cell-restricted LMOD1 coronary artery disease locus. PLoS Genet. 2018, 14, e1007755. [Google Scholar] [CrossRef]
- Ong, R.W.; AlSaman, A.; Selcen, D.; Arabshahi, A.; Yau, K.S.; Ravenscroft, G.; Duff, R.M.; Atkinson, V.; Allcock, R.J.; Laing, N.G. Novel cofilin-2 (CFL2) four base pair deletion causing nemaline myopathy. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1058–1060. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GS | Types | p Value |
|---|---|---|
| Myofibril assembly | GOBP | <0.001 a and <0.001 b |
| Cell communication by electrical coupling | GOBP | <0.001 a and 0.005 b |
| Propanoate metabolism | KEGG | 0.018 a and <0.001 b |
| Butanoate metabolism | KEGG | 0.011 a and <0.001 b |
| Hypertrophic cardiomyopathy hcm | KEGG | <0.001 a and <0.001 b |
| Dilated cardiomyopathy | KEGG | <0.001 a and <0.001 b |
| Arrhythmogenic right ventricular cardiomyopathy arvc | KEGG | <0.001 a and 0.006 b |
| Valine leucine and isoleucine degradation | KEGG | 0.02 a and 0.006 b |
| Adaptive immune response | GOBP | <0.001 c and <0.001 d |
| Leukocyte migration | GOBP | <0.001 c and <0.001 d |
| Positive regulation of leukocyte cell cell adhesion | GOBP | <0.001 c and <0.001 d |
| Positive regulation of cell activation | GOBP | <0.001 c and <0.001 d |
| Leukocyte cell cell adhesion | GOBP | <0.001 c and <0.001 d |
| Cytokine cytokine receptor interaction | KEGG | <0.001 c and <0.001 d |
| Leishmania infection | KEGG | <0.001 c and <0.001 d |
| Intestinal immune network for iga production | KEGG | <0.001 c and <0.001 d |
| B cell receptor signaling pathway | KEGG | <0.001 c and <0.001 d |
| Hematopoietic cell lineage | KEGG | <0.001 c and <0.001 d |
| Toll-like receptor signaling pathway | KEGG | <0.001 c and <0.001 d |
| Genes | Full Name | Related Pathway (Gene Cards) | MGI Phenotype | Phenotypes from GWAS Catalog |
|---|---|---|---|---|
| PTPRC | Receptor-type tyrosine-protein phosphatase C | B Cell Receptor Signaling Pathway | cardiovascular system phenotype | low density lipoprotein cholesterol measurement |
| FCGR2B | Low affinity immunoglobulin gamma Fc region receptor II-b | B Cell Receptor Signaling Pathway | cardiovascular system phenotype | lipid measurement |
| FCER1G | High affinity immunoglobulin epsilon receptor subunit gamma | Innate Immune System | cardiovascular system phenotype | lipoprotein measurement |
| ITGB2 | Integrin Subunit Beta 2 | ERK Signaling | cardiovascular system phenotype | high density lipoprotein cholesterol measurement |
| TYROBP | TYRO protein tyrosine kinase-binding protein | Innate Immune System | immune system phenotype | NA |
| LMOD1 | Leiomodin-1 | Smooth Muscle Contraction | NA | coronary artery disease |
| CFL2 | Cofilin-2 | NA | muscle phenotype | low density lipoprotein cholesterol measurement |
| PLN | Phospholamban | Activation of cAMP-Dependent PKA | cardiovascular system phenotype | resting heart rate |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, L.; Palos-Jasso, A.; Yi, Y.; Qin, M.; Qiu, L.; Yang, X.; Zhang, Y.; Yu, J. Bioinformatic Analysis Revealed the Essential Regulatory Genes and Pathways of Early and Advanced Atherosclerotic Plaque in Humans. Cells 2022, 11, 3976. https://doi.org/10.3390/cells11243976
He L, Palos-Jasso A, Yi Y, Qin M, Qiu L, Yang X, Zhang Y, Yu J. Bioinformatic Analysis Revealed the Essential Regulatory Genes and Pathways of Early and Advanced Atherosclerotic Plaque in Humans. Cells. 2022; 11(24):3976. https://doi.org/10.3390/cells11243976
Chicago/Turabian StyleHe, Luling, Andrea Palos-Jasso, Yao Yi, Manman Qin, Liang Qiu, Xiaofeng Yang, Yifeng Zhang, and Jun Yu. 2022. "Bioinformatic Analysis Revealed the Essential Regulatory Genes and Pathways of Early and Advanced Atherosclerotic Plaque in Humans" Cells 11, no. 24: 3976. https://doi.org/10.3390/cells11243976
APA StyleHe, L., Palos-Jasso, A., Yi, Y., Qin, M., Qiu, L., Yang, X., Zhang, Y., & Yu, J. (2022). Bioinformatic Analysis Revealed the Essential Regulatory Genes and Pathways of Early and Advanced Atherosclerotic Plaque in Humans. Cells, 11(24), 3976. https://doi.org/10.3390/cells11243976