

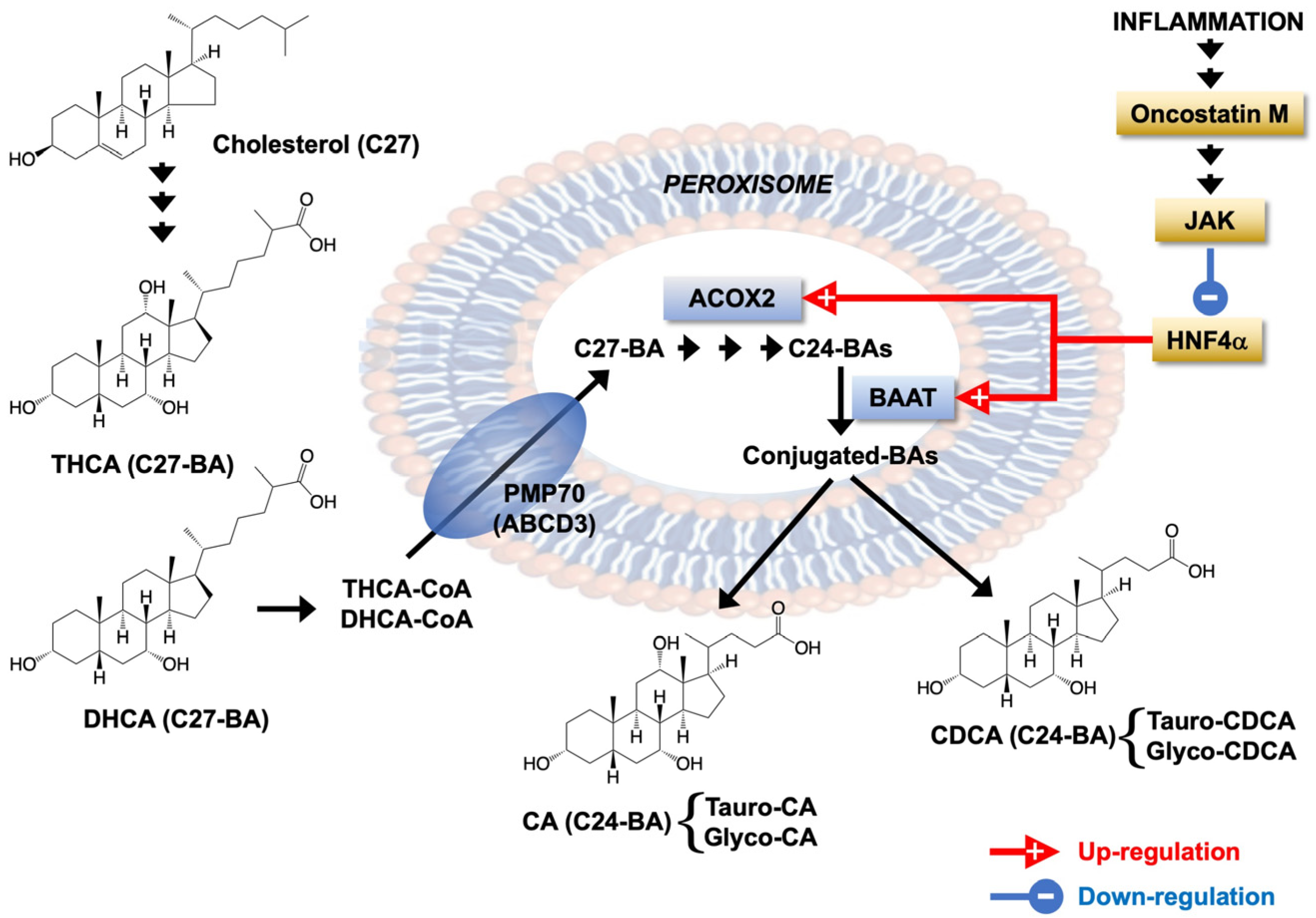
Impact of Liver Inflammation on Bile Acid Side Chain Shortening and Amidation

 , ,
, ,  , ,
, ,  , ,
, ,  , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics
2.2. Cell Lines
2.3. Chemicals
2.4. In Vitro Studies
2.5. Determination of Gene Expression
2.6. Immunoblotting
2.7. Bile Acid Measurement
2.8. In Silico Study
2.9. Validation Study
2.10. Statistical Methods
3. Results
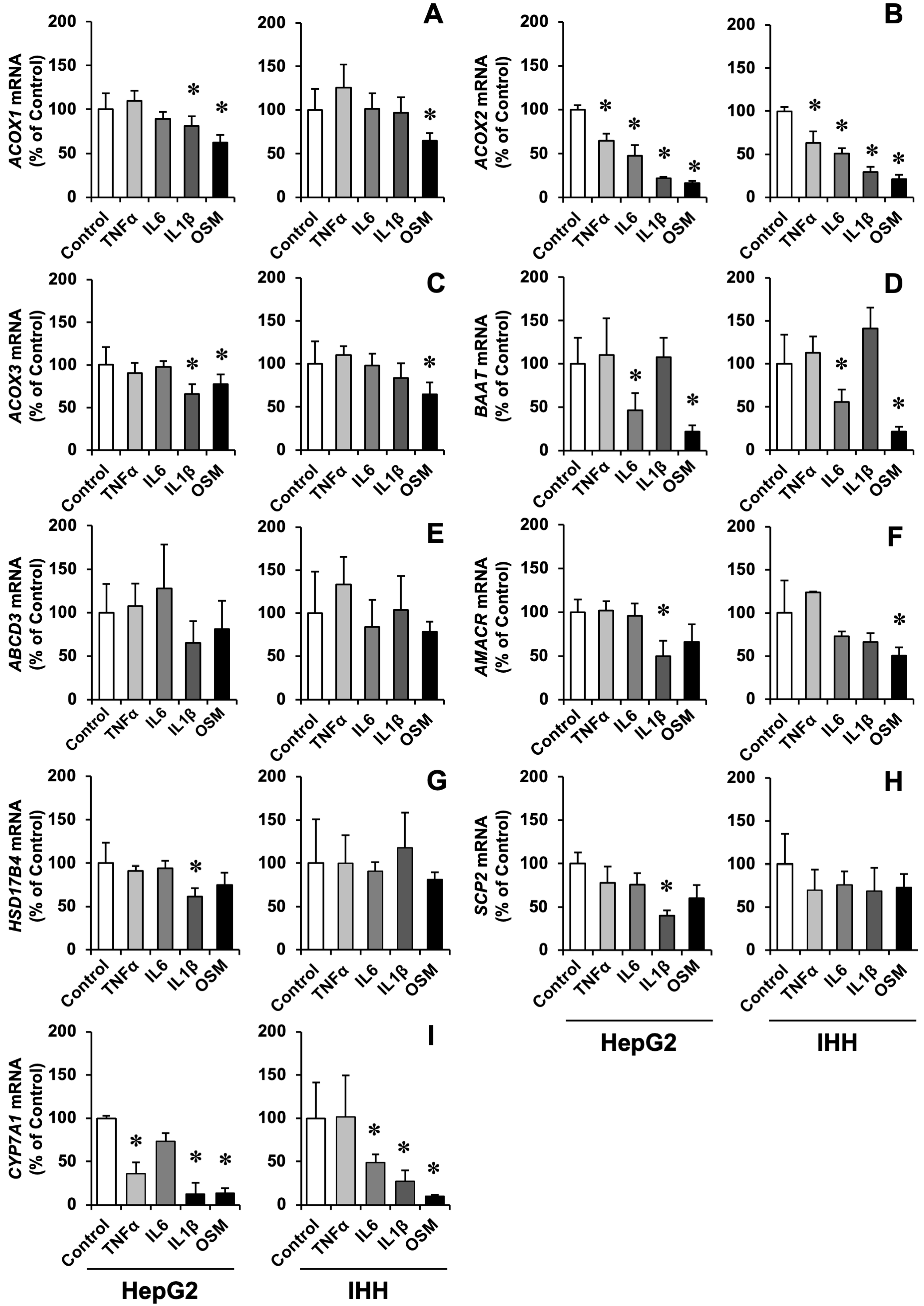
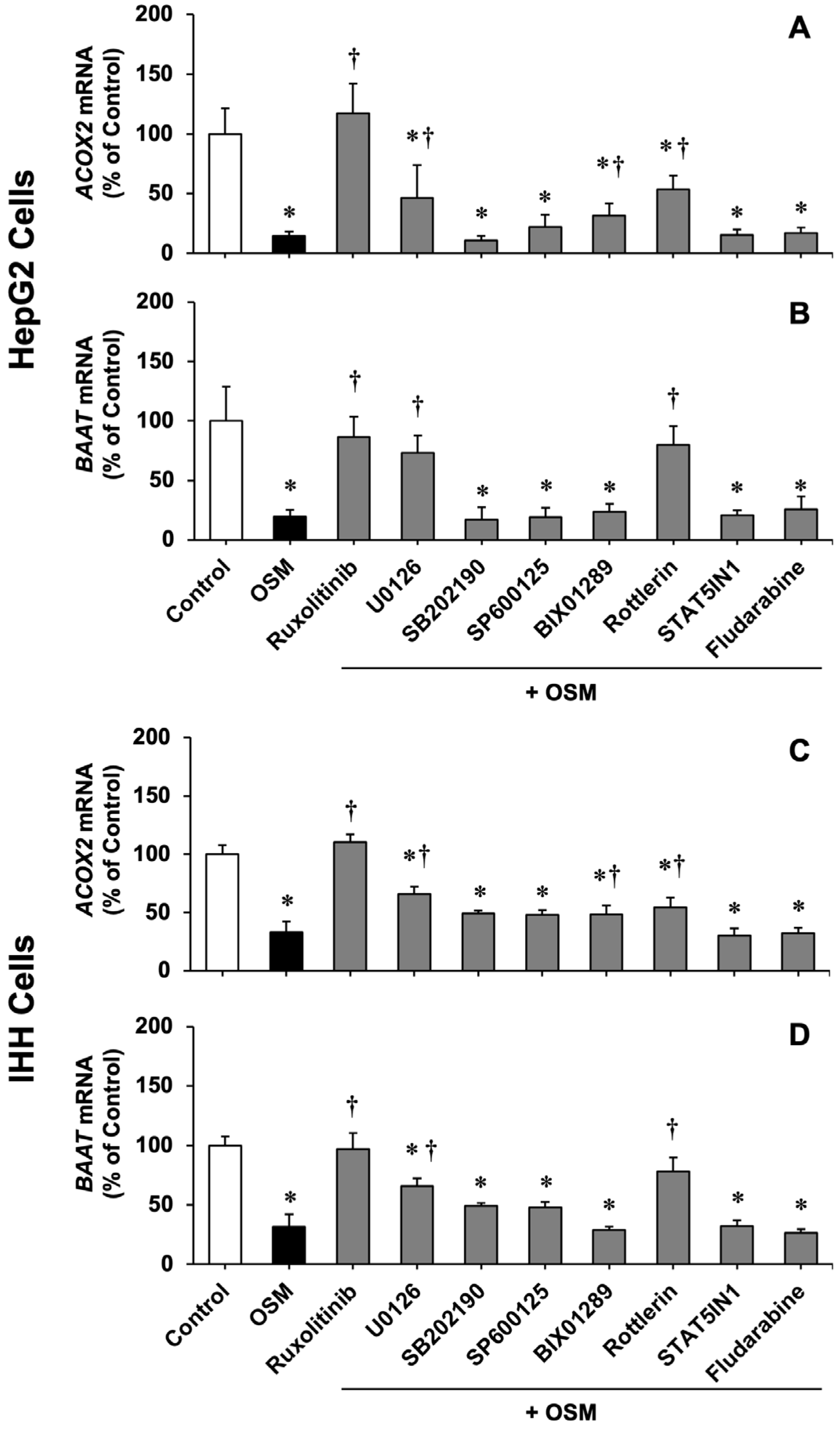
3.1. Cytokine-Induced ACOX2 and BAAT Down-Regulation
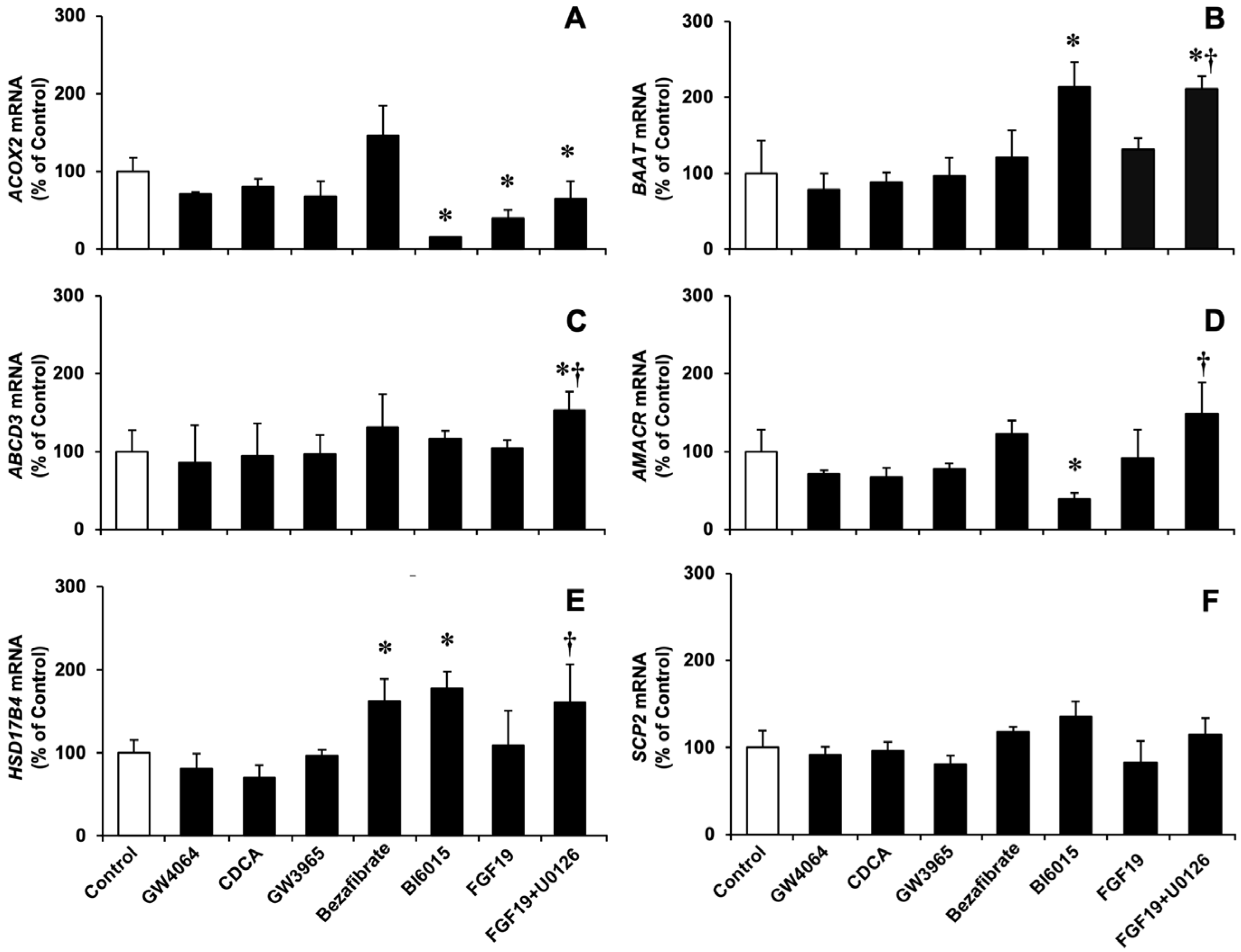
3.2. Role of Major Nuclear and Membrane Receptors Controlling BA Homeostasis
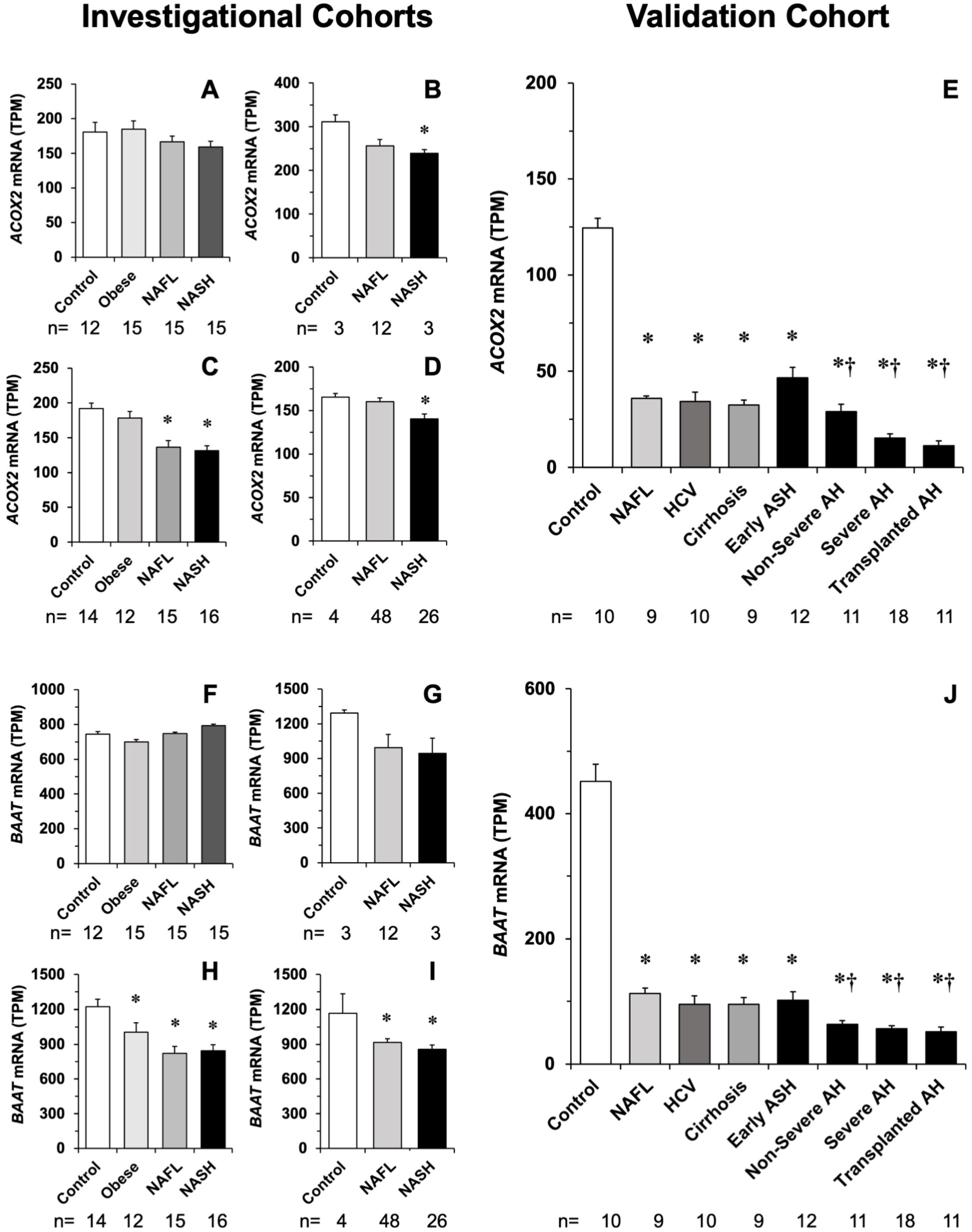
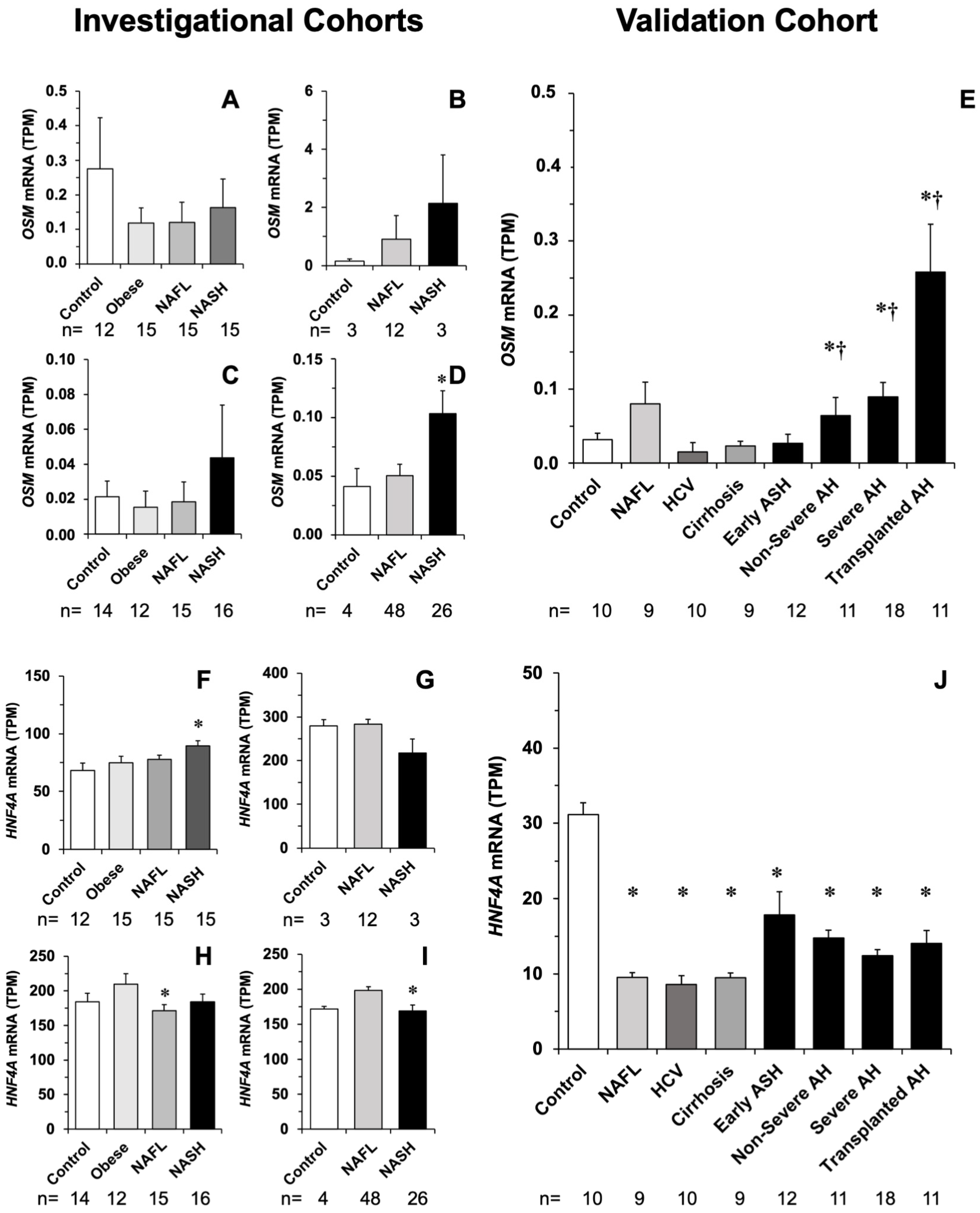
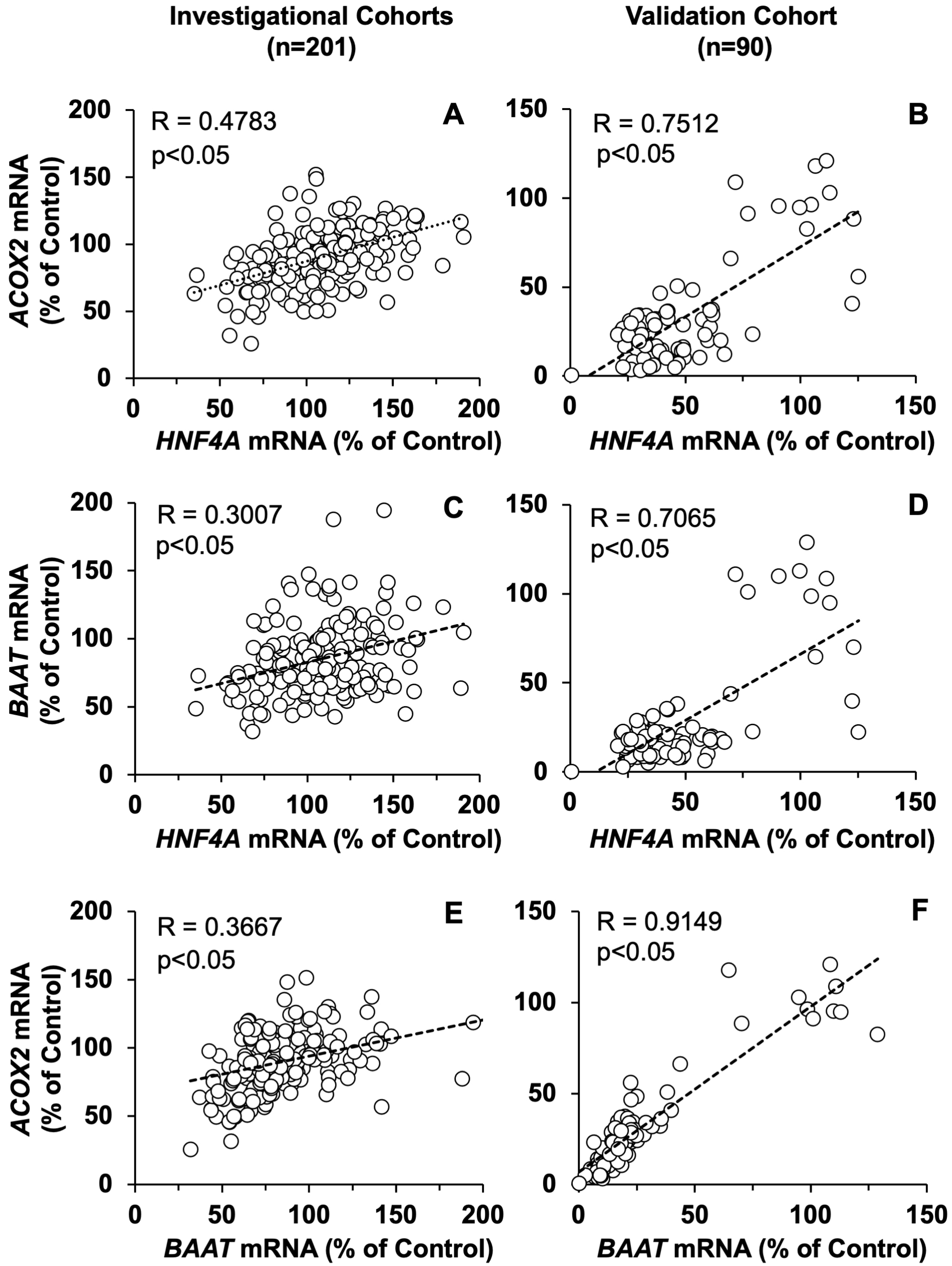
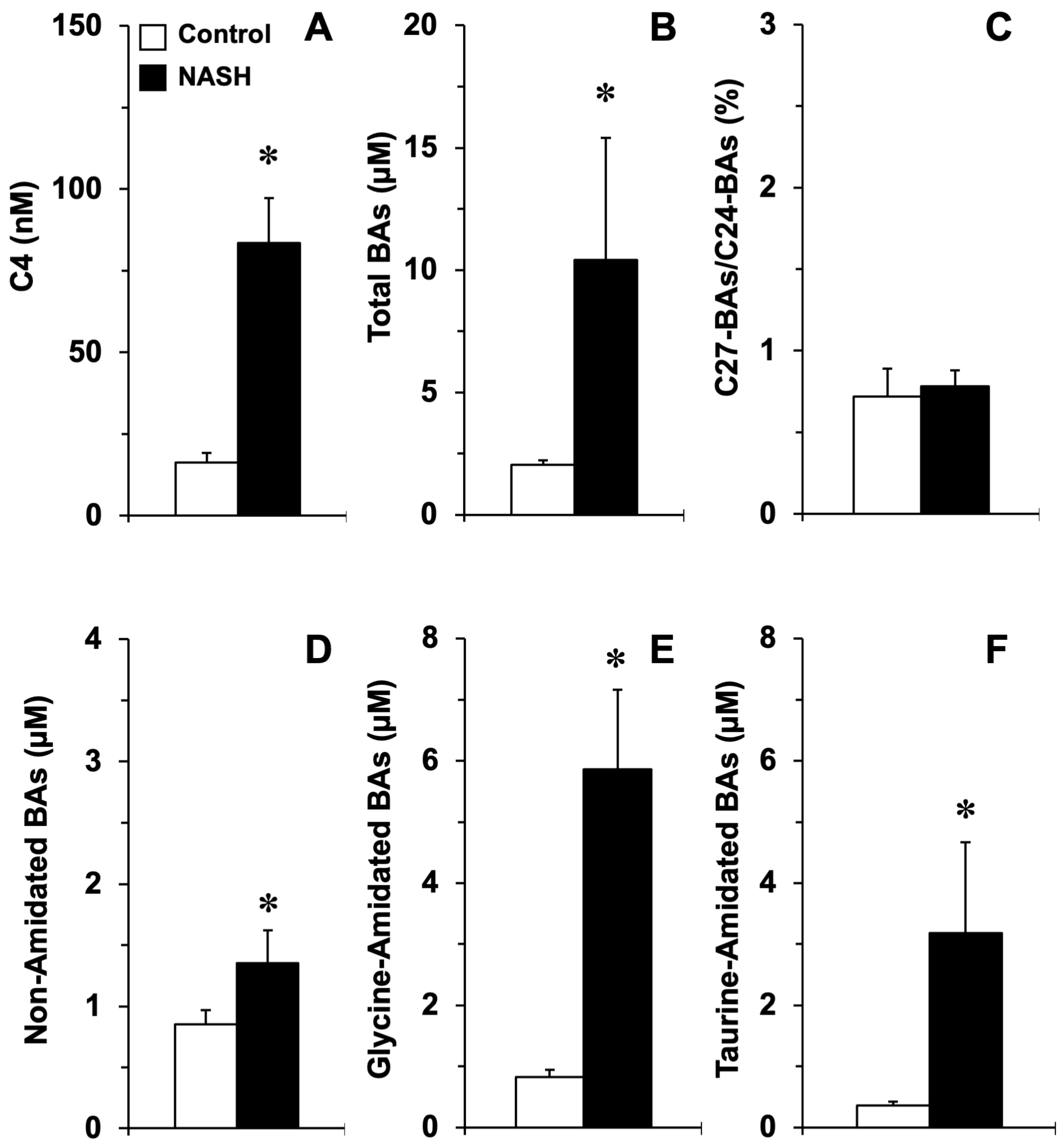
3.3. Impact of Liver Inflammation on ACOX2 and BAAT Expression
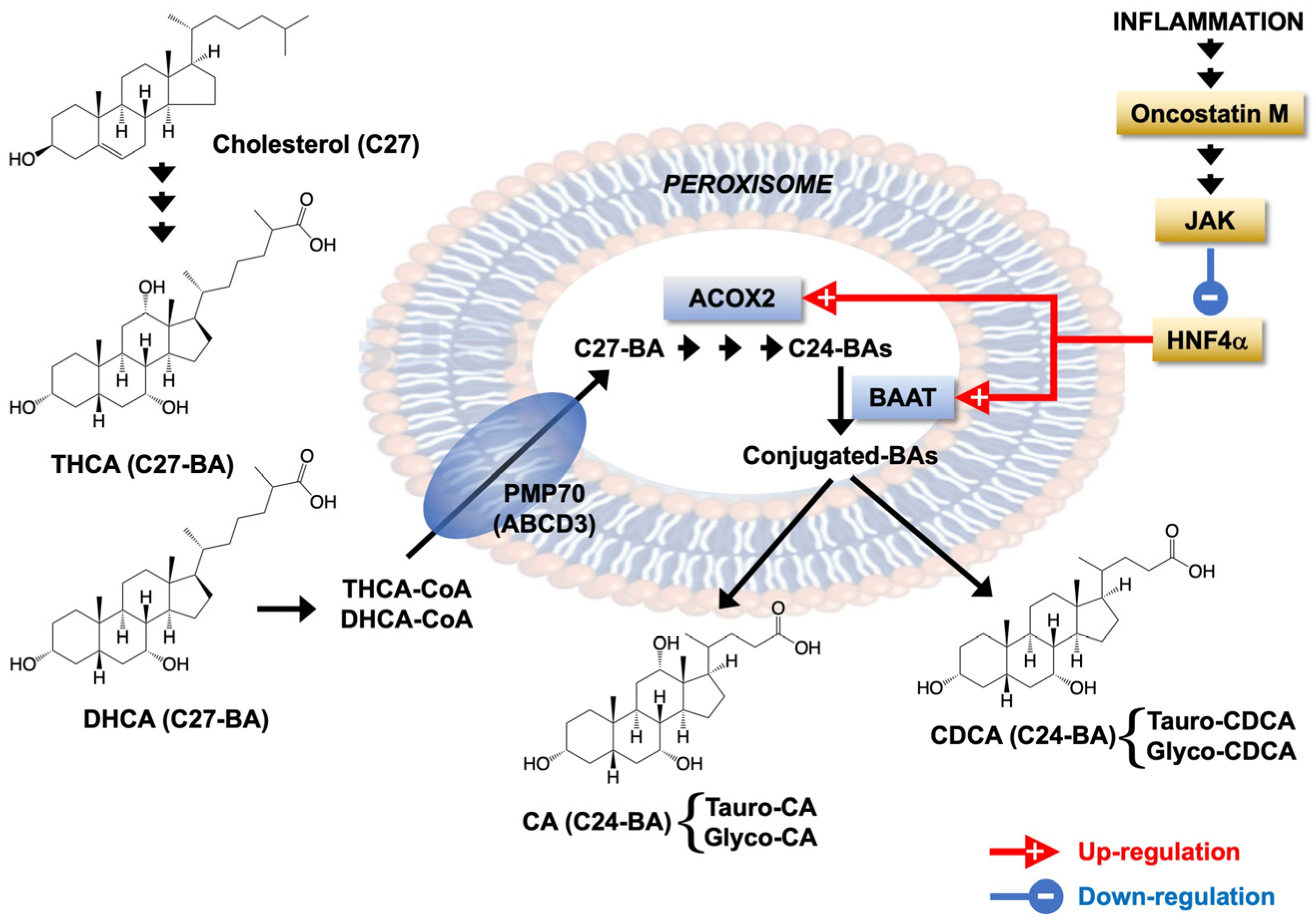
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Puri, P.; Daita, K.; Joyce, A.; Mirshahi, F.; Santhekadur, P.K.; Cazanave, S.; Luketic, V.A.; Siddiqui, M.S.; Boyett, S.; Min, H.K.; et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology 2018, 67, 534–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahn, D.; Geier, A. Bile acids in nonalcoholic steatohepatitis: Pathophysiological driving force or innocent bystanders? Hepatology 2018, 67, 464–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommerfeld, A.; Reinehr, R.; Haussinger, D. Bile acid-induced epidermal growth factor receptor activation in quiescent rat hepatic stellate cells can trigger both proliferation and apoptosis. J. Biol. Chem. 2009, 284, 22173–22183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, J.H.; Wang, S.L.; Davis, R.A. Bile acid induction of cytokine expression by macrophages correlates with repression of hepatic cholesterol 7alpha-hydroxylase. J. Biol. Chem. 2000, 275, 21805–21808. [Google Scholar] [CrossRef] [Green Version]
- Cheema, S.K.; Agellon, L.B. The murine and human cholesterol 7alpha-hydroxylase gene promoters are differentially responsive to regulation by fatty acids mediated via peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 2000, 275, 12530–12536. [Google Scholar] [CrossRef] [Green Version]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Mangelsdorf, D.J. Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Dig. Dis. 2015, 33, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Willson, T.M.; Jones, S.A.; Moore, J.T.; Kliewer, S.A. Chemical genomics: Functional analysis of orphan nuclear receptors in the regulation of bile acid metabolism. Med. Res. Rev. 2001, 21, 513–522. [Google Scholar] [CrossRef]
- Song, K.H.; Li, T.; Owsley, E.; Strom, S.; Chiang, J.Y. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology 2009, 49, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- del Castillo-Olivares, A.; Campos, J.A.; Pandak, W.M.; Gil, G. The role of alpha1-fetoprotein transcription factor/LRH-1 in bile acid biosynthesis: A known nuclear receptor activator that can act as a suppressor of bile acid biosynthesis. J. Biol. Chem. 2004, 279, 16813–16821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Chiang, J.Y. Transcriptional regulation of the human sterol 12alpha-hydroxylase gene (CYP8B1): Roles of heaptocyte nuclear factor 4alpha in mediating bile acid repression. J. Biol. Chem. 2001, 276, 41690–41699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, D.Q.; Bussen, M.; Sehayek, E.; Ananthanarayanan, M.; Shneider, B.L.; Suchy, F.J.; Shefer, S.; Bollileni, J.S.; Gonzalez, F.J.; Breslow, J.L.; et al. Hepatocyte nuclear factor-1alpha is an essential regulator of bile acid and plasma cholesterol metabolism. Nat. Genet. 2001, 27, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Kullak-Ublick, G.A. Hepatocyte nuclear factor 1 alpha: A key mediator of the effect of bile acids on gene expression. Hepatology 2003, 37, 622–631. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar Vallim, T.Q.; Tarling, E.J.; Ahn, H.; Hagey, L.R.; Romanoski, C.E.; Lee, R.G.; Graham, M.J.; Motohashi, H.; Yamamoto, M.; Edwards, P.A. MAFG is a transcriptional repressor of bile acid synthesis and metabolism. Cell Metab. 2015, 21, 298–311. [Google Scholar] [CrossRef] [Green Version]
- Heymann, F.; Tacke, F. Immunology in the liver--from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Li, T.; Jahan, A.; Chiang, J.Y. Bile acids and cytokines inhibit the human cholesterol 7 alpha-hydroxylase gene via the JNK/c-jun pathway in human liver cells. Hepatology 2006, 43, 1202–1210. [Google Scholar] [CrossRef] [Green Version]
- Geier, A.; Dietrich, C.G.; Voigt, S.; Kim, S.K.; Gerloff, T.; Kullak-Ublick, G.A.; Lorenzen, J.; Matern, S.; Gartung, C. Effects of proinflammatory cytokines on rat organic anion transporters during toxic liver injury and cholestasis. Hepatology 2003, 38, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Siewert, E.; Dietrich, C.G.; Lammert, F.; Heinrich, P.C.; Matern, S.; Gartung, C.; Geier, A. Interleukin-6 regulates hepatic transporters during acute-phase response. Biochem. Biophys. Res. Commun. 2004, 322, 232–238. [Google Scholar] [CrossRef]
- Geier, A.; Dietrich, C.G.; Voigt, S.; Ananthanarayanan, M.; Lammert, F.; Schmitz, A.; Trauner, M.; Wasmuth, H.E.; Boraschi, D.; Balasubramaniyan, N.; et al. Cytokine-dependent regulation of hepatic organic anion transporter gene transactivators in mouse liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G831–G841. [Google Scholar] [CrossRef] [PubMed]
- Veto, B.; Bojcsuk, D.; Bacquet, C.; Kiss, J.; Sipeki, S.; Martin, L.; Buday, L.; Balint, B.L.; Aranyi, T. The transcriptional activity of hepatocyte nuclear factor 4 alpha is inhibited via phosphorylation by ERK1/2. PLoS ONE 2017, 12, e0172020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Chiang, J.Y. A novel role of transforming growth factor beta1 in transcriptional repression of human cholesterol 7alpha-hydroxylase gene. Gastroenterology 2007, 133, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Song, K.H.; Ellis, E.; Strom, S.; Chiang, J.Y. Hepatocyte growth factor signaling pathway inhibits cholesterol 7alpha-hydroxylase and bile acid synthesis in human hepatocytes. Hepatology 2007, 46, 1993–2002. [Google Scholar] [CrossRef]
- Wanders, R.J.; Ferdinandusse, S. Peroxisomes, peroxisomal diseases, and the hepatotoxicity induced by peroxisomal metabolites. Curr. Drug. Metab. 2012, 13, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Monte, M.J.; Alonso-Pena, M.; Briz, O.; Herraez, E.; Berasain, C.; Argemi, J.; Prieto, J.; Marin, J.J.G. ACOX2 deficiency: An inborn error of bile acid synthesis identified in an adolescent with persistent hypertransaminasemia. J. Hepatol. 2017, 66, 581–588. [Google Scholar] [CrossRef]
- Vilarinho, S.; Sari, S.; Mazzacuva, F.; Bilguvar, K.; Esendagli-Yilmaz, G.; Jain, D.; Akyol, G.; Dalgic, B.; Gunel, M.; Clayton, P.T.; et al. ACOX2 deficiency: A disorder of bile acid synthesis with transaminase elevation, liver fibrosis, ataxia, and cognitive impairment. Proc. Natl. Acad. Sci. USA 2016, 113, 11289–11293. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandusse, S.; Denis, S.; van Roermund, C.W.T.; Preece, M.A.; Koster, J.; Ebberink, M.S.; Waterham, H.R.; Wanders, R.J.A. A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 952–958. [Google Scholar] [CrossRef]
- Alonso-Pena, M.; Espinosa-Escudero, R.; Herraez, E.; Briz, O.; Cagigal, M.L.; Gonzalez-Santiago, J.M.; Ortega-Alonso, A.; Fernandez-Rodriguez, C.; Bujanda, L.; Calvo Sanchez, M.; et al. Beneficial effect of ursodeoxycholic acid in patients with acyl-CoA oxidase 2 (ACOX2) deficiency-associated hypertransaminasemia. Hepatology 2022, 76, 1259–1274. [Google Scholar] [CrossRef]
- Falany, C.N.; Johnson, M.R.; Barnes, S.; Diasio, R.B. Glycine and taurine conjugation of bile acids by a single enzyme. Molecular cloning and expression of human liver bile acid CoA:amino acid N-acyltransferase. J. Biol. Chem. 1994, 269, 19375–19379. [Google Scholar] [CrossRef]
- Setchell, K.D.; Heubi, J.E.; Shah, S.; Lavine, J.E.; Suskind, D.; Al-Edreesi, M.; Potter, C.; Russell, D.W.; O’Connell, N.C.; Wolfe, B.; et al. Genetic defects in bile acid conjugation cause fat-soluble vitamin deficiency. Gastroenterology 2013, 144, 945–955.e946; quiz e914–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadzic, N.; Bull, L.N.; Clayton, P.T.; Knisely, A.S. Diagnosis in bile acid-CoA: Amino acid N-acyltransferase deficiency. World J. Gastroenterol. 2012, 18, 3322–3326. [Google Scholar] [CrossRef]
- Argemi, J.; Latasa, M.U.; Atkinson, S.R.; Blokhin, I.O.; Massey, V.; Gue, J.P.; Cabezas, J.; Lozano, J.J.; Van Booven, D.; Bell, A.; et al. Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat. Commun. 2019, 10, 3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schippers, I.J.; Moshage, H.; Roelofsen, H.; Muller, M.; Heymans, H.S.; Ruiters, M.; Kuipers, F. Immortalized human hepatocytes as a tool for the study of hepatocytic (de-)differentiation. Cell Biol. Toxicol. 1997, 13, 375–386. [Google Scholar] [CrossRef]
- Markwell, M.A.; Haas, S.M.; Bieber, L.L.; Tolbert, N.E. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal. Biochem 1978, 87, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Becerra, P.; Vaquero, J.; Romero, M.R.; Lozano, E.; Anadon, C.; Macias, R.I.; Serrano, M.A.; Grane-Boladeras, N.; Munoz-Bellvis, L.; Alvarez, L.; et al. No correlation between the expression of FXR and genes involved in multidrug resistance phenotype of primary liver tumors. Mol. Pharm. 2012, 9, 1693–1704. [Google Scholar] [CrossRef]
- Jahn, D.; Sutor, D.; Dorbath, D.; Weiss, J.; Gotze, O.; Schmitt, J.; Hermanns, H.M.; Geier, A. Farnesoid X receptor-dependent and -independent pathways mediate the transcriptional control of human fibroblast growth factor 19 by vitamin A. Biochim. Biophys. Acta 2016, 1859, 381–392. [Google Scholar] [CrossRef]
- Nytofte, N.S.; Serrano, M.A.; Monte, M.J.; Gonzalez-Sanchez, E.; Tumer, Z.; Ladefoged, K.; Briz, O.; Marin, J.J. A homozygous nonsense mutation (c.214C->A) in the biliverdin reductase alpha gene (BLVRA) results in accumulation of biliverdin during episodes of cholestasis. J. Med. Genet. 2011, 48, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Liu, S.; Wang, M.; Shao, Y.; Ding, M. High-performance liquid chromatography-tandem mass spectrometry for the analysis of bile acid profiles in serum of women with intrahepatic cholestasis of pregnancy. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 860, 10–17. [Google Scholar] [CrossRef]
- LeniLek, M.; Vecka, M.; Zizalova, K.; Vitek, L. Comparison of simple extraction procedures in liquid chromatography-mass spectrometry based determination of serum 7alpha-hydroxy-4-cholesten-3-one, a surrogate marker of bile acid synthesis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1033–1034, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Steiner, C.; von Eckardstein, A.; Rentsch, K.M. Quantification of the 15 major human bile acids and their precursor 7alpha-hydroxy-4-cholesten-3-one in serum by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 2870–2880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosch, M.; Kattler, K.; Herrmann, A.; von Schonfels, W.; Nordstrom, K.; Seehofer, D.; Damm, G.; Becker, T.; Zeissig, S.; Nehring, S.; et al. Epigenomic map of human liver reveals principles of zonated morphogenic and metabolic control. Nat. Commun. 2018, 9, 4150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Febbraio, M.A.; Reibe, S.; Shalapour, S.; Ooi, G.J.; Watt, M.J.; Karin, M. Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metab. 2019, 29, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Suppli, M.P.; Rigbolt, K.T.G.; Veidal, S.S.; Heeboll, S.; Eriksen, P.L.; Demant, M.; Bagger, J.I.; Nielsen, J.C.; Oro, D.; Thrane, S.W.; et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G462–G472. [Google Scholar] [CrossRef]
- Hoang, S.A.; Oseini, A.; Feaver, R.E.; Cole, B.K.; Asgharpour, A.; Vincent, R.; Siddiqui, M.; Lawson, M.J.; Day, N.C.; Taylor, J.M.; et al. Gene Expression Predicts Histological Severity and Reveals Distinct Molecular Profiles of Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 12541. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [Green Version]
- Corchete, L.A.; Rojas, E.A.; Alonso-Lopez, D.; De Las Rivas, J.; Gutierrez, N.C.; Burguillo, F.J. Systematic comparison and assessment of RNA-seq procedures for gene expression quantitative analysis. Sci. Rep. 2020, 10, 19737. [Google Scholar] [CrossRef]
- Kechin, A.; Boyarskikh, U.; Kel, A.; Filipenko, M. cutPrimers: A New Tool for Accurate Cutting of Primers from Reads of Targeted Next Generation Sequencing. J. Comput. Biol. 2017, 24, 1138–1143. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Hermanns, H.M. Oncostatin M and interleukin-31: Cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev. 2015, 26, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.E.; Martin, B.; Mellor, L.; Jacob, R.B.; Tawara, K.; McDougal, O.M.; Oxford, J.T.; Jorcyk, C.L. Oncostatin M binds to extracellular matrix in a bioactive conformation: Implications for inflammation and metastasis. Cytokine 2015, 72, 71–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hara, K.A.; Kedda, M.A.; Thompson, P.J.; Knight, D.A. Oncostatin M: An interleukin-6-like cytokine relevant to airway remodelling and the pathogenesis of asthma. Clin. Exp. Allergy 2003, 33, 1026–1032. [Google Scholar] [CrossRef]
- Holt, J.A.; Luo, G.; Billin, A.N.; Bisi, J.; McNeill, Y.Y.; Kozarsky, K.F.; Donahee, M.; Wang, D.Y.; Mansfield, T.A.; Kliewer, S.A.; et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003, 17, 1581–1591. [Google Scholar] [CrossRef] [Green Version]
- Aravindhan, K.; Webb, C.L.; Jaye, M.; Ghosh, A.; Willette, R.N.; DiNardo, N.J.; Jucker, B.M. Assessing the effects of LXR agonists on cellular cholesterol handling: A stable isotope tracer study. J. Lipid Res. 2006, 47, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Wrobel, J.; Steffan, R.; Bowen, S.M.; Magolda, R.; Matelan, E.; Unwalla, R.; Basso, M.; Clerin, V.; Gardell, S.J.; Nambi, P.; et al. Indazole-based liver X receptor (LXR) modulators with maintained atherosclerotic lesion reduction activity but diminished stimulation of hepatic triglyceride synthesis. J. Med. Chem. 2008, 51, 7161–7168. [Google Scholar] [CrossRef]
- Tachibana, K.; Kobayashi, Y.; Tanaka, T.; Tagami, M.; Sugiyama, A.; Katayama, T.; Ueda, C.; Yamasaki, D.; Ishimoto, K.; Sumitomo, M.; et al. Gene expression profiling of potential peroxisome proliferator-activated receptor (PPAR) target genes in human hepatoblastoma cell lines inducibly expressing different PPAR isoforms. Nucl. Recept. 2005, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Ghonem, N.S.; Ananthanarayanan, M.; Soroka, C.J.; Boyer, J.L. Peroxisome proliferator-activated receptor alpha activates human multidrug resistance transporter 3/ATP-binding cassette protein subfamily B4 transcription and increases rat biliary phosphatidylcholine secretion. Hepatology 2014, 59, 1030–1042. [Google Scholar] [CrossRef]
- Ferslew, B.C.; Xie, G.; Johnston, C.K.; Su, M.; Stewart, P.W.; Jia, W.; Brouwer, K.L.; Barritt, A.S.t. Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3318–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varanasi, U.; Chu, R.; Huang, Q.; Castellon, R.; Yeldandi, A.V.; Reddy, J.K. Identification of a peroxisome proliferator-responsive element upstream of the human peroxisomal fatty acyl coenzyme A oxidase gene. J. Biol. Chem. 1996, 271, 2147–2155. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.Y. Hepatocyte nuclear factor 4alpha regulation of bile acid and drug metabolism. Expert Opin. Drug. Metab. Toxicol. 2009, 5, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonzo, J.A.; Ferry, C.H.; Matsubara, T.; Kim, J.H.; Gonzalez, F.J. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4alpha in adult mice. J. Biol. Chem. 2012, 287, 7345–7356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechmann, L.P.; Kocabayoglu, P.; Sowa, J.P.; Sydor, S.; Best, J.; Schlattjan, M.; Beilfuss, A.; Schmitt, J.; Hannivoort, R.A.; Kilicarslan, A.; et al. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology 2013, 57, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Miyazaki, T.; Iwamoto, J.; Hirayama, T.; Morishita, Y.; Monma, T.; Ueda, H.; Mizuno, S.; Sugiyama, F.; Takahashi, S.; et al. Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J. Lipid Res. 2020, 61, 54–69. [Google Scholar] [CrossRef]
- Miyajima, A.; Kinoshita, T.; Tanaka, M.; Kamiya, A.; Mukouyama, Y.; Hara, T. Role of Oncostatin M in hematopoiesis and liver development. Cytokine Growth Factor Rev. 2000, 11, 177–183. [Google Scholar] [CrossRef]
- Levy, M.T.; Trojanowska, M.; Reuben, A. Oncostatin M: A cytokine upregulated in human cirrhosis, increases collagen production by human hepatic stellate cells. J. Hepatol. 2000, 32, 218–226. [Google Scholar] [CrossRef]
- Le Vee, M.; Jouan, E.; Stieger, B.; Lecureur, V.; Fardel, O. Regulation of drug transporter expression by oncostatin M in human hepatocytes. Biochem. Pharmacol. 2011, 82, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Foglia, B.; Sutti, S.; Pedicini, D.; Cannito, S.; Bocca, C.; Maggiora, M.; Bevacqua, M.R.; Rosso, C.; Bugianesi, E.; Albano, E.; et al. Oncostatin M, A Profibrogenic Mediator Overexpressed in Non-Alcoholic Fatty Liver Disease, Stimulates Migration of Hepatic Myofibroblasts. Cells 2019, 9, 28. [Google Scholar] [CrossRef]
- Inoue, Y.; Yu, A.M.; Inoue, J.; Gonzalez, F.J. Hepatocyte nuclear factor 4alpha is a central regulator of bile acid conjugation. J. Biol. Chem. 2004, 279, 2480–2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Robledo, O.; Kinzie, E.; Blanchard, F.; Richards, C.; Miyajima, A.; Baumann, H. Receptor subunit-specific action of oncostatin M in hepatic cells and its modulation by leukemia inhibitory factor. J. Biol. Chem. 2000, 275, 25273–25285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somasundaram, R.; Ruehl, M.; Schaefer, B.; Schmid, M.; Ackermann, R.; Riecken, E.O.; Zeitz, M.; Schuppan, D. Interstitial collagens I, III, and VI sequester and modulate the multifunctional cytokine oncostatin M. J. Biol. Chem. 2002, 277, 3242–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespo, J.; Cayon, A.; Fernandez-Gil, P.; Hernandez-Guerra, M.; Mayorga, M.; Dominguez-Diez, A.; Fernandez-Escalante, J.C.; Pons-Romero, F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso-Peña, M.; Espinosa-Escudero, R.; Hermanns, H.M.; Briz, O.; Herranz, J.M.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Juamperez, J.; Avila, M.; Argemi, J.; et al. Impact of Liver Inflammation on Bile Acid Side Chain Shortening and Amidation. Cells 2022, 11, 3983. https://doi.org/10.3390/cells11243983
Alonso-Peña M, Espinosa-Escudero R, Hermanns HM, Briz O, Herranz JM, Garcia-Ruiz C, Fernandez-Checa JC, Juamperez J, Avila M, Argemi J, et al. Impact of Liver Inflammation on Bile Acid Side Chain Shortening and Amidation. Cells. 2022; 11(24):3983. https://doi.org/10.3390/cells11243983
Chicago/Turabian StyleAlonso-Peña, Marta, Ricardo Espinosa-Escudero, Heike M. Hermanns, Oscar Briz, Jose M. Herranz, Carmen Garcia-Ruiz, Jose C. Fernandez-Checa, Javier Juamperez, Matias Avila, Josepmaria Argemi, and et al. 2022. "Impact of Liver Inflammation on Bile Acid Side Chain Shortening and Amidation" Cells 11, no. 24: 3983. https://doi.org/10.3390/cells11243983
APA StyleAlonso-Peña, M., Espinosa-Escudero, R., Hermanns, H. M., Briz, O., Herranz, J. M., Garcia-Ruiz, C., Fernandez-Checa, J. C., Juamperez, J., Avila, M., Argemi, J., Bataller, R., Crespo, J., Monte, M. J., Geier, A., Herraez, E., & Marin, J. J. G. (2022). Impact of Liver Inflammation on Bile Acid Side Chain Shortening and Amidation. Cells, 11(24), 3983. https://doi.org/10.3390/cells11243983