
Proteomic Landscape of Human Spermatozoa: Optimized Extraction Method and Application
 , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Procedures
2.1. Materials and Chemicals
2.2. Biospecimen Collection
2.3. Protein Extraction
2.4. SDS-PAGE Analysis
2.5. Reduction, Alkylation and Digestion
2.6. Prefractionation of Peptides
2.7. Western Blot Analysis
2.8. LC-MS/MS Analysis
2.9. Data Analysis
2.10. Bioinformatic Analysis
3. Results
3.1. Comprehensive Comparison of the Identified Human Spermatozoa Proteins Using Different Sample Processing Methods
3.2. In-Depth Human Sperm Protein Profiling
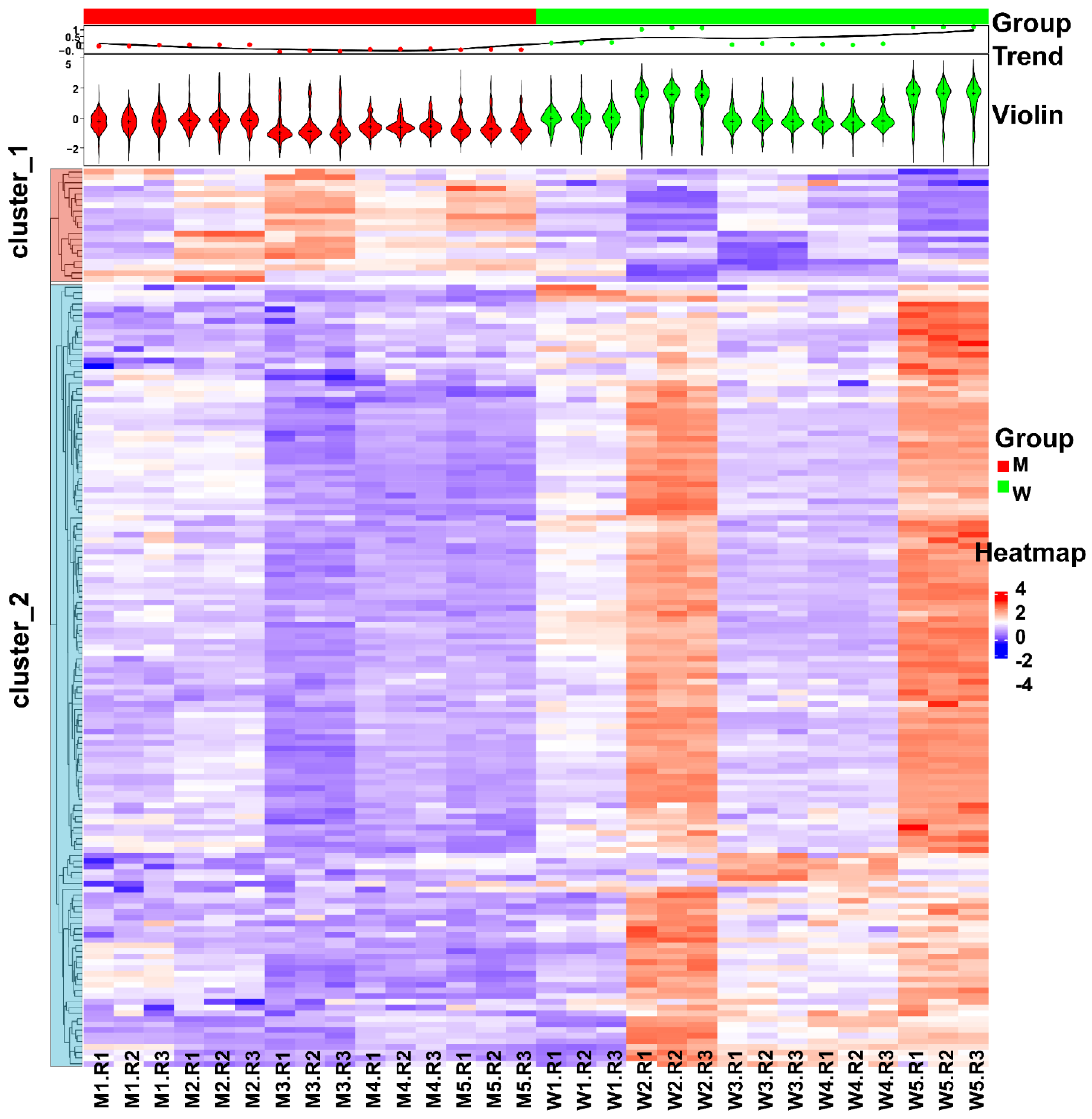
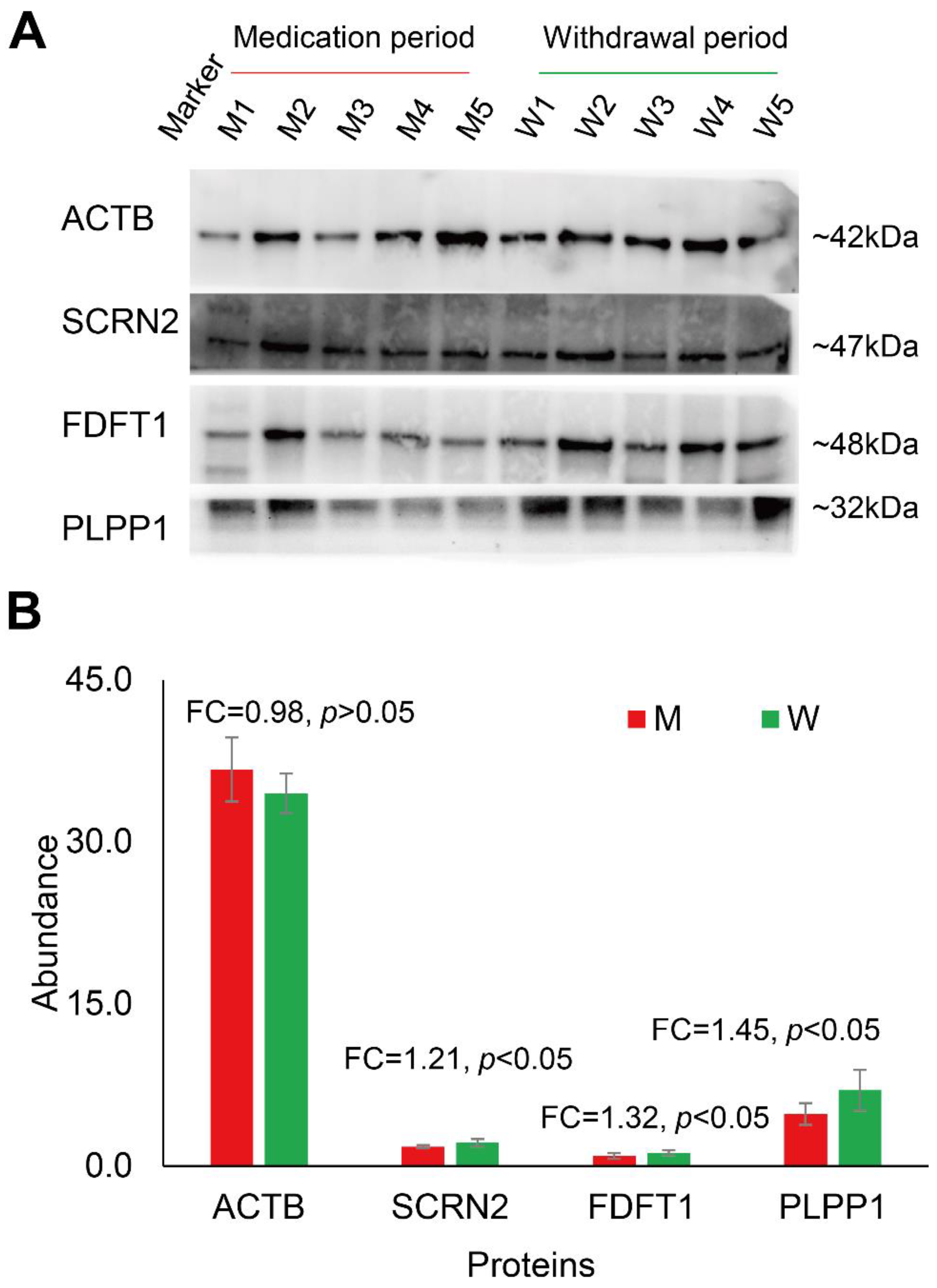
3.3. Changes in Human Spermatozoa Proteins after Antibiotic Therapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, M.; Liu, X.; Chang, G.; Chen, Y.; An, G.; Yan, L.; Gao, S.; Xu, Y.; Cui, Y.; Dong, J.; et al. Single-Cell RNA Sequencing Analysis Reveals Sequential Cell Fate Transition during Human Spermatogenesis. Cell Stem Cell 2018, 23, 599–614.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Guo, Y.; Zhou, T.; Shi, X.; Yu, J.; Yang, Y.; Wu, Y.; Wang, J.; Liu, M.; Chen, X.; et al. In-depth proteomic analysis of the human sperm reveals complex protein compositions. J. Proteom. 2013, 79, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Ostermeier, G.C.; Miller, D.; Huntriss, J.D.; Diamond, M.P.; Krawetz, S.A. Reproductive biology: Delivering spermatozoan RNA to the oocyte. Nature 2004, 429, 154. [Google Scholar] [CrossRef] [PubMed]
- Loppin, B.; Lepetit, D.; Dorus, S.; Couble, P.; Karr, T.L. Origin and neofunctionalization of a Drosophila paternal effect gene essential for zygote viability. Curr. Biol. CB 2005, 15, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, W.; Xu, Y.; Tang, M.; Fang, J.; Sun, H.; Sun, Y.; Gu, M.; Liu, Z.; Zhang, Z.; et al. Proteomic characteristics of human sperm cryopreservation. Proteomics 2014, 14, 298–310. [Google Scholar] [CrossRef]
- Gholami, D.; Ghaffari, S.M.; Shahverdi, A.; Sharafi, M.; Riazi, G.; Fathi, R.; Esmaeili, V.; Hezavehei, M. Proteomic analysis and microtubule dynamicity of human sperm in electromagnetic cryopreservation. J. Cell. Biochem. 2018, 119, 9483–9497. [Google Scholar] [CrossRef]
- Agarwal, A.; Rana, M.; Qiu, E.; AlBunni, H.; Bui, A.D.; Henkel, R. Role of oxidative stress, infection and inflammation in male infertility. Andrologia 2018, 50, e13126. [Google Scholar] [CrossRef]
- Amaral, A.; Castillo, J.; Ramalho-Santos, J.; Oliva, R. The combined human sperm proteome: Cellular pathways and implications for basic and clinical science. Hum. Reprod. Update 2014, 20, 40–62. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Heredia, J.; Estanyol, J.M.; Ballescà, J.; Oliva, R. Proteomic identification of human sperm proteins. Proteomics 2006, 6, 4356–4369. [Google Scholar] [CrossRef]
- Baker, M.A.; Reeves, G.; Hetherington, L.; Müller, J.; Baur, I.; Aitken, R.J. Identification of gene products present in Triton X-100 soluble and insoluble fractions of human spermatozoa lysates using LC-MS/MS analysis. Proteom. Clin. Appl. 2007, 1, 524–532. [Google Scholar] [CrossRef]
- Shen, Z.Q.; Shi, B.; Wang, T.R.; Jiao, J.; Shang, X.J.; Wu, Q.J.; Zhou, Y.M.; Cao, T.F.; Du, Q.; Wang, X.X.; et al. Characterization of the Sperm Proteome and Reproductive Outcomes with in Vitro, Fertilization after a Reduction in Male Ejaculatory Abstinence Period. Mol. Cell. Proteom. 2019, 18, S109–S117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; An, Q.; Zhang, K.; Liu, Y.; Tong, Y.; Xu, J.; Zhou, F.; Wang, X.; Guo, Y.; Lu, W.; et al. Quantitative proteomic characterization of human sperm cryopreservation: Using data-independent acquisition mass spectrometry. BMC Urol. 2019, 19, 133. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Jodar, M.; Oliva, R. The contribution of human sperm proteins to the development and epigenome of the preimplantation embryo. Hum. Reprod. Update 2018, 24, 535–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexovič, M.; Sabo, J.; Longuespée, R. Microproteomic sample preparation. Proteomics 2021, 21, 2000318. [Google Scholar] [CrossRef]
- Carson, S.A.; Kallen, A.N. Diagnosis and Management of Infertility: A Review. JAMA 2021, 326, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, Y.; Ni, A.; Shi, L.; Wang, P.; Isa, A.M.; Ge, P.; Jiang, L.; Fan, J.; Ma, H.; et al. Seminal Plasma Proteome as an Indicator of Sperm Dysfunction and Low Sperm Motility in Chickens. Mol. Cell. Proteom. 2020, 19, 1035–1046. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Cui, Y.; Zhang, X.; Lou, J.; Zhou, J.; Bei, H.; Wei, R. Proteomic profile of human spermatozoa in healthy and asthenozoospermic individuals. Reprod. Biol. Endocrinol. RBE 2018, 16, 16. [Google Scholar] [CrossRef] [Green Version]
- Murdica, V.; Cermisoni, G.C.; Zarovni, N.; Salonia, A.; Viganò, P.; Vago, R. Proteomic analysis reveals the negative modulator of sperm function glycodelin as over-represented in semen exosomes isolated from asthenozoospermic patients. Hum. Reprod. 2019, 34, 1416–1427. [Google Scholar] [CrossRef]
- Wu, Y.; Yuan, Y.; Chen, L.; Wang, M.; Yang, Y.; Wang, Y.; Quan, C.; Chen, D.; Chen, Y.; Huang, X.; et al. Quantitative Proteomic Analysis of Human Seminal Plasma from Normozoospermic and Asthenozoospermic Individuals. Biomed. Res. Int. 2019, 2019, 2735038. [Google Scholar] [CrossRef] [Green Version]
- Netherton, J.; Ogle, R.A.; Hetherington, L.; Silva Balbin Villaverde, A.I.; Hondermarck, H.; Baker, M.A. Proteomic Analysis Reveals that Topoisomerase 2A is Associated with Defective Sperm Head Morphology. Mol. Cell. Proteom. 2020, 19, 444–455. [Google Scholar] [CrossRef]
- Arenaza, I.U.; Osinalde, N.; Akimov, V.; Puglia, M.; Candenas, L.; Pinto, F.M.; Muñoa-Hoyos, I.; Gianzo, M.; Matorras, R.; Irazusta, J.; et al. Phosphoproteomic and Functional Analyses Reveal Sperm-specific Protein Changes Downstream of Kappa Opioid Receptor in Human Spermatozoa. Mol. Cell. Proteom. 2019, 18, S118–S131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahat, A.M.; Rizzoto, G.; Kastelic, J.P. Amelioration of heat stress-induced damage to testes and sperm quality. Theriogenology 2020, 158, 84–96. [Google Scholar] [CrossRef]
- Xin, M.; You, S.; Xu, Y.; Shi, W.; Zhu, B.; Shen, J.; Wu, J.; Li, C.; Chen, Z.; Su, Y.; et al. Precision Glycoproteomics Reveals Distinctive N-Glycosylation in Human Spermatozoa. Mol. Cell. Proteom. 2022, 21, 100214. [Google Scholar] [CrossRef] [PubMed]
- Martin-Hidalgo, D.; Serrano, R.; Zaragoza, C.; Garcia-Marin, L.J.; Bragado, M.J. Human sperm phosphoproteome reveals differential phosphoprotein signatures that regulate human sperm motility. J. Proteom. 2020, 215, 103654. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Meyer, K.J.; Iinishi, A.; Favre-Godal, Q.; Green, R.; Manuse, S.; Caboni, M.; Mori, M.; Niles, S.; Ghiglieri, M.; et al. A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576, 459–464. [Google Scholar] [CrossRef]
- Ahmadi, M.H.; Mirsalehian, A.; Sadighi Gilani, M.A.; Bahador, A.; Afraz, K. Association of asymptomatic Chlamydia trachomatis infection with male infertility and the effect of antibiotic therapy in improvement of semen quality in infected infertile men. Andrologia 2018, 50, e12944. [Google Scholar] [CrossRef]
- Wang, M.W.; Yang, Z.; Chen, X.; Zhou, S.H.; Huang, G.L.; Sun, J.N.; Jiang, H.; Xu, W.M.; Lin, H.C.; Yu, X.; et al. Activation of PTH1R alleviates epididymitis and orchitis through Gq and β-arrestin-1 pathways. Proc. Natl. Acad. Sci. USA 2021, 118, e2107363118. [Google Scholar] [CrossRef]
- Kim, S.; Covington, A.; Pamer, E.G. The intestinal microbiota: Antibiotics, colonization resistance, and enteric pathogens. Immunol. Rev. 2017, 279, 90–105. [Google Scholar] [CrossRef]
- Hernández-Avilés, C.; Love, C.C.; Serafini, R.; Ramírez-Agámez, L.; Kelley, D.E.; de Andino, E.M.; Teague, S.R.; LaCaze, K.A.; Brinsko, S.P.; Varner, D.D. Inclusion of supplemental antibiotics (amikacin-penicillin) in a commercial extender for stallion semen: Effects on sperm quality, bacterial growth, and fertility following cooled storage. Theriogenology 2020, 158, 209–217. [Google Scholar] [CrossRef]
- Ghoneim, I.M.; Al-Mubarak, A.H.; Fayez, M.M.; Waheed, M.M.; El-Bahr, S.M. Impact of antibiotics on spermatozoa quality and bacterial load of chilled-stored camels (Camelus dromedarius) semen. Trop. Anim. Health Prod. 2021, 54, 21. [Google Scholar] [CrossRef]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagari, R.M.; Frazzoni, L.; Marasco, G.; Fuccio, L.; Bazzoli, F. Treatment of Helicobacter pylori infection: A clinical practice update. Minerva. Med. 2021, 112, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Zheng, S.; Su, T.; Cheng, J.; Mao, Y.; Zhong, Y.; Liu, Y.; Chen, J.; Zhao, W.; Lin, T.; et al. Comparative N-Glycoproteomics Analysis of Clinical Samples Via Different Mass Spectrometry Dissociation Methods. Front. Chem. 2022, 10, 839470. [Google Scholar] [CrossRef]
- Zhang, F.; Ge, W.; Ruan, G.; Cai, X.; Guo, T. Data-Independent Acquisition Mass Spectrometry-Based Proteomics and Software Tools: A Glimpse in 2020. Proteomics 2020, 20, e1900276. [Google Scholar] [CrossRef]
- Danis, R.B.; Samplaski, M.K. Sperm Morphology: History, Challenges, and Impact on Natural and Assisted Fertility. Curr. Urol. Rep. 2019, 20, 43. [Google Scholar] [CrossRef] [PubMed]
- Menkveld, R. Sperm morphology assessment using strict (tygerberg) criteria. Methods Mol. Biol. 2013, 927, 39–50. [Google Scholar] [PubMed]
- Bang, G.; Lee, H.; Kim, H.; Han, E.H.; Park, Y.H.; Kim, J.Y. Comparison of protein characterization using In solution and S-Trap digestion methods for proteomics. Biochem. Biophys. Res. Commun. 2022, 589, 197–203. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Mayne, J.; Ning, Z.; Stintzi, A.; Figeys, D. Assessing the impact of protein extraction methods for human gut metaproteomics. J. Proteom. 2018, 180, 120–127. [Google Scholar] [CrossRef]
- Takeya, K.; Kaneko, T.; Miyazu, M.; Takai, A. Addition of urea and thiourea to electrophoresis sample buffer improves efficiency of protein extraction from TCA/acetone-treated smooth muscle tissues for phos-tag SDS-PAGE. Electrophoresis 2018, 39, 326–333. [Google Scholar] [CrossRef]
- Jorge, S.; Araújo, J.E.; Pimentel-Santos, F.M.; Branco, J.C.; Santos, H.M.; Lodeiro, C.; Capelo, J.L. Unparalleled sample treatment throughput for proteomics workflows relying on ultrasonic energy. Talanta 2018, 178, 1067–1076. [Google Scholar] [CrossRef]
- Hahn, J.; Moritz, M.; Voß, H.; Pelczar, P.; Huber, S.; Schlüter, H. Tissue Sampling and Homogenization in the Sub-Microliter Scale with a Nanosecond Infrared Laser (NIRL) for Mass Spectrometric Proteomics. Int. J. Mol. Sci. 2021, 22, 10833. [Google Scholar] [CrossRef] [PubMed]
- Gilany, K.; Minai-Tehrani, A.; Amini, M.; Agharezaee, N.; Arjmand, B. The Challenge of Human Spermatozoa Proteome: A Systematic Review. J. Reprod. Infertil. 2017, 18, 267–279. [Google Scholar] [PubMed]
- Adhikari, S.; Nice, E.C.; Deutsch, E.W.; Lane, L.; Omenn, G.S.; Pennington, S.R.; Paik, Y.-K.; Overall, C.M.; Corrales, F.J.; Cristea, I.M.; et al. A high-stringency blueprint of the human proteome. Nat. Commun. 2020, 11, 5301. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.T. Single-cell Proteomics: Progress and Prospects. Mol. Cell. Proteom. 2020, 19, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Drazic, A.; Aksnes, H.; Marie, M.; Boczkowska, M.; Varland, S.; Timmerman, E.; Foyn, H.; Glomnes, N.; Rebowski, G.; Impens, F.; et al. NAA80 is actin’s N-terminal acetyltransferase and regulates cytoskeleton assembly and cell motility. Proc. Natl. Acad. Sci. USA 2018, 115, 4399–4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lin, Z.; Bustin, K.A.; McKnight, N.R.; Parsons, W.H.; Matthews, M.L. Discovery of Potent and Selective Inhibitors against Protein-Derived Electrophilic Cofactors. J. Am. Chem. Soc. 2022, 144, 5377–5388. [Google Scholar] [CrossRef]
- Smyth, S.S.; Sciorra, V.A.; Sigal, Y.J.; Pamuklar, Z.; Wang, Z.; Xu, Y.; Prestwich, G.D.; Morris, A.J. Lipid phosphate phosphatases regulate lysophosphatidic acid production and signaling in platelets: Studies using chemical inhibitors of lipid phosphate phosphatase activity. J. Biol. Chem. 2003, 278, 43214–43223. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, M.; Su, T.; Wang, S.; Chen, J.; Lin, T.; Cheng, Q.; Chen, Y.; Gong, M.; Yang, H.; Li, F.; et al. Proteomic Landscape of Human Spermatozoa: Optimized Extraction Method and Application. Cells 2022, 11, 4064. https://doi.org/10.3390/cells11244064
Luo M, Su T, Wang S, Chen J, Lin T, Cheng Q, Chen Y, Gong M, Yang H, Li F, et al. Proteomic Landscape of Human Spermatozoa: Optimized Extraction Method and Application. Cells. 2022; 11(24):4064. https://doi.org/10.3390/cells11244064
Chicago/Turabian StyleLuo, Mengqi, Tao Su, Shisheng Wang, Jianhai Chen, Tianhai Lin, Qingyuan Cheng, Younan Chen, Meng Gong, Hao Yang, Fuping Li, and et al. 2022. "Proteomic Landscape of Human Spermatozoa: Optimized Extraction Method and Application" Cells 11, no. 24: 4064. https://doi.org/10.3390/cells11244064