.png)
D4Z4 Methylation Levels Combined with a Machine Learning Pipeline Highlight Single CpG Sites as Discriminating Biomarkers for FSHD Patients

 ,
,  , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
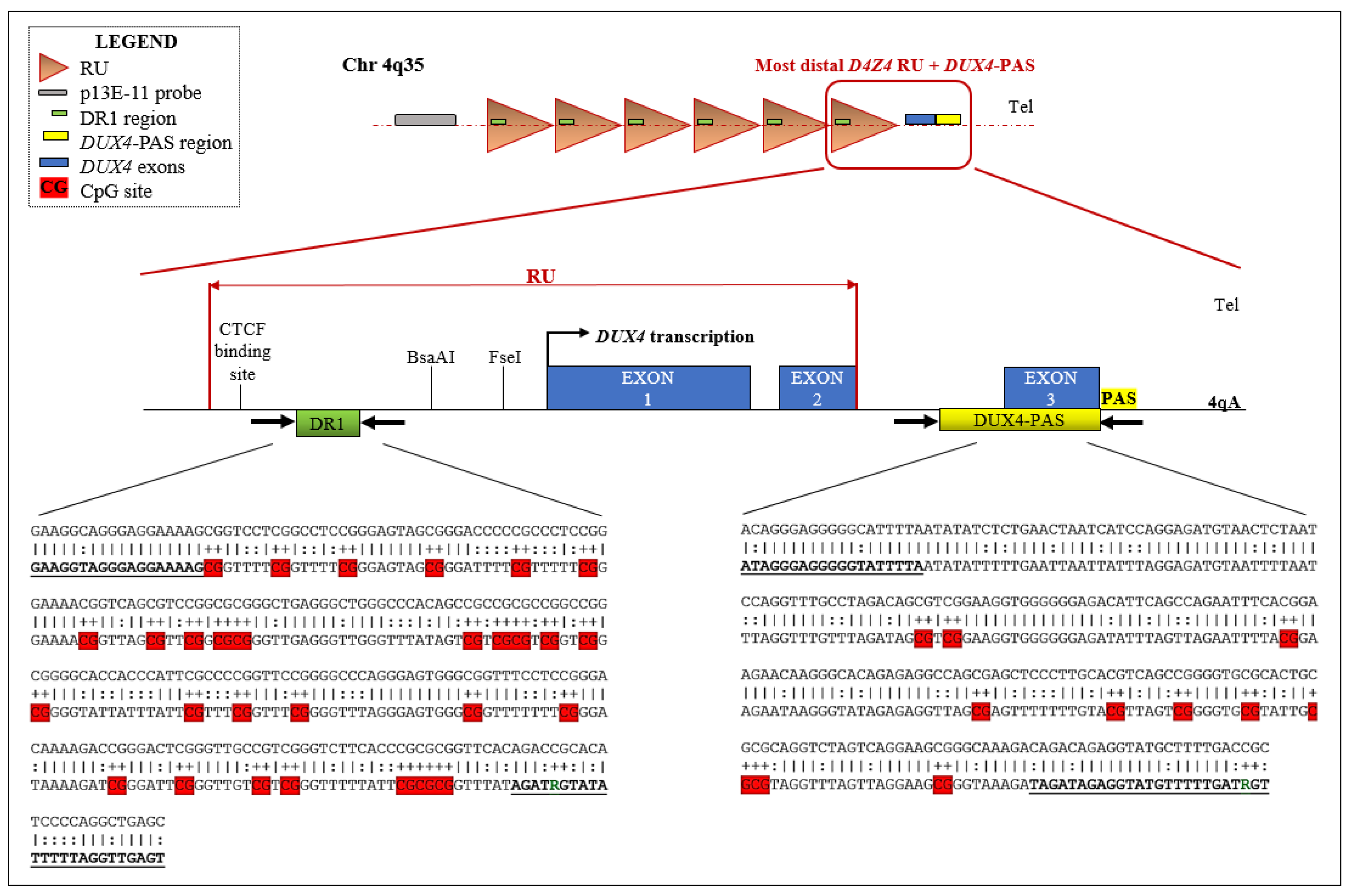
2.1. Selection of the Cohort
2.2. Analysis of DNA Methylation and 4q Subtelomeric Variant Typing
2.3. Statistical Analysis
2.4. Machine Learning Pipeline for Classification
2.4.1. FSHD vs. CTRL
2.4.2. FSHDlow-RU vs. FSHDhigh-RU and FSHDvar+ vs. FSHDvar−
3. Results
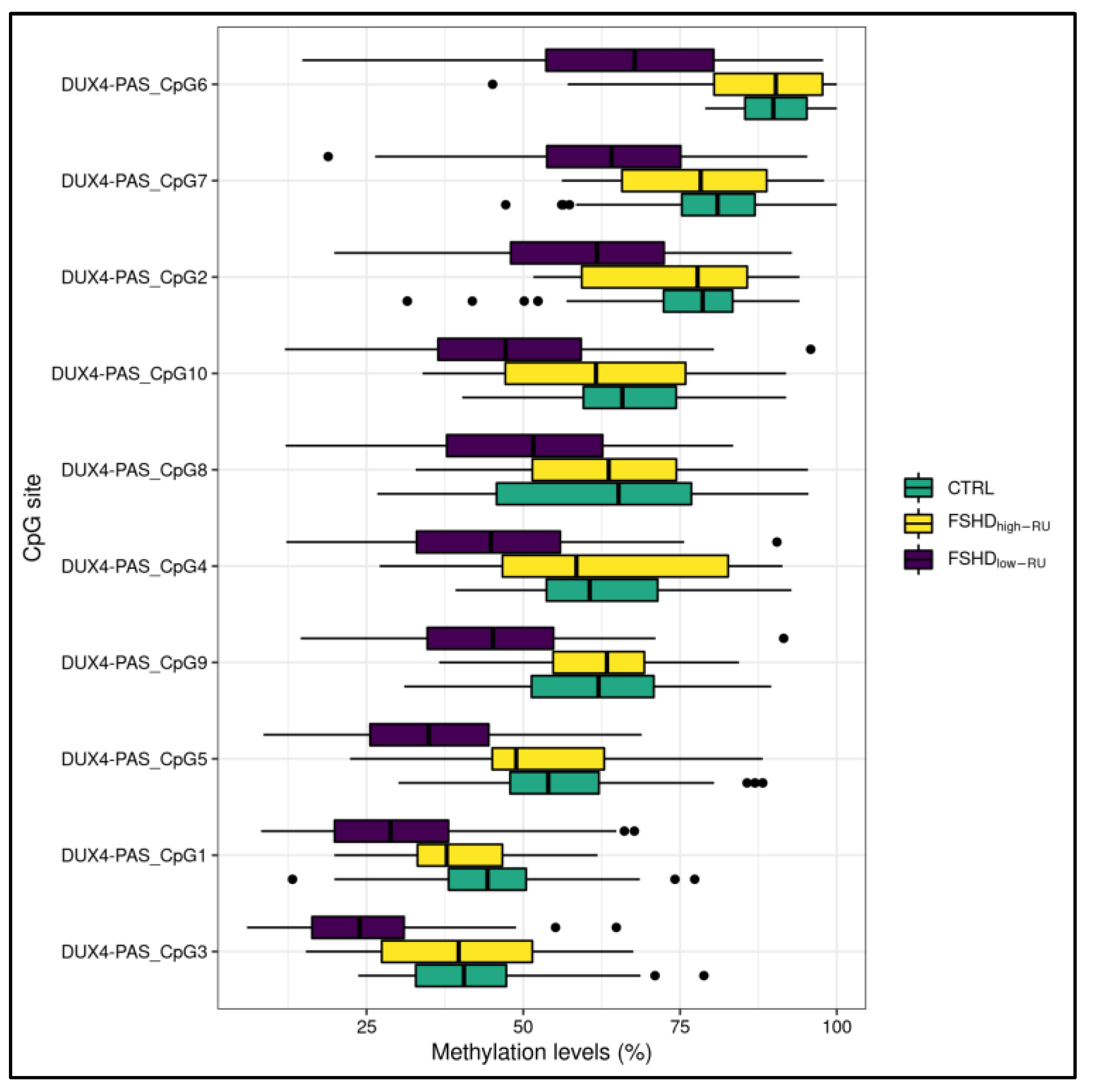
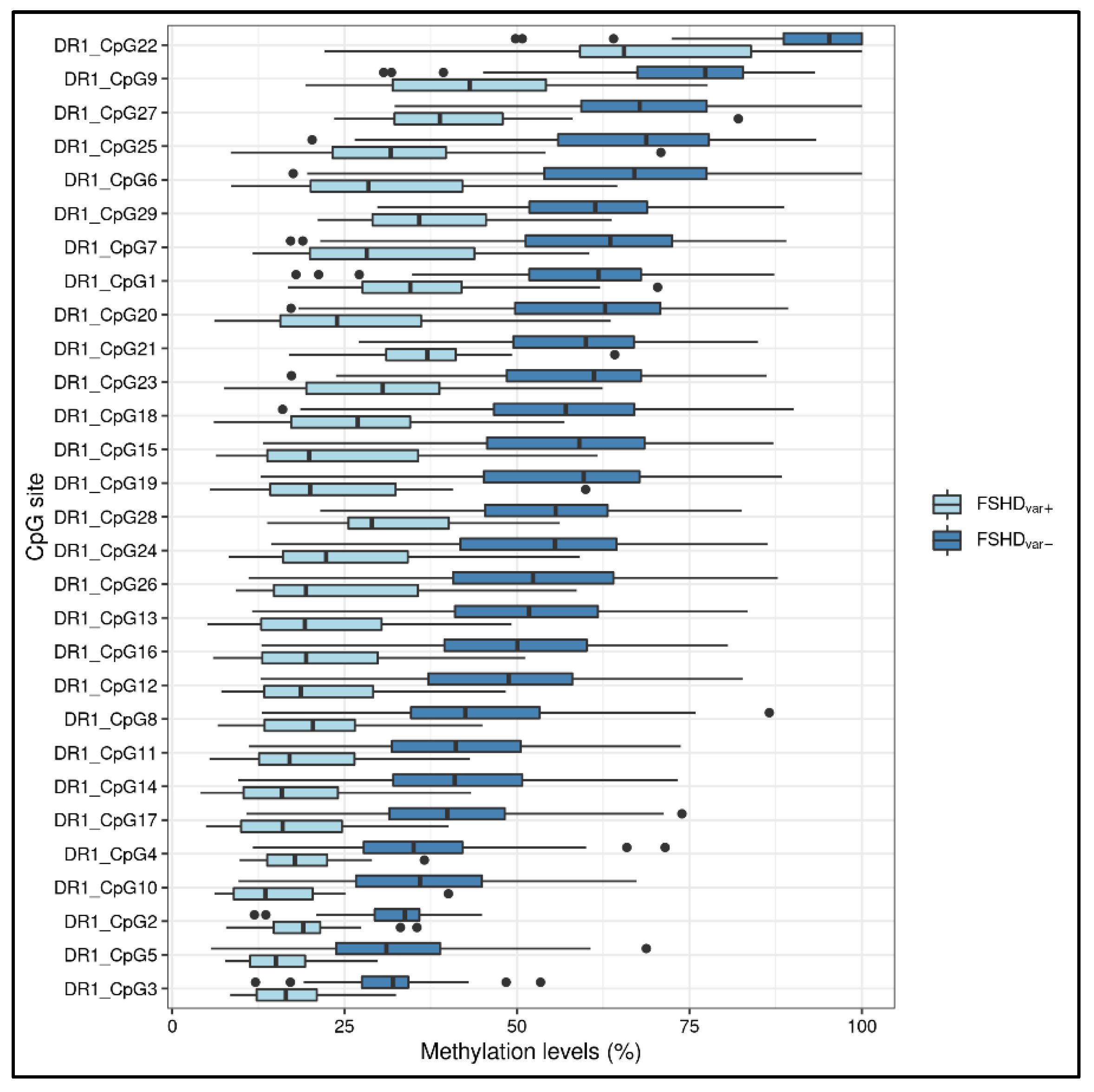
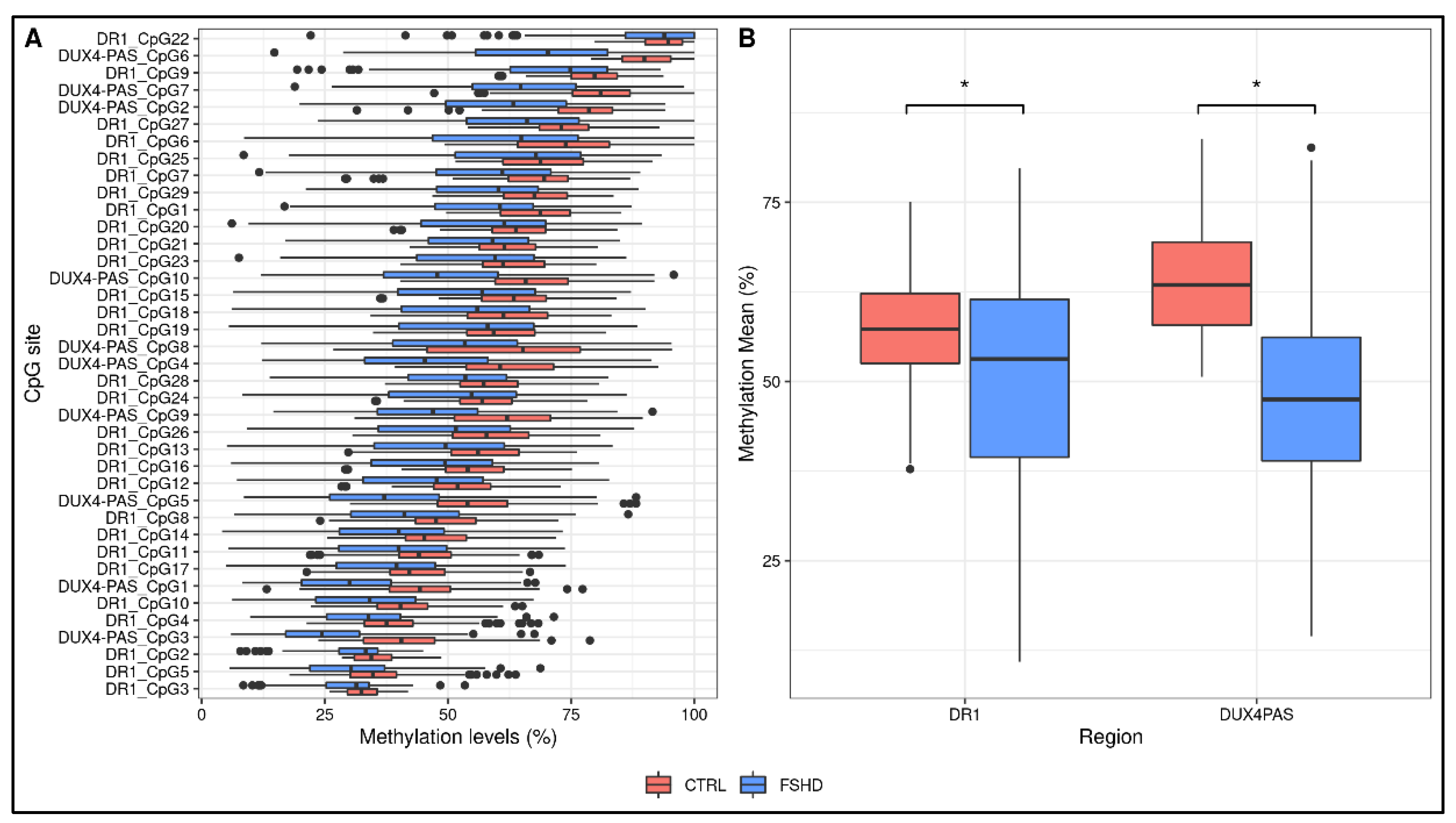
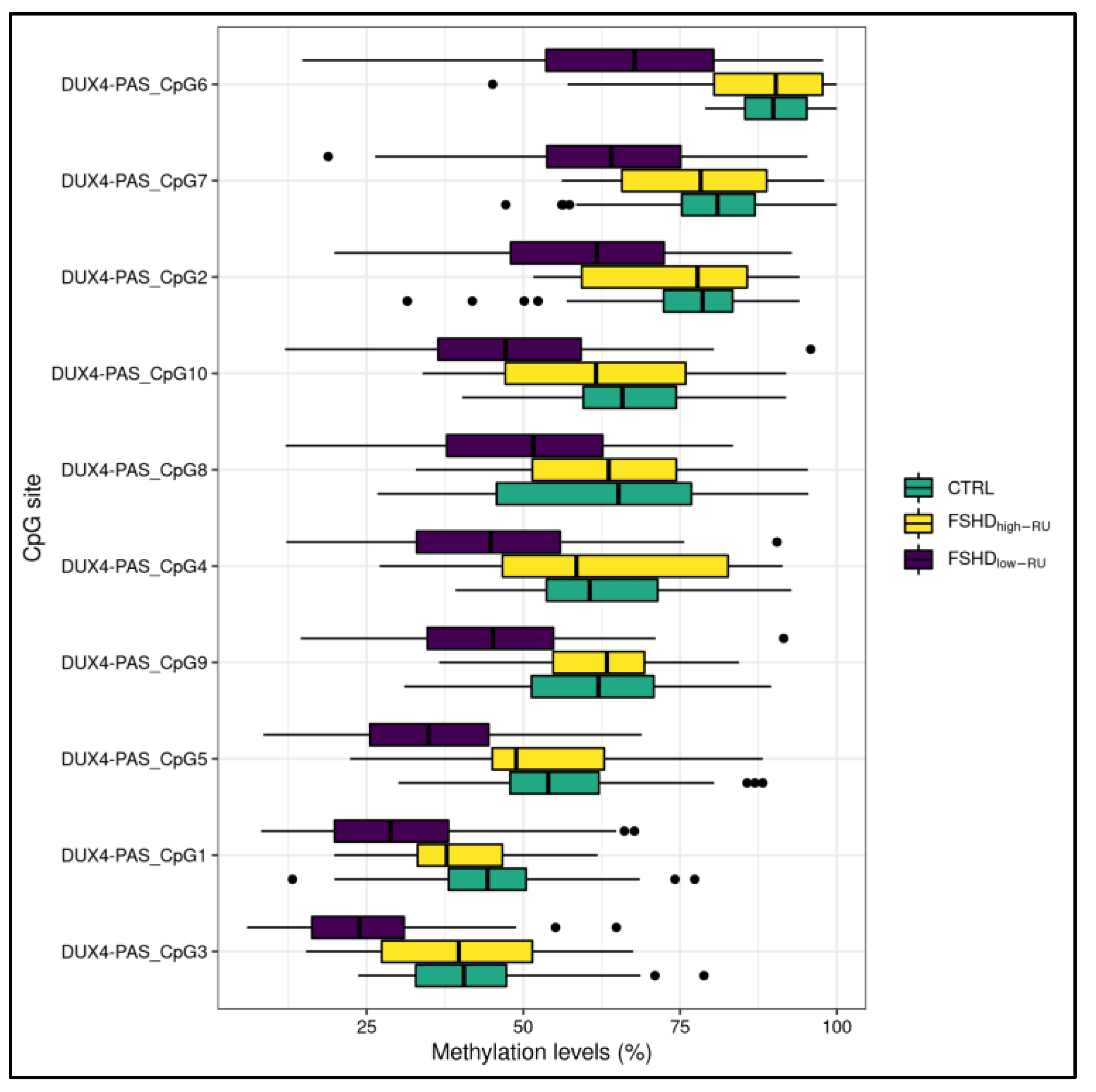
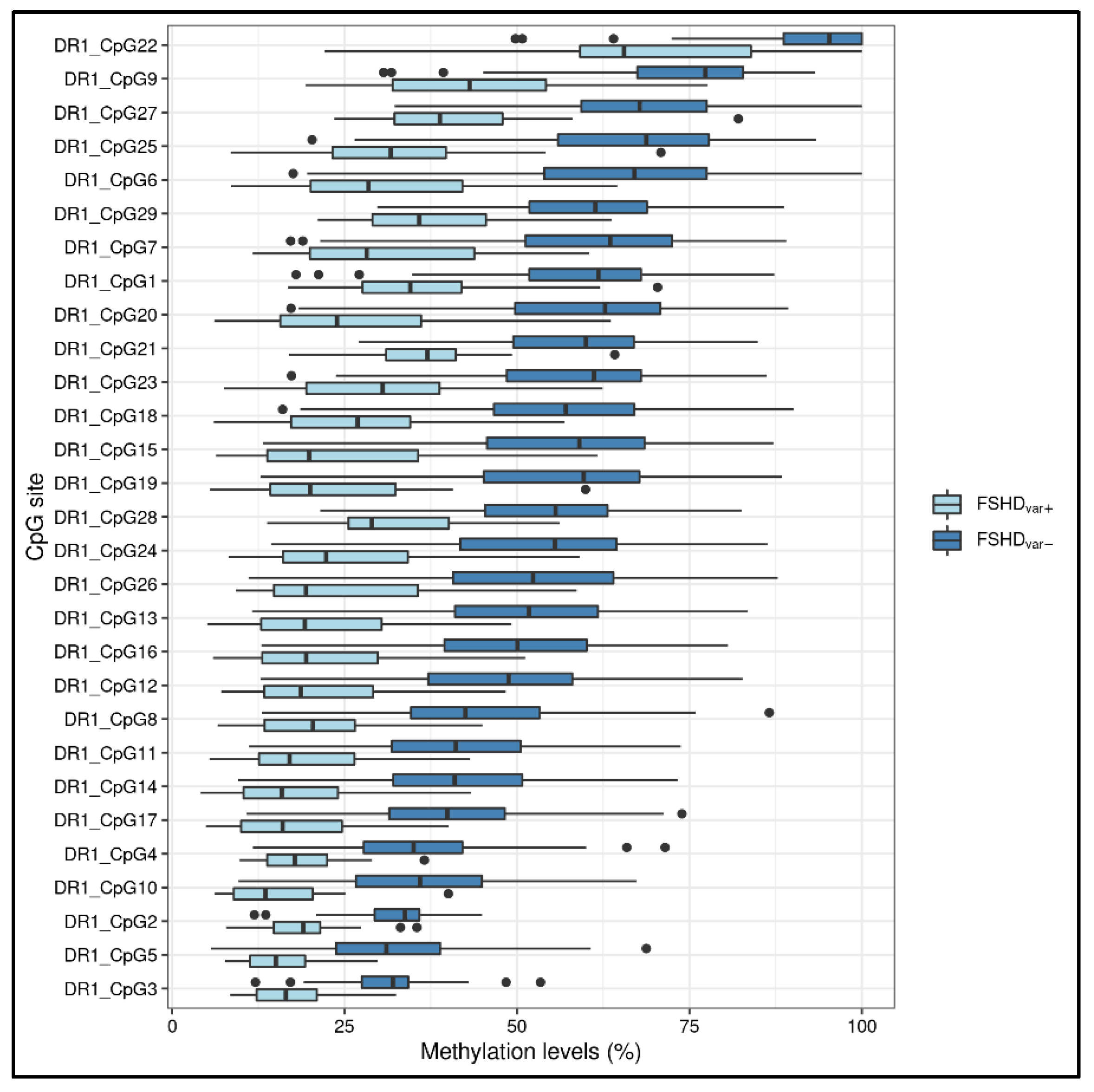
3.1. Statistical Analysis
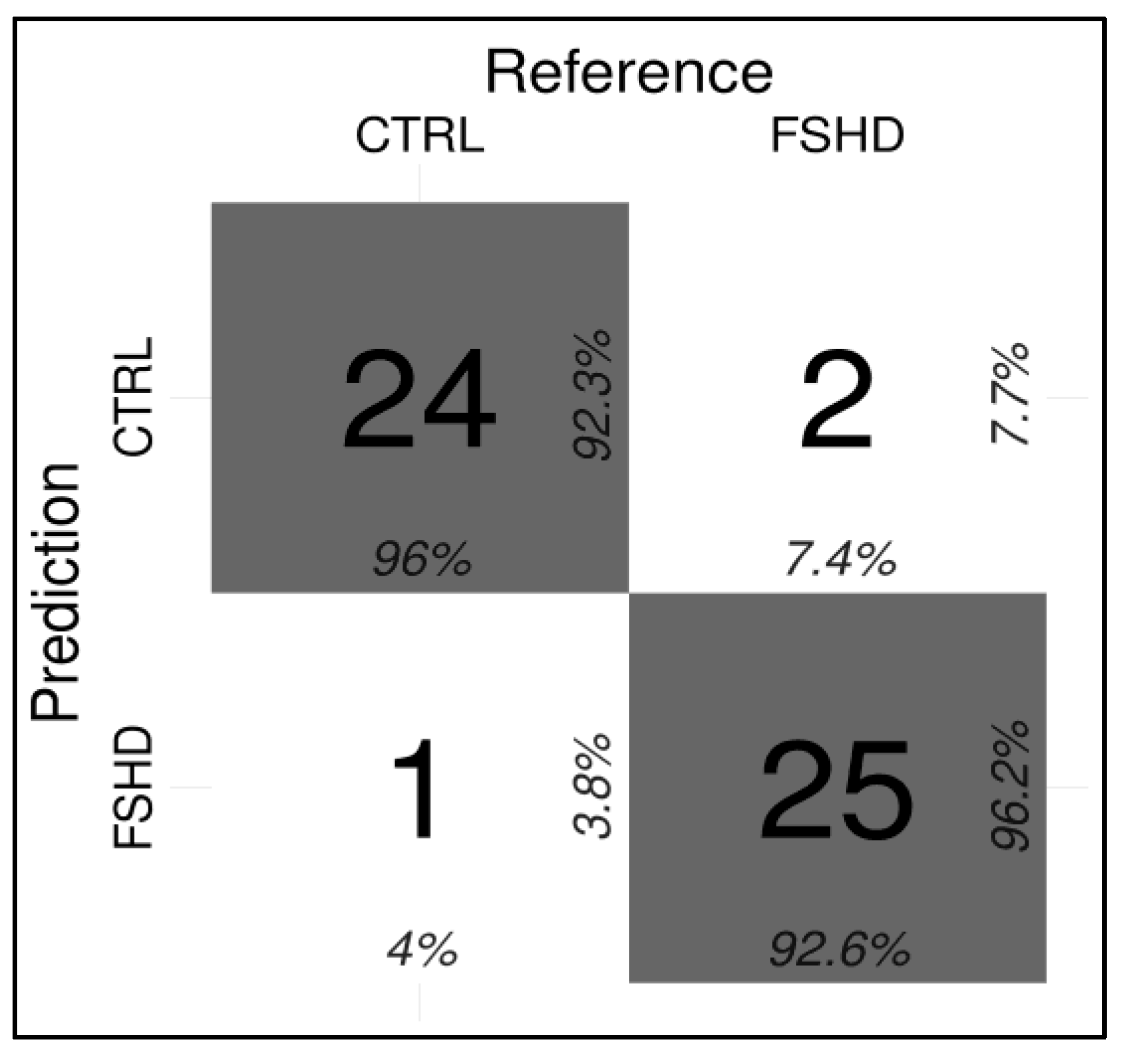
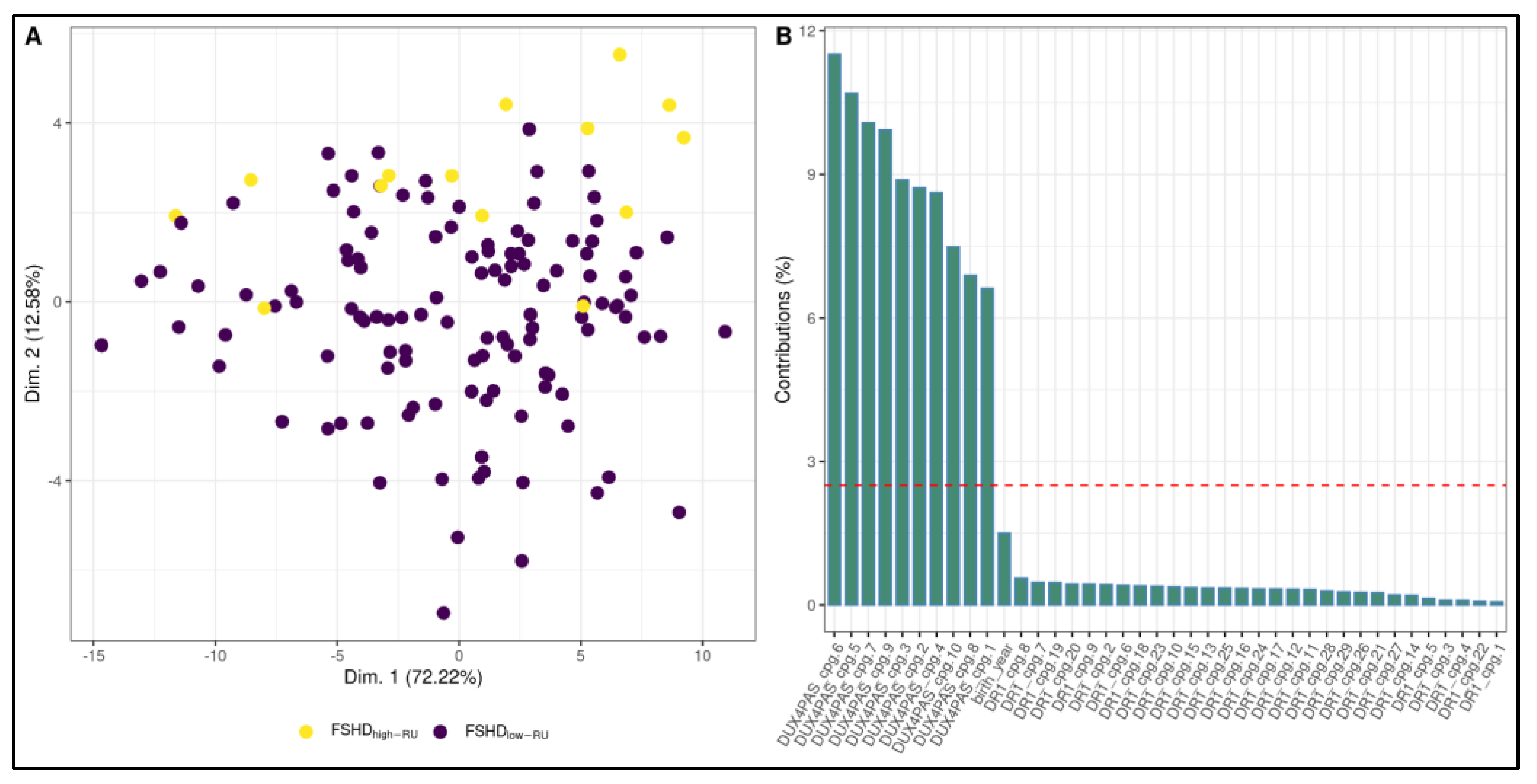
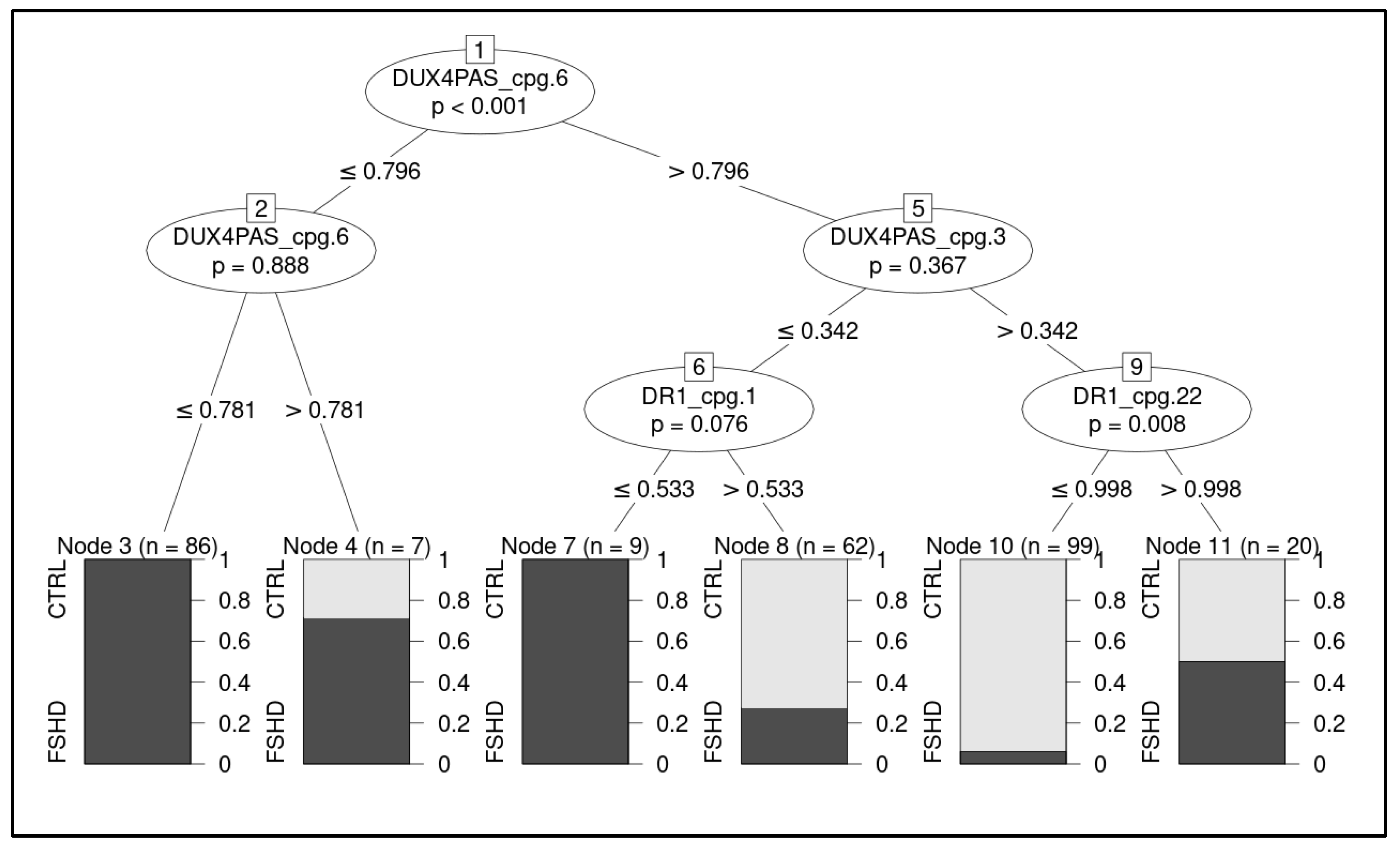
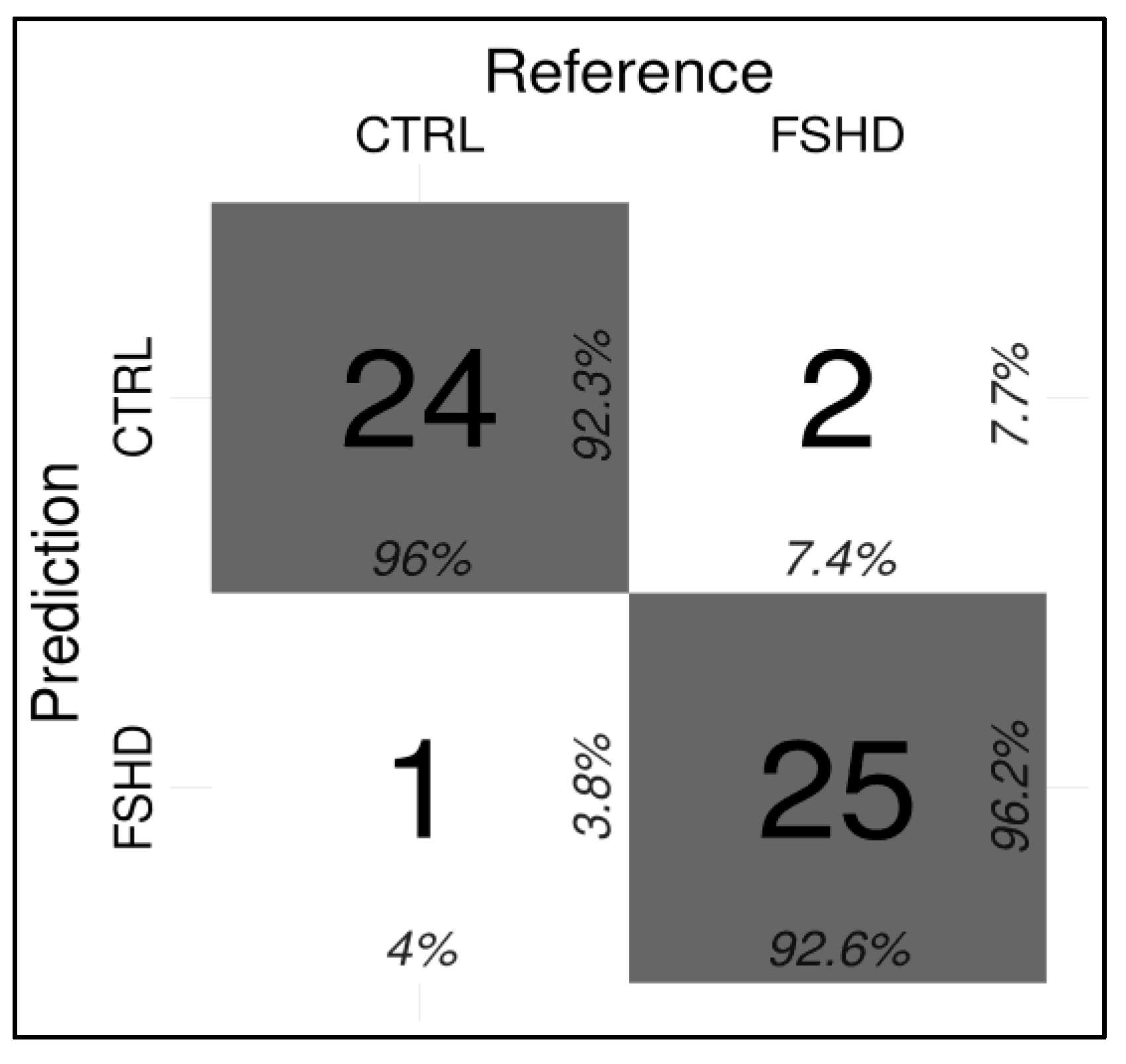
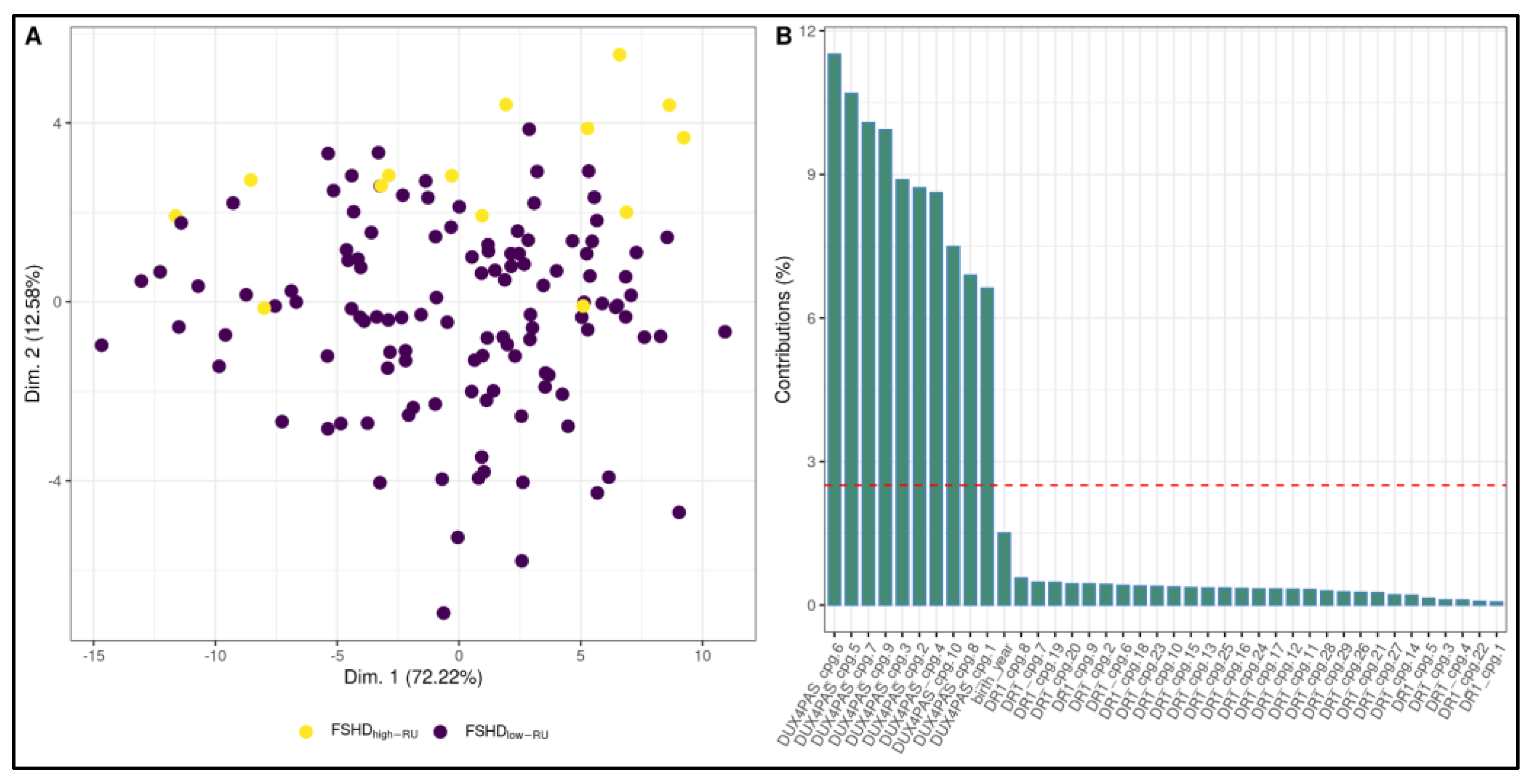
3.2. Development of a ML-Based Classifier for the Discrimination of FSHD Subjects
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Himeda, C.L.; Jones, P.L. The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy. Annu. Rev. Genom. Hum. Genet. 2019, 20, 265–291. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Strafella, C.; Caputo, V.; Galota, R.; Errichiello, V.; Scutifero, M.; Petillo, R.; Marella, G.; Arcangeli, M.; Colantoni, L.; et al. Digenic Inheritance of Shortened Repeat Units of the D4Z4 Region and a Loss-of-Function Variant in SMCHD1 in a Family With FSHD. Front. Neurol. 2018, 9, 1027. [Google Scholar] [CrossRef] [PubMed]
- Strafella, C.; Caputo, V.; Galota, R.; Campoli, G.; Bax, C.; Colantoni, L.; Minozzi, G.; Orsini, C.; Politano, L.; Tasca, G.; et al. The Variability of SMCHD1 Gene in FSHD Patients: Evidence of New Mutations. Hum. Mol. Genet. 2019, 28, 3912–3920. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.F.; Drew, A.P.; Nicholson, G.A.; Corbett, A.; Kumar, K.R. Facioscapulohumeral Muscular Dystrophy Type 2: An Update on the Clinical, Genetic, and Molecular Findings. Neuromuscul. Disord. 2021, 31, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, K.; Šikrová, D.; Mitsuhashi, S.; Masuda, H.; Sekiguchi, Y.; Sugiyama, A.; Shibuya, K.; Lemmers, R.J.L.F.; Goossens, R.; Ogawa, M.; et al. Homozygous Nonsense Variant in LRIF1 Associated with Facioscapulohumeral Muscular Dystrophy. Neurology 2020, 94, e2441–e2447. [Google Scholar] [CrossRef]
- Sacconi, S.; Lemmers, R.J.L.F.; Balog, J.; van der Vliet, P.J.; Lahaut, P.; van Nieuwenhuizen, M.P.; Straasheijm, K.R.; Debipersad, R.D.; Vos-Versteeg, M.; Salviati, L.; et al. The FSHD2 Gene SMCHD1 Is a Modifier of Disease Severity in Families Affected by FSHD1. Am. J. Hum. Genet. 2013, 93, 744–751. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.; Rost, S.; El Hajj, N.; Ferbert, A.; Deschauer, M.; Walter, M.C.; Schoser, B.; Tacik, P.; Kress, W.; Müller, C.R. Diagnostic Approach for FSHD Revisited: SMCHD1 Mutations Cause FSHD2 and Act as Modifiers of Disease Severity in FSHD1. Eur. J. Hum. Genet. 2015, 23, 808–816. [Google Scholar] [CrossRef]
- van den Boogaard, M.L.; Lemmers, R.J.L.F.; Balog, J.; Wohlgemuth, M.; Auranen, M.; Mitsuhashi, S.; van der Vliet, P.J.; Straasheijm, K.R.; van den Akker, R.F.P.; Kriek, M.; et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016, 98, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Sacconi, S.; Camaño, P.; de Greef, J.C.; Lemmers, R.J.L.F.; Salviati, L.; Boileau, P.; Lopez de Munain Arregui, A.; van der Maarel, S.M.; Desnuelle, C. Patients with a Phenotype Consistent with Facioscapulohumeral Muscular Dystrophy Display Genetic and Epigenetic Heterogeneity. J. Med. Genet. 2012, 49, 41–46. [Google Scholar] [CrossRef]
- Lemmers, R.J.L.F.; Goeman, J.J.; van der Vliet, P.J.; van Nieuwenhuizen, M.P.; Balog, J.; Vos-Versteeg, M.; Camano, P.; Ramos Arroyo, M.A.; Jerico, I.; Rogers, M.T.; et al. Inter-Individual Differences in CpG Methylation at D4Z4 Correlate with Clinical Variability in FSHD1 and FSHD2. Hum. Mol. Genet. 2015, 24, 659–669. [Google Scholar] [CrossRef]
- Zampatti, S.; Colantoni, L.; Strafella, C.; Galota, R.M.; Caputo, V.; Campoli, G.; Pagliaroli, G.; Carboni, S.; Mela, J.; Peconi, C.; et al. Facioscapulohumeral Muscular Dystrophy (FSHD) Molecular Diagnosis: From Traditional Technology to the NGS Era. Neurogenetics 2019, 20, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.R.; Eng, C.M. Next-Generation Sequencing to Diagnose Suspected Genetic Disorders. N. Engl. J. Med. 2018, 379, 1353–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, T.I.; King, O.D.; Himeda, C.L.; Homma, S.; Chen, J.C.J.; Beermann, M.L.; Yan, C.; Emerson, C.P.; Miller, J.B.; Wagner, K.R.; et al. Individual Epigenetic Status of the Pathogenic D4Z4 Macrosatellite Correlates with Disease in Facioscapulohumeral Muscular Dystrophy. Clin. Epigenet. 2015, 7, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaillard, M.-C.; Roche, S.; Dion, C.; Tasmadjian, A.; Bouget, G.; Salort-Campana, E.; Vovan, C.; Chaix, C.; Broucqsault, N.; Morere, J.; et al. Differential DNA Methylation of the D4Z4 Repeat in Patients with FSHD and Asymptomatic Carriers. Neurology 2014, 83, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Calandra, P.; Cascino, I.; Lemmers, R.J.L.F.; Galluzzi, G.; Teveroni, E.; Monforte, M.; Tasca, G.; Ricci, E.; Moretti, F.; van der Maarel, S.M.; et al. Allele-Specific DNA Hypomethylation Characterises FSHD1 and FSHD2. J. Med. Genet. 2016, 53, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Roche, S.; Dion, C.; Broucqsault, N.; Laberthonnière, C.; Gaillard, M.-C.; Robin, J.D.; Lagarde, A.; Puppo, F.; Vovan, C.; Chaix, C.; et al. Methylation Hotspots Evidenced by Deep Sequencing in Patients with Facioscapulohumeral Dystrophy and Mosaicism. Neurol. Genet. 2019, 5, e372. [Google Scholar] [CrossRef] [Green Version]
- Gould, T.; Jones, T.I.; Jones, P.L. Precise Epigenetic Analysis Using Targeted Bisulfite Genomic Sequencing Distinguishes FSHD1, FSHD2, and Healthy Subjects. Diagnostics 2021, 11, 1469. [Google Scholar] [CrossRef]
- Nikolic, A.; Jones, T.I.; Govi, M.; Mele, F.; Maranda, L.; Sera, F.; Ricci, G.; Ruggiero, L.; Vercelli, L.; Portaro, S.; et al. Interpretation of the Epigenetic Signature of Facioscapulohumeral Muscular Dystrophy in Light of Genotype-Phenotype Studies. Int. J. Mol. Sci. 2020, 21, 2635. [Google Scholar] [CrossRef]
- Erdmann, H.; Scharf, F.; Gehling, S.; Benet-Pagès, A.; Jakubiczka, S.; Becker, K.; Seipelt, M.; Kleefeld, F.; Knop, K.C.; Prott, E.C.; et al. Methylation of the 4q35 D4Z4 Repeat Defines Disease Status in Facioscapulohumeral Muscular Dystrophy. Brain 2022, awac336. [Google Scholar] [CrossRef]
- Hiramuki, Y.; Kure, Y.; Saito, Y.; Ogawa, M.; Ishikawa, K.; Mori-Yoshimura, M.; Oya, Y.; Takahashi, Y.; Kim, D.-S.; Arai, N.; et al. Simultaneous Measurement of the Size and Methylation of Chromosome 4qA-D4Z4 Repeats in Facioscapulohumeral Muscular Dystrophy by Long-Read Sequencing. J. Transl. Med. 2022, 20, 517. [Google Scholar] [CrossRef]
- Hartweck, L.M.; Anderson, L.J.; Lemmers, R.J.; Dandapat, A.; Toso, E.A.; Dalton, J.C.; Tawil, R.; Day, J.W.; van der Maarel, S.M.; Kyba, M. A Focal Domain of Extreme Demethylation within D4Z4 in FSHD2. Neurology 2013, 80, 392–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasca, G.; Monforte, M.; Ottaviani, P.; Pelliccioni, M.; Frusciante, R.; Laschena, F.; Ricci, E. Magnetic Resonance Imaging in a Large Cohort of Facioscapulohumeral Muscular Dystrophy Patients: Pattern Refinement and Implications for Clinical Trials. Ann. Neurol. 2016, 79, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Ricci, E.; Galluzzi, G.; Deidda, G.; Cacurri, S.; Colantoni, L.; Merico, B.; Piazzo, N.; Servidei, S.; Vigneti, E.; Pasceri, V.; et al. Progress in the Molecular Diagnosis of Facioscapulohumeral Muscular Dystrophy and Correlation between the Number of KpnI Repeats at the 4q35 Locus and Clinical Phenotype. Ann. Neurol. 1999, 45, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Giacomucci, G.; Monforte, M.; Diaz-Manera, J.; Mul, K.; Fernandez Torrón, R.; Maggi, L.; Marini Bettolo, C.; Dahlqvist, J.R.; Haberlova, J.; Camaño, P.; et al. Deep Phenotyping of Facioscapulohumeral Muscular Dystrophy Type 2 by Magnetic Resonance Imaging. Eur. J. Neurol. 2020, 27, 2604–2615. [Google Scholar] [CrossRef]
- Monforte, M.; Bortolani, S.; Torchia, E.; Cristiano, L.; Laschena, F.; Tartaglione, T.; Ricci, E.; Tasca, G. Diagnostic Magnetic Resonance Imaging Biomarkers for Facioscapulohumeral Muscular Dystrophy Identified by Machine Learning. J. Neurol. 2021, 269, 2055–2063. [Google Scholar] [CrossRef]
- Kuhn, M. Caret: Classification and Regression Training; Astrophysics Source Code Library, 2015. 1505.003. Available online: https://www.semanticscholar.org/paper/caret%3A-Classification-and-Regression-Training-Kuhn/258c7e3242b91e02e092e77e058f6275ba52b12d (accessed on 20 September 2022).
- Kuhn, M.; Johnson, K. Applied Predictive Modeling; Springer: New York, NY, USA, 2013; ISBN 978-1-4614-6848-6. [Google Scholar]
- Greco, A.; Goossens, R.; van Engelen, B.; van der Maarel, S.M. Consequences of Epigenetic Derepression in Facioscapulohumeral Muscular Dystrophy. Clin. Genet. 2020, 97, 799–814. [Google Scholar] [CrossRef]
- Jones, T.I.; Yan, C.; Sapp, P.C.; McKenna-Yasek, D.; Kang, P.B.; Quinn, C.; Salameh, J.S.; King, O.D.; Jones, P.L. Identifying Diagnostic DNA Methylation Profiles for Facioscapulohumeral Muscular Dystrophy in Blood and Saliva Using Bisulfite Sequencing. Clin. Epigenet. 2014, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- van Overveld, P.G.M.; Lemmers, R.J.F.L.; Sandkuijl, L.A.; Enthoven, L.; Winokur, S.T.; Bakels, F.; Padberg, G.W.; van Ommen, G.-J.B.; Frants, R.R.; van der Maarel, S.M. Hypomethylation of D4Z4 in 4q-Linked and Non-4q-Linked Facioscapulohumeral Muscular Dystrophy. Nat. Genet. 2003, 35, 315–317. [Google Scholar] [CrossRef]
- Ricci, G.; Mele, F.; Govi, M.; Ruggiero, L.; Sera, F.; Vercelli, L.; Bettio, C.; Santoro, L.; Mongini, T.; Villa, L.; et al. Large Genotype-Phenotype Study in Carriers of D4Z4 Borderline Alleles Provides Guidance for Facioscapulohumeral Muscular Dystrophy Diagnosis. Sci. Rep. 2020, 10, 21648. [Google Scholar] [CrossRef]
- Scionti, I.; Greco, F.; Ricci, G.; Govi, M.; Arashiro, P.; Vercelli, L.; Berardinelli, A.; Angelini, C.; Antonini, G.; Cao, M.; et al. Large-Scale Population Analysis Challenges the Current Criteria for the Molecular Diagnosis of Fascioscapulohumeral Muscular Dystrophy. Am. J. Hum. Genet. 2012, 90, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Huichalaf, C.; Micheloni, S.; Ferri, G.; Caccia, R.; Gabellini, D. DNA Methylation Analysis of the Macrosatellite Repeat Associated with FSHD Muscular Dystrophy at Single Nucleotide Level. PLoS ONE 2014, 9, e115278. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, S.; Briand-Suleau, A.; Gros, M.; Baudoin, C.; Lemmers, R.J.L.F.; Rondeau, S.; Lagha, N.; Nigumann, P.; Cambieri, C.; Puma, A.; et al. FSHD1 and FSHD2 Form a Disease Continuum. Neurology 2019, 92, e2273–e2285. [Google Scholar] [CrossRef] [PubMed]
- Briganti, G.; Le Moine, O. Artificial Intelligence in Medicine: Today and Tomorrow. Front. Med. 2020, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Caputo, V.; Megalizzi, D.; Fabrizio, C.; Termine, A.; Colantoni, L.; Caltagirone, C.; Giardina, E.; Cascella, R.; Strafella, C. Update on the Molecular Aspects and Methods Underlying the Complex Architecture of FSHD. Cells 2022, 11, 2687. [Google Scholar] [CrossRef]
- Fabrizio, C.; Termine, A.; Caputo, V.; Megalizzi, D.; Zampatti, S.; Falsini, B.; Cusumano, A.; Eandi, C.M.; Ricci, F.; Giardina, E.; et al. WARE: Wet AMD Risk-Evaluation Tool as a Clinical Decision-Support System Integrating Genetic and Non-Genetic Factors. J. Pers. Med. 2022, 12, 1034. [Google Scholar] [CrossRef]
- Zampatti, S.; Fabrizio, C.; Ragazzo, M.; Campoli, G.; Caputo, V.; Strafella, C.; Pellicano, C.; Cascella, R.; Spalletta, G.; Petrosini, L.; et al. Precision Medicine into Clinical Practice: A Web-Based Tool Enables Real-Time Pharmacogenetic Assessment of Tailored Treatments in Psychiatric Disorders. J. Pers. Med. 2021, 11, 851. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cooper-Knock, J.; Weimer, A.K.; Shi, M.; Moll, T.; Marshall, J.N.G.; Harvey, C.; Nezhad, H.G.; Franklin, J.; Souza, C.D.S.; et al. Genome-Wide Identification of the Genetic Basis of Amyotrophic Lateral Sclerosis. Neuron 2022, 110, 992.e11–1008.e11. [Google Scholar] [CrossRef]
- Marzola, F.; van Alfen, N.; Doorduin, J.; Meiburger, K.M. Deep Learning Segmentation of Transverse Musculoskeletal Ultrasound Images for Neuromuscular Disease Assessment. Comput. Biol. Med. 2021, 135, 104623. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Cohort | n | Mean Age (±SD) | F:M Ratio |
|---|---|---|---|---|
| FSHD | Training group | 133 | 51.4 (±17.6) | 45:55 |
| CTRL | Training group | 150 | 55.7 (±15.8) | 36:64 |
| FSHD | Test set | 27 | 56.0 (±16.7) | 45:55 |
| CTRL | Test set | 25 | 50.0 (±14.7) | 52:48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caputo, V.; Megalizzi, D.; Fabrizio, C.; Termine, A.; Colantoni, L.; Bax, C.; Gimenez, J.; Monforte, M.; Tasca, G.; Ricci, E.; et al. D4Z4 Methylation Levels Combined with a Machine Learning Pipeline Highlight Single CpG Sites as Discriminating Biomarkers for FSHD Patients. Cells 2022, 11, 4114. https://doi.org/10.3390/cells11244114
Caputo V, Megalizzi D, Fabrizio C, Termine A, Colantoni L, Bax C, Gimenez J, Monforte M, Tasca G, Ricci E, et al. D4Z4 Methylation Levels Combined with a Machine Learning Pipeline Highlight Single CpG Sites as Discriminating Biomarkers for FSHD Patients. Cells. 2022; 11(24):4114. https://doi.org/10.3390/cells11244114
Chicago/Turabian StyleCaputo, Valerio, Domenica Megalizzi, Carlo Fabrizio, Andrea Termine, Luca Colantoni, Cristina Bax, Juliette Gimenez, Mauro Monforte, Giorgio Tasca, Enzo Ricci, and et al. 2022. "D4Z4 Methylation Levels Combined with a Machine Learning Pipeline Highlight Single CpG Sites as Discriminating Biomarkers for FSHD Patients" Cells 11, no. 24: 4114. https://doi.org/10.3390/cells11244114
APA StyleCaputo, V., Megalizzi, D., Fabrizio, C., Termine, A., Colantoni, L., Bax, C., Gimenez, J., Monforte, M., Tasca, G., Ricci, E., Caltagirone, C., Giardina, E., Cascella, R., & Strafella, C. (2022). D4Z4 Methylation Levels Combined with a Machine Learning Pipeline Highlight Single CpG Sites as Discriminating Biomarkers for FSHD Patients. Cells, 11(24), 4114. https://doi.org/10.3390/cells11244114