
Expression of Fibrosis-Related Genes in Liver and Kidney Fibrosis in Comparison to Inflammatory Bowel Diseases


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Bioinformatics Analysis
2.2. Selected Gene Expression Analysis
2.2.1. Patients’ Selection
- 11 patients with kidney fibrosis due to diabetic nephropathy and 9 patients as controls without fibrosis, with thin glomerular basement membrane, with no other abnormalities;
- 10 patients with liver cirrhosis and 7 deceased patients with morphologically normal liver (pre-transplantation screening biopsies of potential donor livers);
- 20 patients with CD of the colon (14 samples of colon with fibrosis, 14 samples of colon with inflammation without fibrosis), 10 patients with UC and 11 patients with colorectal carcinoma (samples of morphologically normal colon, taken from uninvolved margins of resection specimens).
2.2.2. Tissue Acquisition
2.2.3. RNA Isolation
2.2.4. Reverse Transcription and Quantitative PCR
2.2.5. Statistical Analysis of qPCR Experiments
3. Results
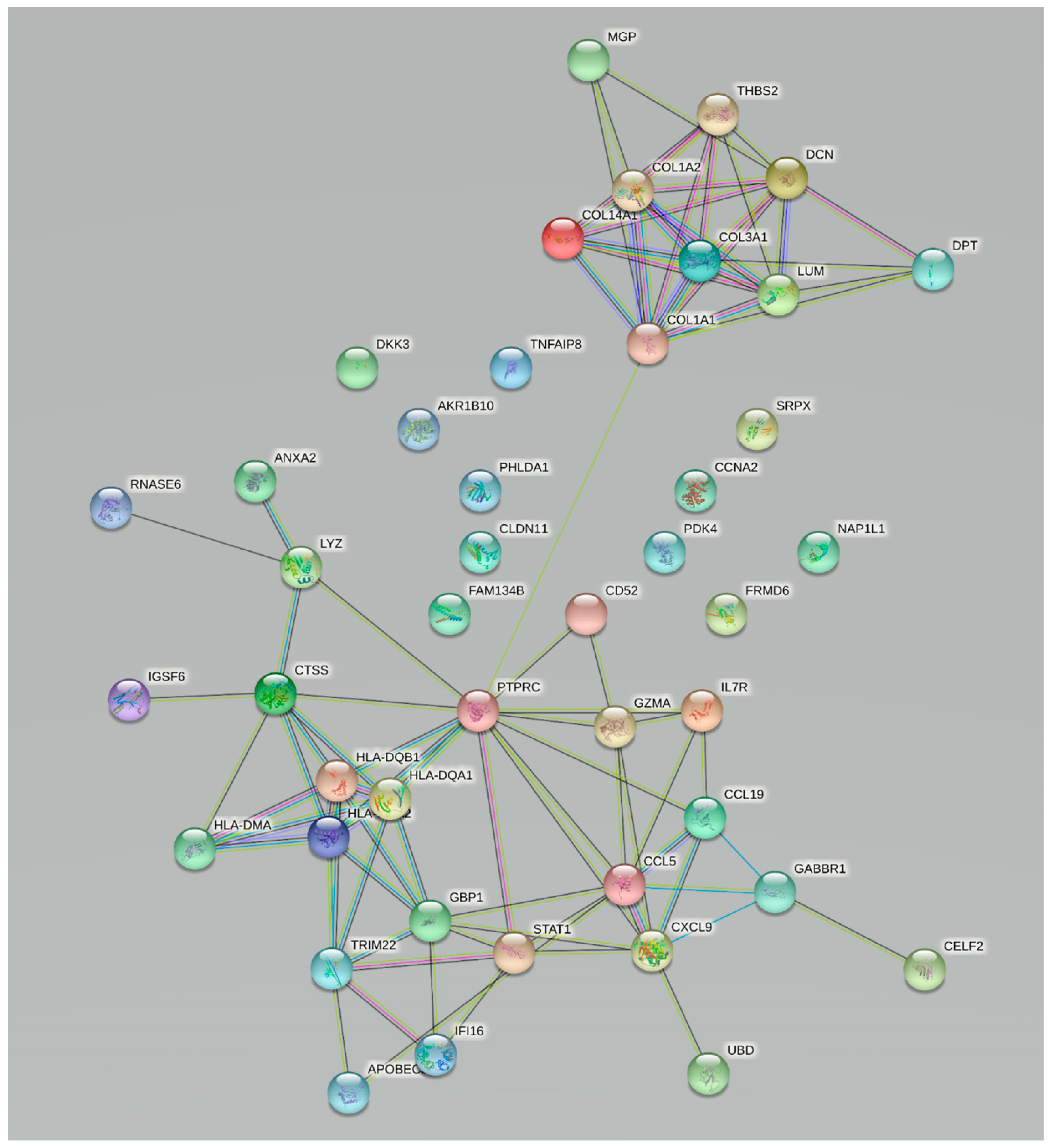
3.1. Bioinformatics Analysis
3.2. Validation of Changes in Gene Expression
3.2.1. Patient Population
3.2.2. Expression of Selected Fibrosis-Related Genes in the Liver and Kidney
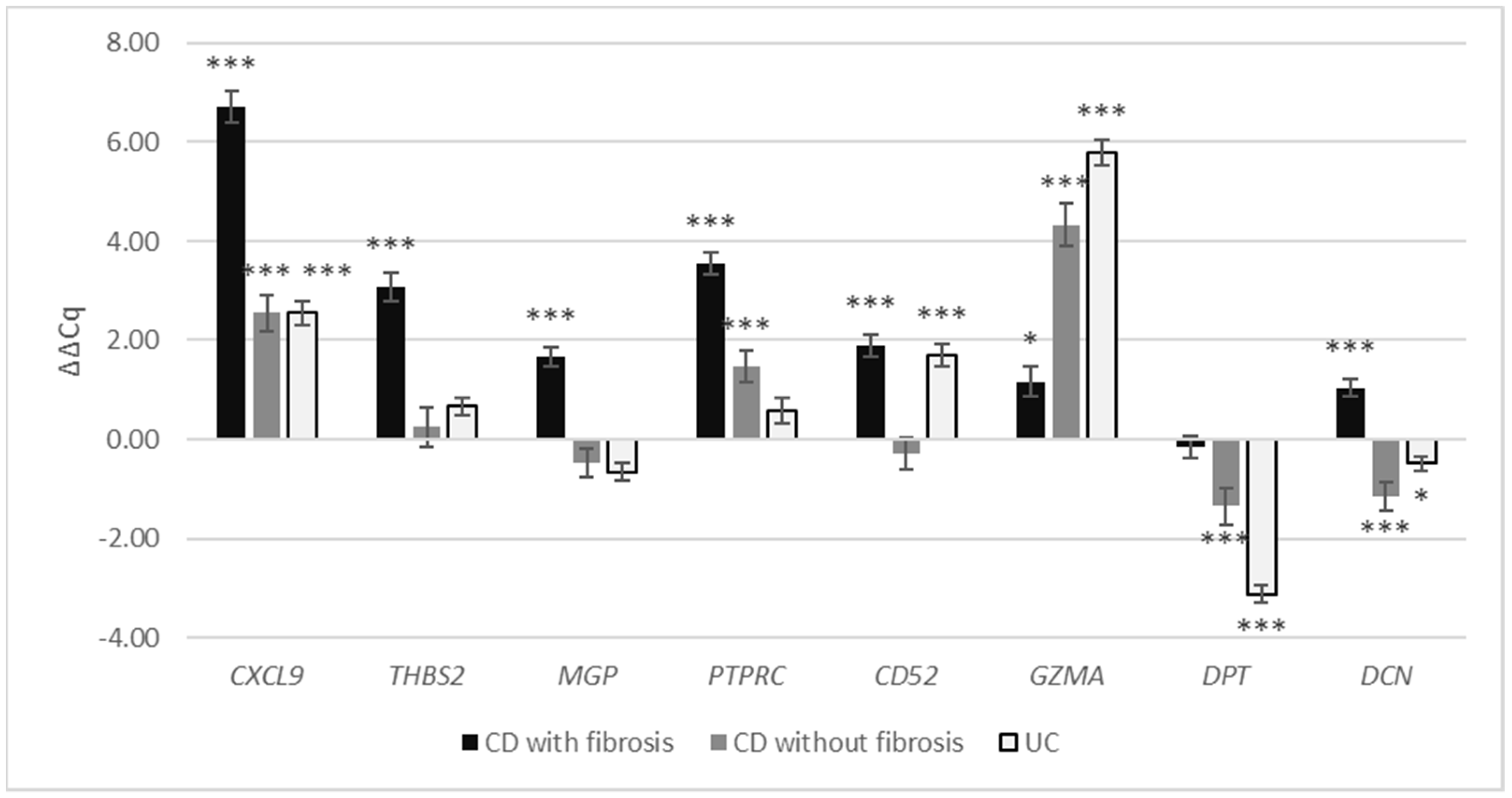
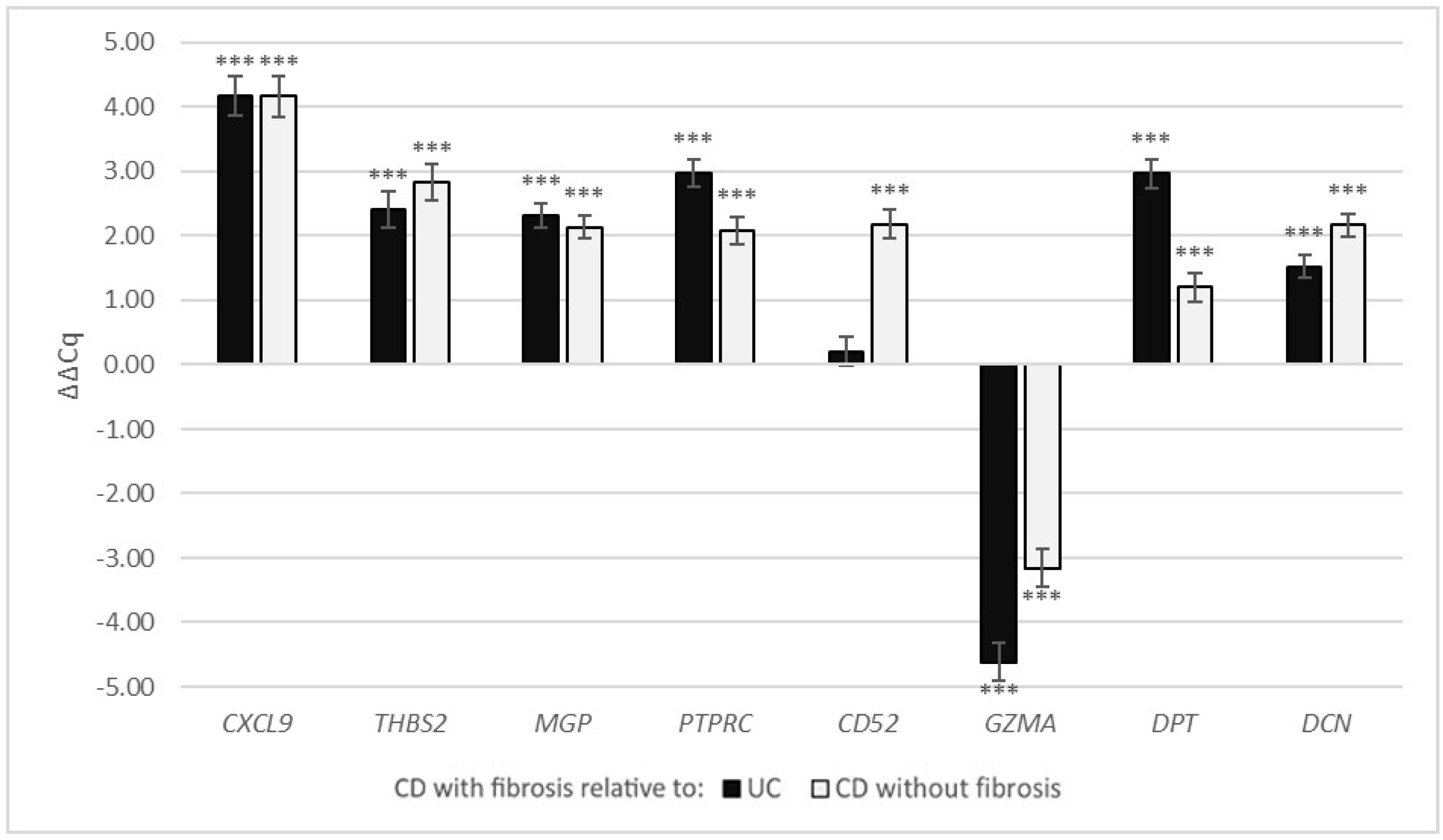
3.2.3. Expression of Selected Fibrosis-Related Genes in the Crohn’s Disease and Ulcerative Colitis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rieder, F.; Latella, G.; Magro, F.; Yuksel, E.S.; Higgins, P.D.R.; Di Sabatino, A.; de Bruyn, J.R.; Rimola, J.; Brito, J.; Bettenworth, D.; et al. European Crohn’s and Colitis Organisation topical review on prediction, diagnosis and management of fibrostenosing Crohn’s disease. J. Crohn’s Colitis 2016, 10, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Gordon, I.O.; Bettenworth, D.; Bokemeyer, A.; Srivastava, A.; Rosty, C.; de Hertogh, G.; Robert, M.E.; Valasek, M.A.; Mao, R.; Kurada, S.; et al. Histopathology scoring systems of stenosis associated with small bowel Crohn’s disease: A systematic review. Gastroenterology 2020, 158, 137–150.e1. [Google Scholar] [CrossRef]
- Thia, K.T.; Sandborn, W.J.; Harmsen, W.S.; Zinsmeister, A.R.; Loftus, E.V. Risk factors associated with progression to intestinal complications of Crohn’s disease in a population-based cohort. Gastroenterology 2010, 139, 1147–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latella, G.; Di Gregorio, J.; Flati, V.; Rieder, F.; Lawrance, I.C. Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand. J. Gastroenterol. 2014, 50, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Bemelman, W.A.; Warusavitarne, J.; Sampietro, G.M.; Serclova, Z.; Zmora, O.; Luglio, G.; de Buck van Overstraeten, A.; Burke, J.P.; Buskens, C.J.; Francesco, C.; et al. ECCO-ESCP consensus on surgery for Crohn’s disease. J. Crohn’s Colitis 2017, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bettenworth, D. Assessment of stricturing Crohn’s disease: Current clinical practice and future avenues. World J. Gastroenterol. 2016, 22, 1008. [Google Scholar] [CrossRef]
- Gordon, I.O.; Agrawal, N.; Goldblum, J.R.; Fiocchi, C.; Rieder, F. Fibrosis in ulcerative colitis. Inflamm. Bowel Dis. 2014, 20, 2198–2206. [Google Scholar] [CrossRef]
- Gordon, I.O.; Agrawal, N.; Willis, E.; Goldblum, J.R.; Lopez, R.; Allende, D.; Liu, X.; Patil, D.Y.; Yerian, L.; El-Khider, F.; et al. Fibrosis in ulcerative colitis is directly linked to severity and chronicity of mucosal inflammation. Aliment. Pharmacol. Ther. 2018, 47, 922–939. [Google Scholar] [CrossRef]
- de Bruyn, J.R.; Meijer, S.L.; Wildenberg, M.E.; Bemelman, W.A.; van den Brink, G.R.; D’Haens, G.R. Development of fibrosis in acute and longstanding ulcerative colitis. J. Crohn’s Colitis 2015, 9, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Lawrance, I.C.; Rogler, G.; Bamias, G.; Breynaert, C.; Florholmen, J.; Pellino, G.; Reif, S.; Speca, S.; Latella, G. Cellular and molecular mediators of intestinal fibrosis. J. Crohn’s Colitis 2015, 11, 1491–1503. [Google Scholar] [CrossRef] [Green Version]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Latella, G.; Rieder, F. Intestinal fibrosis. Curr. Opin. Gastroenterol. 2017, 33, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, S.E.; Peduto, L. The perivascular origin of pathological fibroblasts. J. Clin. Investig. 2018, 128, 54–63. [Google Scholar] [CrossRef]
- Sun, M.; Kisseleva, T. Reversibility of liver fibrosis. Clin. Res. Hepatol. Gastroenterol. 2015, 39, S60–S63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Asp. Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Decaris, M.L.; Gatmaitan, M.; FlorCruz, S.; Luo, F.; Li, K.; Holmes, W.E.; Hellerstein, M.K.; Turner, S.M.; Emson, C.L. Proteomic analysis of altered extracellular matrix turnover in bleomycin-induced pulmonary fibrosis. Mol. Cell. Proteom. 2014, 13, 1741–1752. [Google Scholar] [CrossRef] [Green Version]
- Bettenworth, D.; Rieder, F. Reversibility of stricturing Crohn’s disease—Fact or fiction? Inflamm. Bowel Dis. 2016, 22, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Buck van Overstraeten, A.; Vermeire, S.; Vanbeckevoort, D.; Rimola, J.; Ferrante, M.; Van Assche, G.; Wolthuis, A.; D’Hoore, A. Modified side-to-side isoperistaltic strictureplasty over the ileocaecal valve: An alternative to ileocaecal resection in extensive terminal ileal Crohn’s disease. J. Crohn’s Colitis 2016, 10, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy-analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Singh, U.; Venkataraman, C.; Singh, R.; Lillard, J., Jr. CXCR3 axis: Role in inflammatory bowel disease and its therapeutic implication. Endocrine Metab. Immune Disord. Targets 2007, 7, 111–123. [Google Scholar] [CrossRef]
- Zhao, H.; Xi, H.; Wei, B.; Cai, A.; Wang, T.; Wang, Y.; Zhao, X.; Song, Y.; Chen, L. Expression of decorin in intestinal tissues of mice with inflammatory bowel disease and its correlation with autophagy. Exp. Ther. Med. 2016, 12, 3885–3892. [Google Scholar] [CrossRef] [Green Version]
- Sahebally, S.M.; Burke, J.P.; Chang, K.H.; Kiernan, M.G.; O’Connell, P.R.; Coffey, J.C. Circulating fibrocytes and Crohn’s disease. Br. J. Surg. 2013, 100, 1549–1556. [Google Scholar] [CrossRef]
- Haberman, Y.; Minar, P.; Karns, R.; Dexheimer, P.J.; Ghandikota, S.; Tegge, S.; Shapiro, D.; Shuler, B.; Venkateswaran, S.; Braun, T.; et al. Mucosal inflammatory and wound healing gene programmes reveal targets for stricturing behaviour in paediatric Crohn’s disease. J. Crohn’s Colitis 2021, 15, 273–286. [Google Scholar] [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.-J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Lin, C.-F.; Su, C.-J.; Liu, J.-H.; Chen, S.-T.; Huang, H.-L.; Pan, S.-L. Potential effects of CXCL9 and CCL20 on cardiac fibrosis in patients with myocardial infarction and isoproterenol-treated rats. J. Clin. Med. 2019, 8, 659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouroumalis, A.; Nibbs, R.J.; Aptel, H.; Wright, K.L.; Kolios, G.; Ward, S.G. The chemokines CXCL9, CXCL10, and CXCL11 differentially stimulate Gα i -independent signaling and actin responses in human intestinal myofibroblasts. J. Immunol. 2005, 175, 5403–5411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nukui, Y.; Yamana, T.; Masuo, M.; Tateishi, T.; Kishino, M.; Tateishi, U.; Tomita, M.; Hasegawa, T.; Aritsu, T.; Miyazaki, Y. Serum CXCL9 and CCL17 as biomarkers of declining pulmonary function in chronic bird-related hypersensitivity pneumonitis. PLoS ONE 2019, 14, e0220462. [Google Scholar] [CrossRef] [Green Version]
- Worrell, J.C.; Walsh, S.M.; Fabre, A.; Kane, R.; Hinz, B.; Keane, M.P. CXCR3A promotes the secretion of the antifibrotic decoy receptor sIL-13Rα2 by pulmonary fibroblasts. Am. J. Physiol. Physiol. 2020, 319, C1059–C1069. [Google Scholar] [CrossRef] [PubMed]
- Luther, J.; Gala, M.; Patel, S.J.; Dave, M.; Borren, N.; Xavier, R.J.; Ananthakrishnan, A.N. Loss of response to anti-Tumor Necrosis Factor alpha therapy in Crohn’s disease is not associated with emergence of novel inflammatory pathways. Dig. Dis. Sci. 2018, 63, 738–745. [Google Scholar] [CrossRef]
- Chen, W.; Wu, X.; Yan, X.; Xu, A.; Yang, A.; You, H. Multitranscriptome analyses reveal prioritized genes specifically associated with liver fibrosis progression independent of etiology. Am. J. Physiol. Liver Physiol. 2019, 316, G744–G754. [Google Scholar] [CrossRef]
- Shichino, S.; Abe, J.; Ueha, S.; Otsuji, M.; Tsukui, T.; Kosugi-Kanaya, M.; Shand, F.H.W.; Hashimoto, S.; Suzuki, H.I.; Morikawa, T.; et al. Reduced supply of monocyte-derived macrophages leads to a transition from nodular to diffuse lesions and tissue cell activation in silica-induced pulmonary fibrosis in mice. Am. J. Pathol. 2015, 185, 2923–2938. [Google Scholar] [CrossRef] [Green Version]
- Rödder, S.; Scherer, A.; Raulf, F.; Berthier, C.C.; Hertig, A.; Couzi, L.; Durrbach, A.; Rondeau, E.; Marti, H.-P. Renal allografts with IF/TA display distinct expression profiles of metzincins and related genes. Am. J. Transplant. 2009, 9, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Brouwer, B.; Piscaer, I.; Von Der Thusen, J.H.; Grutters, J.C.; Schutgens, R.E.G.; Wouters, E.F.M.; Janssen, R. Should vitamin K be supplemented instead of antagonised in patients with idiopathic pulmonary fibrosis? Expert Rev. Respir. Med. 2018, 12, 169–175. [Google Scholar] [CrossRef]
- Miyata, K.N.; Nast, C.C.; Dai, T.; Dukkipati, R.; LaPage, J.A.; Troost, J.P.; Schurgers, L.J.; Kretzler, M.; Adler, S.G. Renal matrix Gla protein expression increases progressively with CKD and predicts renal outcome. Exp. Mol. Pathol. 2018, 105, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Holmes, N. CD45: All is not yet crystal clear. Immunology 2006, 117, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.E.; Cowper, S.E.; Bucala, R. The role of circulating fibrocytes in fibrosis. Curr. Rheumatol. Rep. 2006, 8, 145–150. [Google Scholar] [CrossRef]
- Lin, R.J.; Su, Z.Z.; Liang, S.M.; Chen, Y.Y.; Shu, X.R.; Nie, R.Q.; Wang, J.F.; Xie, S.L. Role of circulating fibrocytes in cardiac fibrosis. Chin. Med. J. 2016, 129, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cong, M.; Park, T.J.; Scholten, D.; Brenner, D.A.; Kisseleva, T. Contribution of bone marrow-derived fibrocytes to liver fibrosis. Hepatobiliary Surg. Nutr. 2015, 4, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Rolla, S.; Maglione, A.; De Mercanti, S.F.; Clerico, M. The meaning of immune reconstitution after alemtuzumab therapy in multiple sclerosis. Cells 2020, 9, 1396. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.L.; Noble, P.W.; White, B.; Wise, R.; Liu, M.C.; Bochner, B.S. Extensive surface phenotyping of alveolar macrophages in interstitial lung disease. Clin. Immunol. 2000, 94, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Zeglinski, M.R.; Granville, D.J. Granzymes in cardiovascular injury and disease. Cell. Signal. 2020, 76, 109804. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.; Ding, B.; Zhang, Y.; Xia, Q.; Li, Y.; Zhu, B. Proteomic profiling change during the early development of silicosis disease. J. Thorac. Dis. 2016, 8, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.T.; Hiroyasu, S.; Granville, D.J. Granzyme B as a therapeutic target for wound healing. Expert Opin. Ther. Targets 2019, 23, 745–754. [Google Scholar] [CrossRef]
- Shen, Y.; Cheng, F.; Sharma, M.; Merkulova, Y.; Raithatha, S.A.; Parkinson, L.G.; Zhao, H.; Westendorf, K.; Bohunek, L.; Bozin, T.; et al. Granzyme B deficiency protects against angiotensin II–induced cardiac fibrosis. Am. J. Pathol. 2016, 186, 87–100. [Google Scholar] [CrossRef]
- Okamoto, O.; Fujiwara, S. Dermatopontin, a novel player in the biology of the extracellular matrix. Connect. Tissue Res. 2006, 47, 177–189. [Google Scholar] [CrossRef]
- Zavvos, V.; Buxton, A.T.; Evans, C.; Lambie, M.; Davies, S.J.; Topley, N.; Wilkie, M.; Summers, A.; Brenchley, P.; Goumenos, D.S.; et al. A prospective, proteomics study identified potential biomarkers of encapsulating peritoneal sclerosis in peritoneal effluent. Kidney Int. 2017, 92, 988–1002. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.B.; Russell, J.D.; Trupin, K.M.; Gayden, A.E.; Opalenik, S.R.; Nanney, L.B.; Broquist, A.H.; Raju, L.; Williams, S.M. Epigenetically altered wound healing in keloid fibroblasts. J. Investig. Dermatol. 2010, 130, 2489–2496. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, P.; Lalloyer, F.; Baugé, E.; Pawlak, M.; Gheeraert, C.; Dehondt, H.; Vanhoutte, J.; Woitrain, E.; Hennuyer, N.; Mazuy, C.; et al. Interspecies NASH disease activity whole-genome profiling identifies a fibrogenic role of PPARα-regulated dermatopontin. JCI Insight 2017, 2, e92264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ge, Y.; Cheng, Q.; Zhang, Q.; Fang, L.; Zheng, J. Decorin is a pivotal effector in the extracellular matrix and tumour microenvironment. Oncotarget 2018, 9, 5480–5491. [Google Scholar] [CrossRef] [Green Version]
- Baghy, K.; Iozzo, R.V.; Kovalszky, I. Decorin–TGFβ axis in hepatic fibrosis and cirrhosis. J. Histochem. Cytochem. 2012, 60, 262–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, O.; Fujiwara, S.; Abe, M.; Sato, Y. Dermatopontin interacts with transforming growth factor beta and enhances its biological activity. Biochem. J. 1999, 337, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.N.; Hyde, D.M.; Braun, R.K.; Gaarde, W.; Harper, J.R.; Pierschbacher, M.D. Antifibrotic effect of decorin in a bleomycin hamster model of lung fibrosis. Biochem. Pharmacol. 1997, 54, 1205–1216. [Google Scholar] [CrossRef]
- Border, W.A.; Noble, N.A.; Yamamoto, T.; Harper, J.R.; Yamaguchi, Y.U.; Pierschbacher, M.D.; Ruoslahti, E. Natural inhibitor of transforming growth factor-beta protects against scarring in experimental kidney disease. Nature 1992, 360, 361–364. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kidney | Liver | |||
|---|---|---|---|---|
| Gene | logFC | p-Value | logFC | p-Value |
| PTPRC | 2.96 | 1.30 × 10−5 | 1.28 | 5.78 × 10−8 |
| CXCL9 | 2.54 | 4.06 × 10−4 | 1.95 | 2.75 × 10−8 |
| CD52 | 2.53 | 3.39 × 10−6 | 1.33 | 4.58 × 10−9 |
| GZMA | 1.89 | 2.29 × 10−5 | 1.13 | 2.48 × 10−8 |
| DCN | 1.64 | 7.75 × 10−5 | 1.11 | 2.58 × 10−13 |
| THBS2 | 1.47 | 4.57 × 10−5 | 1.90 | 1.58 × 10−15 |
| DPT | 1.33 | 2.32 × 10−3 | 1.26 | 4.09 × 10−8 |
| MGP | 1.01 | 9.31 × 10−3 | 1.89 | 8.65 × 10−12 |
| Number of Patients | Age in Years (St. Dev) | Gender (M:F) | |
|---|---|---|---|
| Normal liver | 7 | 59.2 (21.0) | 2:1 1 |
| Liver fibrosis | 10 | 62.7 (10.9) | 7:3 |
| Normal kidney | 9 | 34.6 (18.7) | 2:7 |
| Kidney fibrosis | 11 | 65.8 (10.9) | 9:2 |
| Normal bowel | 11 | 78.9 (8.7) | 4:7 |
| Crohn’s disease with fibrosis | 14 | 36.1 (14.4) | 9:5 |
| Crohn’s disease without fibrosis | 14 | 35.8 (15.9) | 9:5 |
| Ulcerative colitis | 10 | 42.2 (20.5) | 4:6 |
| Liver | Kidney | |||
|---|---|---|---|---|
| Gene | ΔΔCq | p-Value | ΔΔCq | p-Value |
| CXCL9 | 0.76 | 0.206 | 3.10 | <0.001 |
| THBS2 | 1.46 | 0.031 | 2.11 | 0.001 |
| MGP | 3.43 | 0.001 | 1.70 | 0.012 |
| PTPRC | 3.06 | 0.002 | 2.60 | 0.002 |
| CD52 | 2.39 | <0.001 | 3.83 | <0.001 |
| GZMA | 0.43 | 0.310 | −0.21 | 0.635 |
| DPT | 4.68 | <0.001 | −1.31 | 0.080 |
| DCN | 0.81 | 0.056 | 1.45 | <0.001 |
| CD with Fibrosis | CD without Fibrosis | UC | ||||
|---|---|---|---|---|---|---|
| Gene | ΔΔCq | p-Value | ΔΔCq | p-Value | ΔΔCq | p-Value |
| CXCL9 | 6.73 | <0.001 | 2.56 | 0.001 | 2.55 | <0.001 |
| THBS2 | 3.08 | <0.001 | 0.25 | 0.574 | 0.66 | 0.051 |
| MGP | 1.66 | <0.001 | −0.47 | 0.252 | −0.66 | 0.134 |
| PTPRC | 3.55 | <0.001 | 1.46 | <0.001 | 0.58 | 0.091 |
| CD52 | 1.90 | <0.001 | −0.28 | 0.432 | 1.69 | <0.001 |
| GZMA | 1.17 | 0.030 | 4.33 | <0.001 | 5.78 | <0.001 |
| DPT | −0.16 | 0.905 | −1.36 | 0.001 | −3.13 | <0.001 |
| DCN | 1.03 | <0.001 | −1.14 | 0.001 | −0.49 | 0.019 |
| UC | CD without Fibrosis | |||
|---|---|---|---|---|
| Gene | ΔΔCq | p-Value | ΔΔCq | p-Value |
| CXCL9 | 4.18 | <0.001 | 4.17 | <0.001 |
| THBS2 | 2.41 | <0.001 | 2.83 | <0.001 |
| MGP | 2.32 | <0.001 | 2.13 | <0.001 |
| PTPRC | 2.97 | <0.001 | 2.09 | <0.001 |
| CD52 | 0.21 | 0.305 | 2.18 | <0.001 |
| GZMA | −4.62 | <0.001 | −3.16 | <0.001 |
| DPT | 2.97 | <0.001 | 1.20 | 0.001 |
| DCN | 1.52 | <0.001 | 2.17 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jerala, M.; Hauptman, N.; Kojc, N.; Zidar, N. Expression of Fibrosis-Related Genes in Liver and Kidney Fibrosis in Comparison to Inflammatory Bowel Diseases. Cells 2022, 11, 314. https://doi.org/10.3390/cells11030314
Jerala M, Hauptman N, Kojc N, Zidar N. Expression of Fibrosis-Related Genes in Liver and Kidney Fibrosis in Comparison to Inflammatory Bowel Diseases. Cells. 2022; 11(3):314. https://doi.org/10.3390/cells11030314
Chicago/Turabian StyleJerala, Miha, Nina Hauptman, Nika Kojc, and Nina Zidar. 2022. "Expression of Fibrosis-Related Genes in Liver and Kidney Fibrosis in Comparison to Inflammatory Bowel Diseases" Cells 11, no. 3: 314. https://doi.org/10.3390/cells11030314