
The Role of the Lysosomal Cl−/H+ Antiporter ClC-7 in Osteopetrosis and Neurodegeneration

Abstract
:1. Introduction
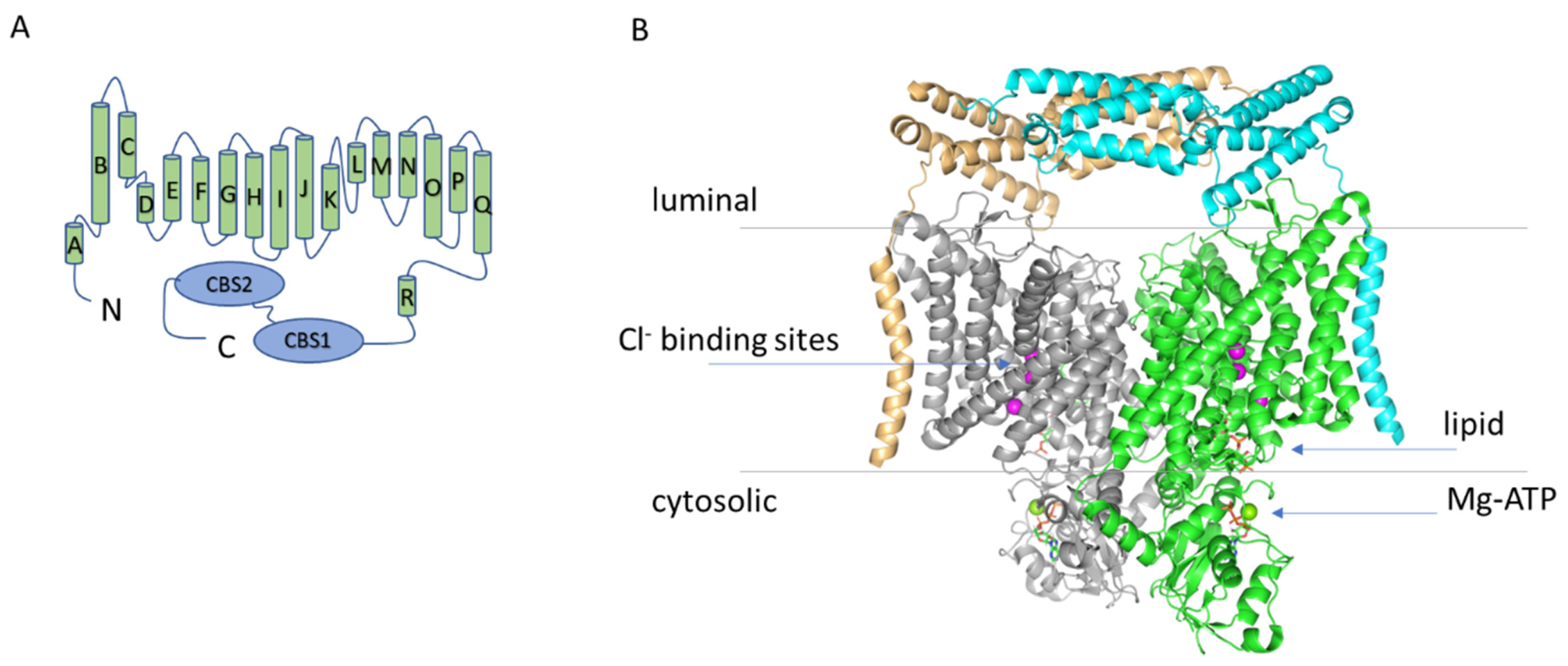
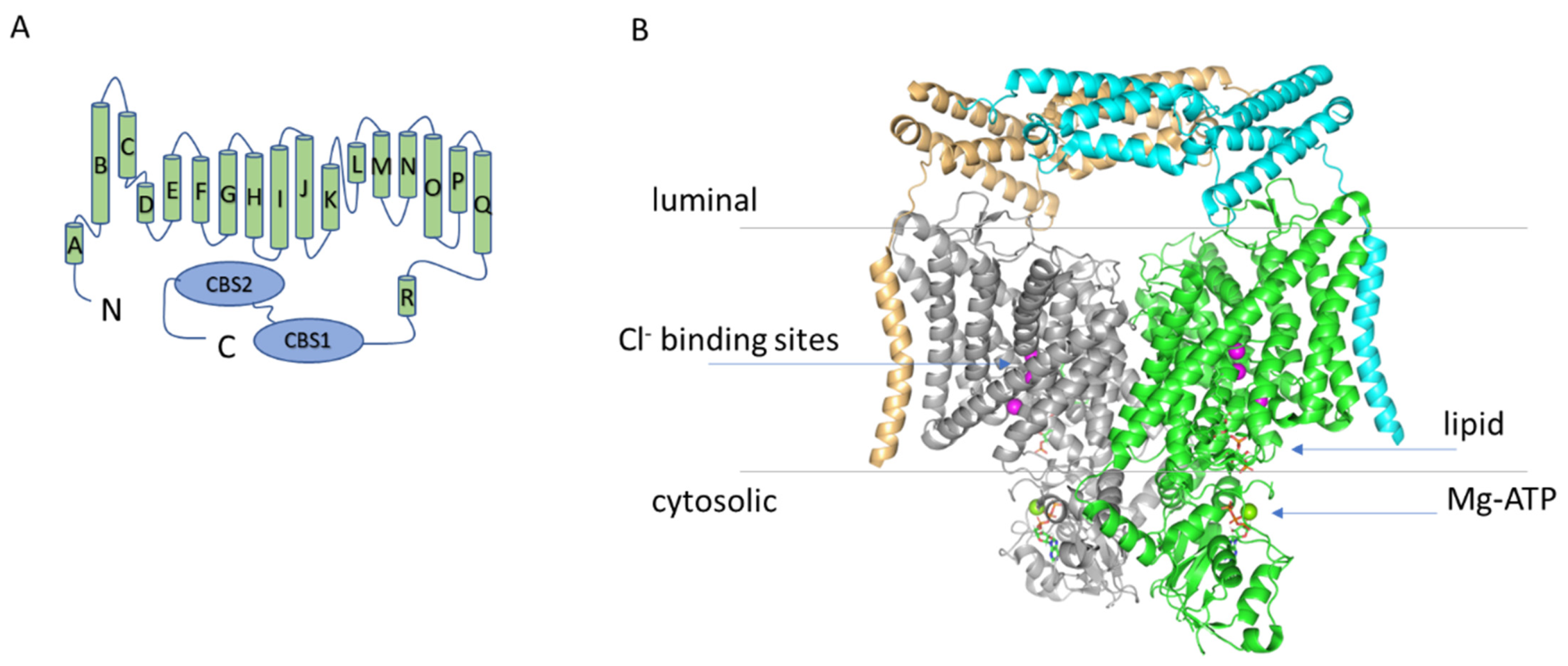
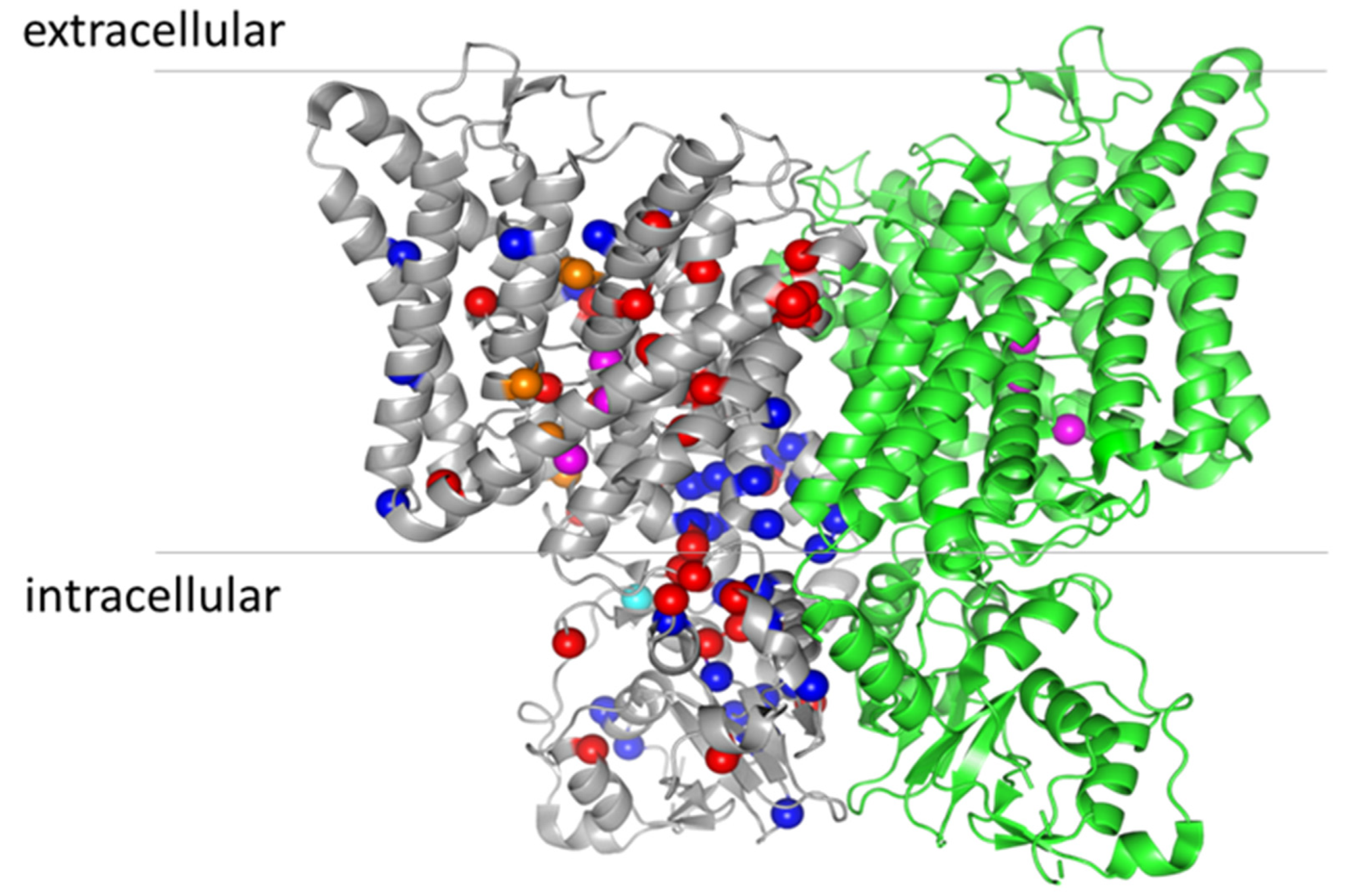
2. The Structure of ClC-7
Phosphatidylinositol Binding Site
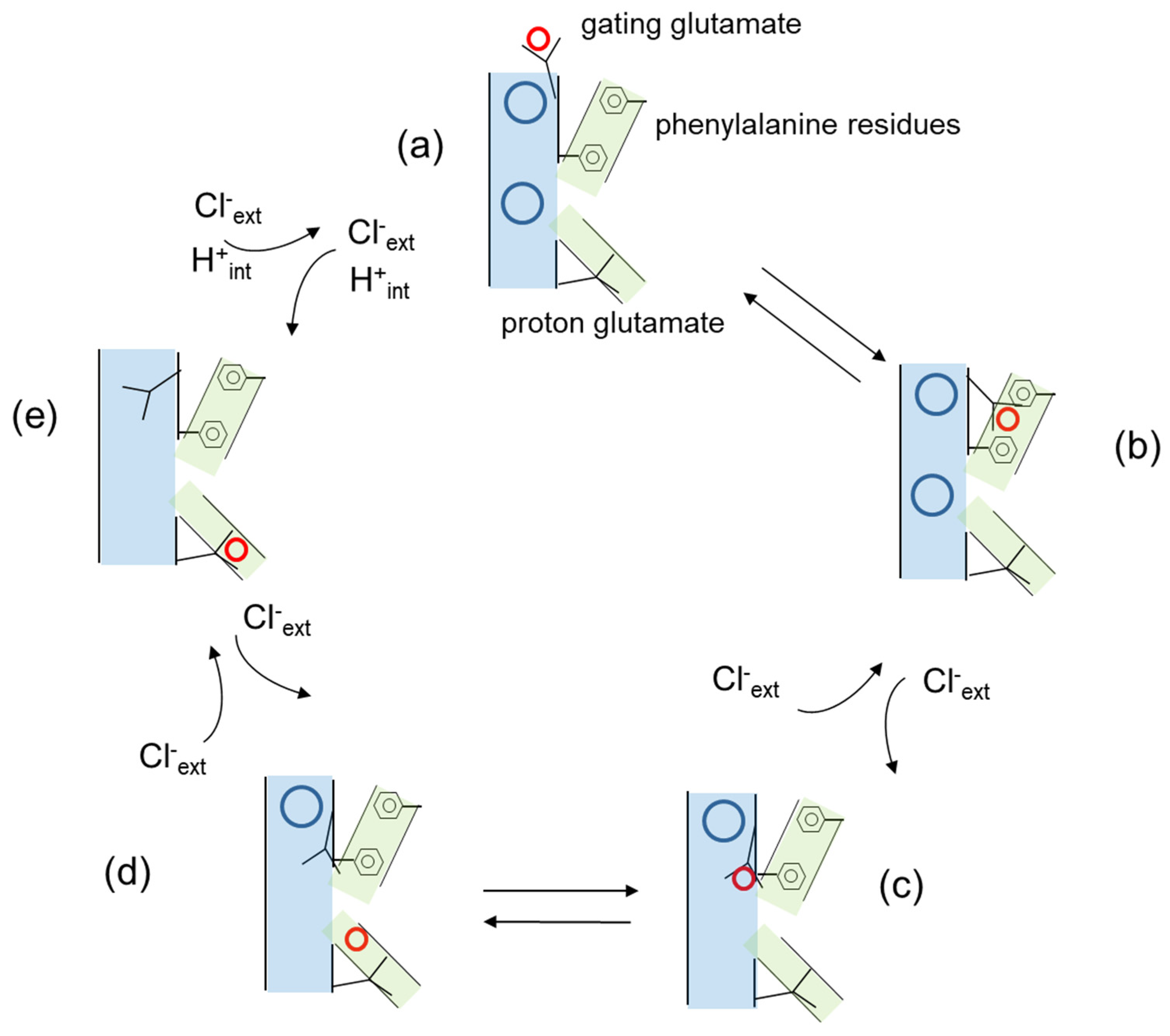
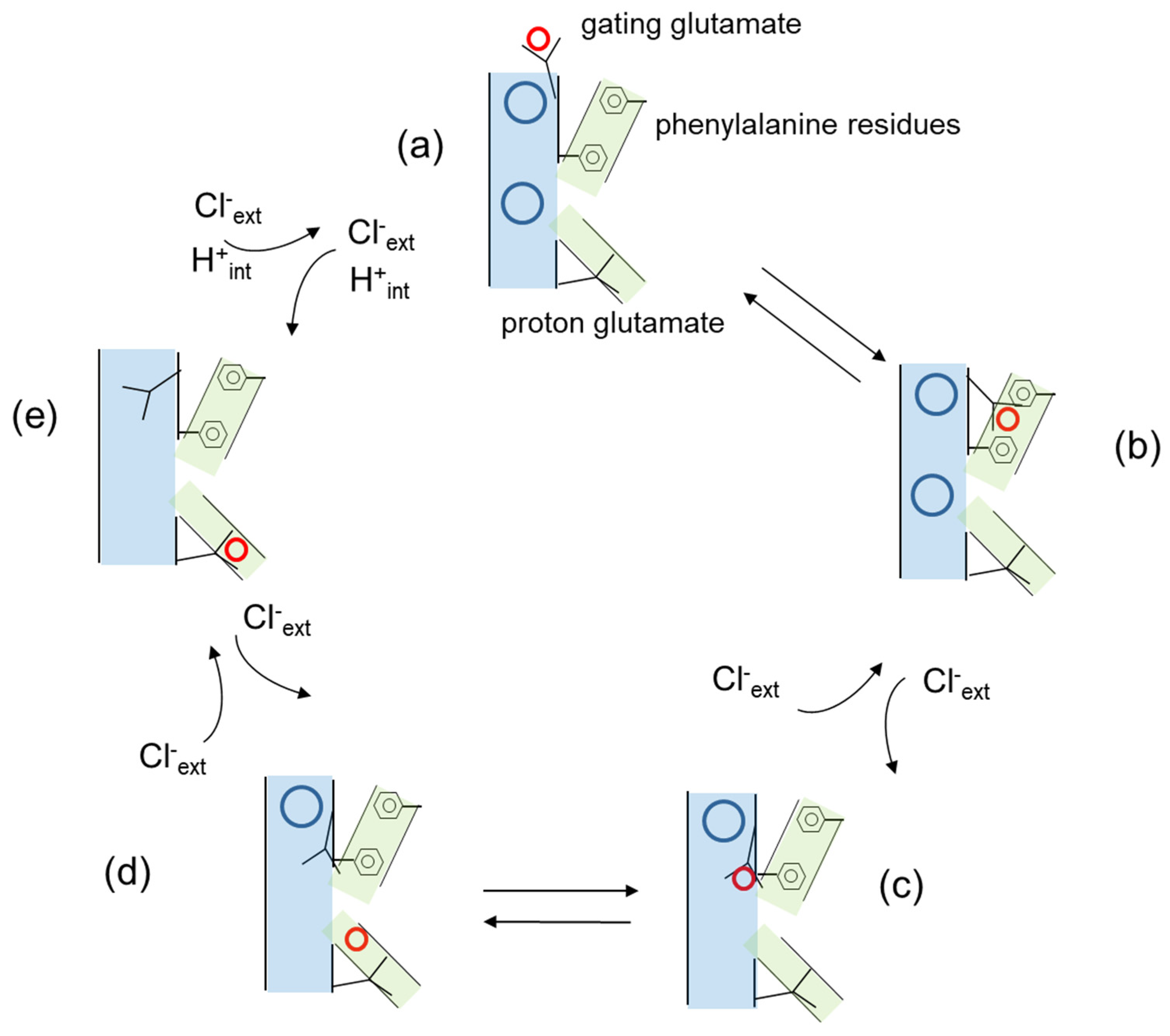
3. Cl−/H+ Exchange and Transport Mechanism in CLC Antiporters
4. Electrophysiological Properties of ClC-7
4.1. The Role of the Proton Glutamate
4.2. Transient Capacitive Currents
5. Osteopetrosis
5.1. Autosomal Dominant Osteopetrosis Type II (ADO II)
5.2. Autosomal Recessive Osteopetrosis (ARO)
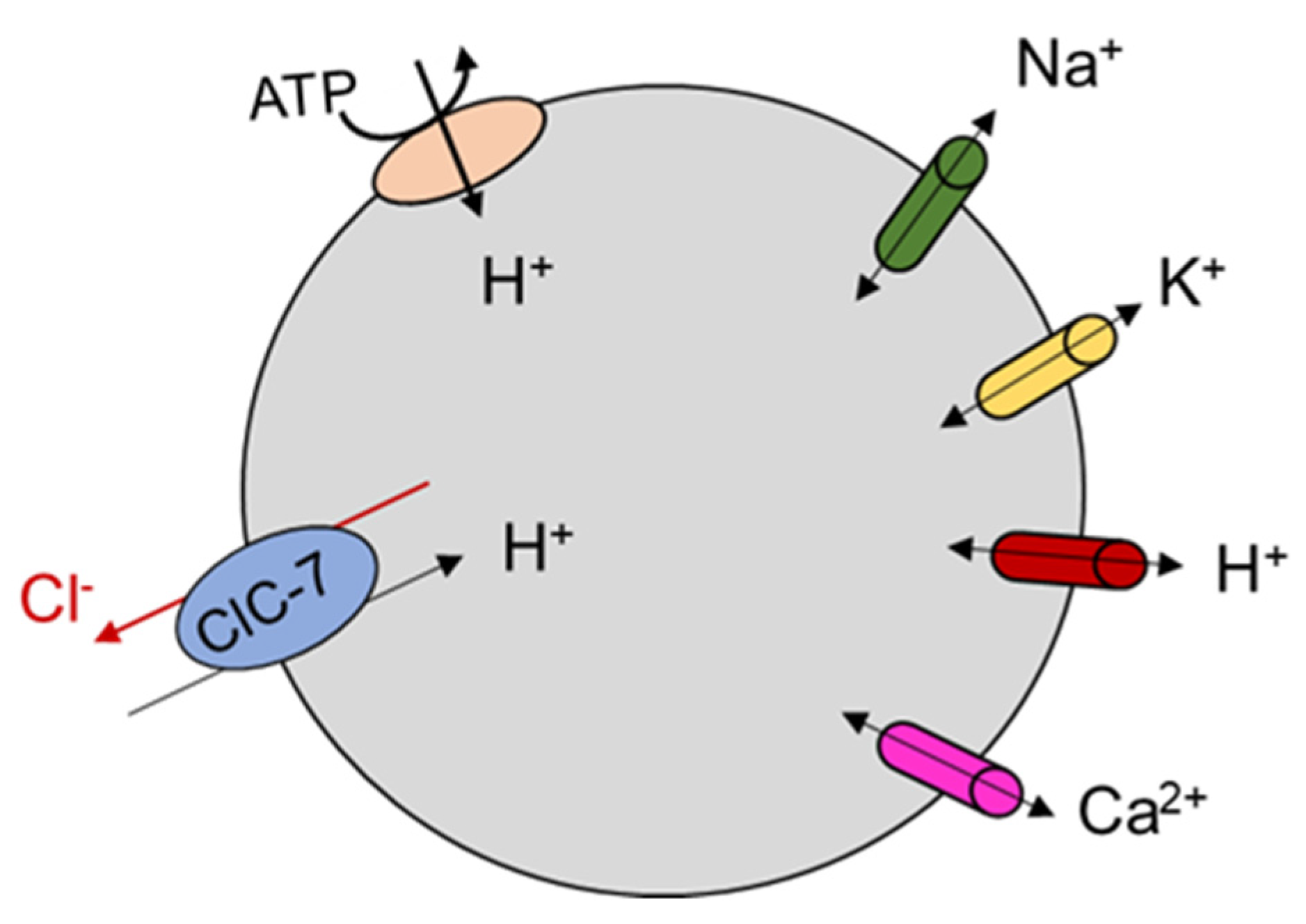
6. The Physiological Role of ClC-7 and Ostm1
ClC-7 Molecular Role from Animal and Cellular Models
7. Lysosomal Storage Disease and Neurodegeneration
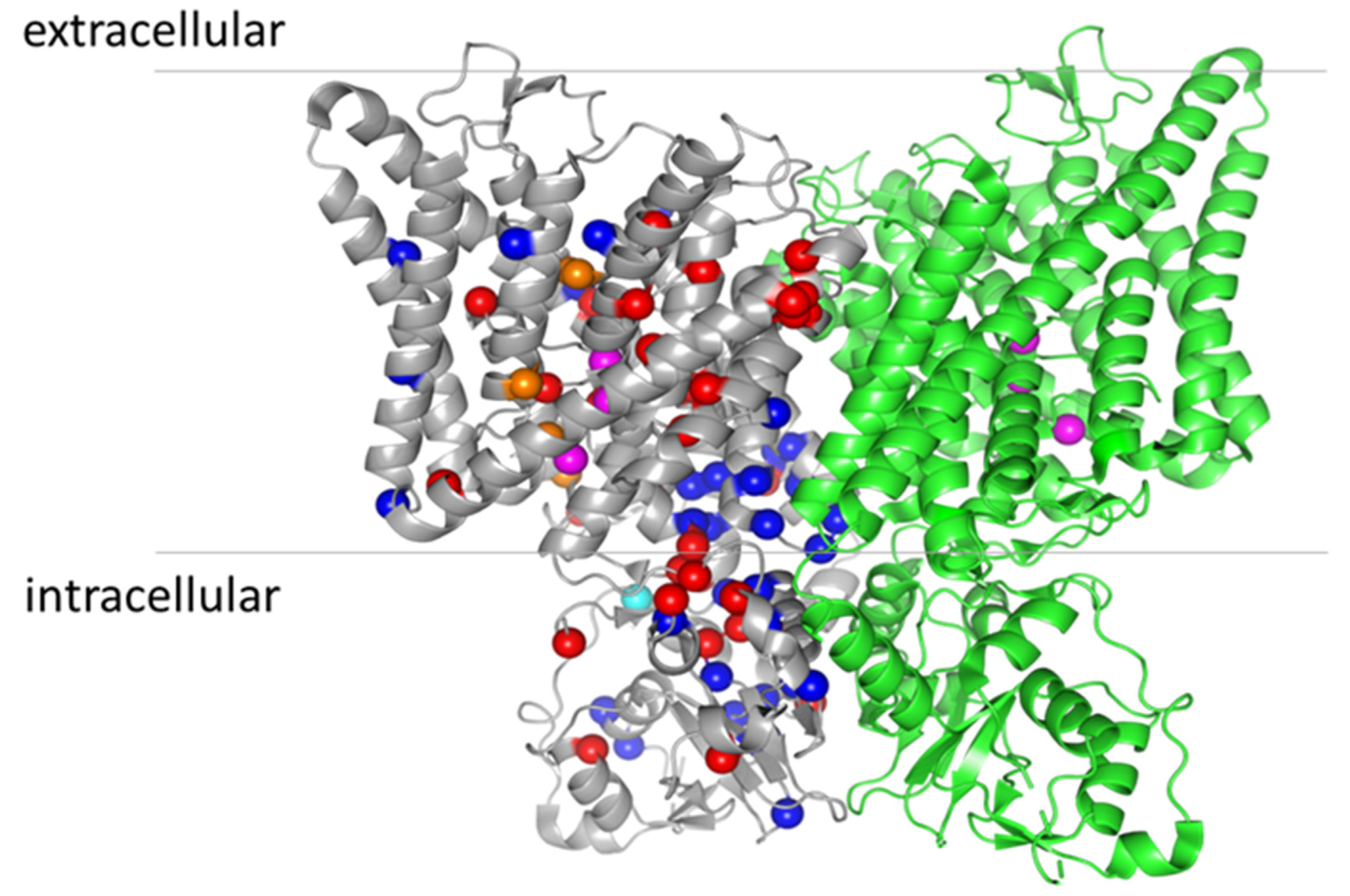
8. Structure–Function Analysis of ClC-7
Funding
Conflicts of Interest
References
- Jentsch, T.J.; Pusch, M. CLC Chloride Channels and Transporters: Structure, Function, Physiology, and Disease. Physiol. Rev. 2018, 98, 1493–1590. [Google Scholar] [CrossRef]
- Zifarelli, G.; Pusch, M. CLC chloride channels and transporters: A biophysical and physiological perspective. Rev. Physiol. Biochem. Pharmacol. 2006, 158, 23–76. [Google Scholar] [CrossRef]
- Brandt, S.; Jentsch, T. ClC-6 and ClC-7 are two novel broadly expressed members of the CLC chloride channel family. FEBS Lett. 1995, 377, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Zifarelli, G.; Pusch, M. CLC transport proteins in plants. FEBS Lett. 2009, 584, 2122–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Liu, Y.; Zhang, B.; Zhou, J.; Li, T.; Liu, Z.; Li, Y.; Yang, M. Molecular insights into the human CLC-7/Ostm1 transporter. Sci. Adv. 2020, 6, eabb4747. [Google Scholar] [CrossRef] [PubMed]
- Schrecker, M.; Korobenko, J.; Hite, R.K. Cryo-EM structure of the lysosomal chloride-proton exchanger CLC-7 in complex with OSTM1. eLife 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Kornak, U.; Kasper, D.; Bösl, M.R.; Kaiser, E.; Schweizer, M.; Schulz, A.; Friedrich, W.; Delling, G.; Jentsch, T.J. Loss of the ClC-7 Chloride Channel Leads to Osteopetrosis in Mice and Man. Cell 2001, 104, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Kasper, D.; Planells-Cases, R.; Fuhrmann, J.; Scheel, O.; Zeitz, O.; Ruether, K.; Schmitt, A.; Poët, M.; Steinfeld, R.; Schweizer, M.; et al. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 2005, 24, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, J.C.; Lange, P.F.; Wartosch, L.; Jentsch, T.J. ClC-7 requires Ostm1 as a beta-subunit to support bone resorption and lysosomal function. GBM Annu. Spring Meet. Mosb. 2007, 2007, 220–223. [Google Scholar] [CrossRef]
- Chalhoub, N.; Benachenhou, N.; Rajapurohitam, V.; Pata, M.; Ferron, M.; Frattini, A.; Villa, A.; Vacher, J. Grey-lethal mutation induces severe malignant autosomal recessive osteopetrosis in mouse and human. Nat. Med. 2003, 9, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Leisle, L.; Ludwig, C.F.; Wagner, F.A.; Jentsch, T.J.; Stauber, T. ClC-7 is a slowly voltage-gated 2Cl(-)/1H(+)-exchanger and requires Ostm1 for transport activity. EMBO J. 2011, 30, 2140–2152. [Google Scholar] [CrossRef] [Green Version]
- Graves, A.R.; Curran, P.K.; Smith, C.L.; Mindell, J.A. The Cl-/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature 2008, 453, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Weinert, S.; Jabs, S.; Supanchart, C.; Schweizer, M.; Gimber, N.; Richter, M.; Rademann, J.; Stauber, T.; Kornak, U.; Jentsch, T.J. Lysosomal Pathology and Osteopetrosis upon Loss of H + -Driven Lysosomal Cl—Accumulation. Science 2010, 328, 1401–1403. [Google Scholar] [CrossRef] [Green Version]
- Zifarelli, G. A tale of two CLCs: Biophysical insights toward understanding ClC-5 and ClC-7 function in endosomes and lysosomes. J. Physiol. 2015, 593, 4139–4150. [Google Scholar] [CrossRef] [Green Version]
- Wartosch, L.; Fuhrmann, J.C.; Schweizer, M.; Stauber, T.; Jentsch, T.J. Lysosomal degradation of endocytosed proteins depends on the chloride transport protein ClC-7. FASEB J. 2009, 23, 4056–4068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, S.; He, H.; Stauber, T. Neurodegeneration Upon Dysfunction of Endosomal/Lysosomal CLC Chloride Transporters. Front. Cell Dev. Biol. 2021, 9, 639231. [Google Scholar] [CrossRef]
- Damkier, H.H.; Christensen, H.L.; Christensen, I.B.; Wu, Q.; Fenton, R.A.; Praetorius, J. The murine choroid plexus epithelium expresses the 2Cl(-)/H(+) exchanger ClC-7 and Na(+)/H(+) exchanger NHE6 in the luminal membrane domain. Am. J. Physiol. Cell Physiol. 2018, 314, C439–C448. [Google Scholar] [CrossRef] [Green Version]
- Piret, S.; Gorvin, C.; Trinh, A.; Taylor, J.; Lise, S.; Taylor, J.C.; Ebeling, P.R.; Thakker, R.V. Autosomal dominant osteopetrosis associated with renal tubular acidosis is due to a CLCN7 mutation. Am. J. Med. Genet. Part A 2016, 170, 2988–2992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumdar, A.; Capetillo-Zarate, E.; Cruz, D.; Gouras, G.K.; Maxfield, F.R. Degradation of Alzheimer’s amyloid fibrils by microglia requires delivery of ClC-7 to lysosomes. Mol. Biol. Cell 2011, 22, 1664–1676. [Google Scholar] [CrossRef]
- Dutzler, R.; Campbell, E.B.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature 2002, 415, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Dutzler, R.; Campbell, E.B.; MacKinnon, R. Gating the Selectivity Filter in ClC Chloride Channels. Science 2003, 300, 108–112. [Google Scholar] [CrossRef]
- Feng, L.; Campbell, E.B.; Hsiung, Y.; MacKinnon, R. Structure of a Eukaryotic CLC Transporter Defines an Intermediate State in the Transport Cycle. Science 2010, 330, 635–641. [Google Scholar] [CrossRef] [Green Version]
- Park, E.; Campbell, E.B.; MacKinnon, E.P.E.B.C.R. Structure of a CLC chloride ion channel by cryo-electron microscopy. Nat. Cell Biol. 2017, 541, 500–505. [Google Scholar] [CrossRef]
- Park, E.; MacKinnon, R. Structure of the CLC-1 chloride channel from Homo sapiens. eLife 2018, 7, e36629. [Google Scholar] [CrossRef]
- Wang, K.; Preisler, S.S.; Zhang, L.; Cui, Y.; Missel, J.W.; Grønberg, C.; Gotfryd, K.; Lindahl, E.; Andersson, M.; Calloe, K.; et al. Structure of the human ClC-1 chloride channel. PLoS Biol. 2019, 17, e3000218. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Cheng, R.C.; Maduke, M.C.; Tajkhorshid, E. Water access points and hydration pathways in CLC H+/Cl- transporters. Proc. Natl. Acad. Sci. USA 2014, 111, 1819–1824. [Google Scholar] [CrossRef] [Green Version]
- Leisle, L.; Xu, Y.; Fortea, E.; Lee, S.; Galpin, J.D.; Vien, M.; Ahern, C.A.; Accardi, A.; Bernèche, S. Divergent Cl- and H+ pathways underlie transport coupling and gating in CLC exchangers and channels. eLife 2020, 9, 9. [Google Scholar] [CrossRef]
- Wang, D.; Voth, G.A. Proton transport pathway in the ClC Cl-/H+ antiporter. Biophys. J. 2009, 97, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.; Savaresi, S.; Forster, I.C.; Dutzler, R. Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nat. Struct. Mol. Biol. 2007, 14, 60–67. [Google Scholar] [CrossRef]
- Accardi, A.; Miller, C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl- channels. Nature 2004, 427, 803–807. [Google Scholar] [CrossRef]
- Zifarelli, G.; Pusch, M. Conversion of the 2 Cl(-)/1 H(+) antiporter ClC-5 in a NO(3)(-)/H(+) antiporter by a single point mutation. Embo. J. 2009, 28, 175–182. [Google Scholar] [CrossRef]
- Miller, C.J. ClC chloride channels viewed through a transporter lens. Nature 2006, 440, 484–489. [Google Scholar] [CrossRef]
- Gadsby, D.C. Ion channels versus ion pumps: The principal difference, in principle. Nat. Rev. Mol. Cell Biol. 2009, 10, 344–352. [Google Scholar] [CrossRef] [Green Version]
- Accardi, A.; Walden, M.; Nguitragool, W.; Jayaram, H.; Williams, C.; Miller, C. Separate ion pathways in a Cl-/H+ exchanger. J. Gen. Physiol. 2005, 126, 563–570. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.H.; Shane, T.; Miller, C. Intracellular proton access in a Cl(-)/H(+) antiporter. PLoS Biol. 2012, 10, e1001441. [Google Scholar] [CrossRef]
- Chavan, T.S.; Cheng, R.C.; Jiang, T.; Mathews, I.I.; Stein, R.A.; Koehl, A.; Mchaourab, H.S.; Tajkhorshid, E.; Maduke, M. A CLC-ec1 mutant reveals global conformational change and suggests a unifying mechanism for the CLC Cl(-)/H(+) transport cycle. eLife 2020, 9, e53479. [Google Scholar] [CrossRef] [Green Version]
- Picollo, A.; Malvezzi, M.; Houtman, J.; Accardi, A. Basis of substrate binding and conservation of selectivity in the CLC family of channels and transporters. Nat. Struct. Mol. Biol. 2009, 16, 1294–1301. [Google Scholar] [CrossRef]
- Bergsdorf, E.-Y.; Zdebik, A.A.; Jentsch, T.J. Residues Important for Nitrate/Proton Coupling in Plant and Mammalian CLC Transporters. J. Biol. Chem. 2009, 284, 11184–11193. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Campbell, E.B.; MacKinnon, R. Molecular mechanism of proton transport in CLC Cl-/H+ exchange transporters. Proc. Natl. Acad. Sci. USA. 2012, 109, 11699–11704. [Google Scholar] [CrossRef] [Green Version]
- Stauber, T.; Jentsch, T.J. Sorting Motifs of the Endosomal/Lysosomal CLC Chloride Transporters. J. Biol. Chem. 2010, 285, 34537–34548. [Google Scholar] [CrossRef] [Green Version]
- Pusch, M.; Zifarelli, G. Large transient capacitive currents in wild-type lysosomal Cl-/H+ antiporter ClC-7 and residual transport activity in the proton glutamate mutant E312A. J. Gen. Physiol. 2021, 153, e202012583. [Google Scholar] [CrossRef]
- Zdebik, A.A.; Zifarelli, G.; Bergsdorf, E.-Y.; Soliani, P.; Scheel, O.; Jentsch, T.; Pusch, M. Determinants of Anion-Proton Coupling in Mammalian Endosomal CLC Proteins. J. Biol. Chem. 2008, 283, 4219–4227. [Google Scholar] [CrossRef] [Green Version]
- Neagoe, I.; Stauber, T.; Fidzinski, P.; Bergsdorf, E.-Y.; Jentsch, T.J. The Late Endosomal ClC-6 Mediates Proton/Chloride Countertransport in Heterologous Plasma Membrane Expression. J. Biol. Chem. 2010, 285, 21689–21697. [Google Scholar] [CrossRef] [Green Version]
- Guzman, R.E.; Grieschat, M.; Fahlke, C.; Alekov, A.K. ClC-3 Is an Intracellular Chloride/Proton Exchanger with Large Voltage-Dependent Nonlinear Capacitance. ACS Chem. Neurosci. 2013, 4, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Weinert, S.; Jabs, S.; Hohensee, S.; Chan, W.L.; Kornak, U.; Jentsch, T.J. Transport activity and presence of ClC-7/Ostm1 complex account for different cellular functions. EMBO Rep. 2014, 15, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.J.; Lippiat, J.D. Voltage-dependent charge movement associated with activation of the CLC-5 2Cl-/1H+ exchanger. Faseb. J. 2010, 24, 3696–3705. [Google Scholar] [CrossRef]
- Alekov, A.; Fahlke, C. Channel-like slippage modes in the human anion/proton exchanger ClC-4. J. Gen. Physiol. 2009, 133, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Zifarelli, G.; De Stefano, S.; Zanardi, I.; Pusch, M. On the Mechanism of Gating Charge Movement of ClC-5, a Human Cl−/H+ Antiporter. Biophys. J. 2012, 102, 2060–2069. [Google Scholar] [CrossRef] [Green Version]
- De Stefano, S.; Pusch, M.; Zifarelli, G. A single point mutation reveals gating of the human ClC-5 Cl-/H+ antiporter. J. Physiol. 2013, 591, 5879–5893. [Google Scholar] [CrossRef] [Green Version]
- Sobacchi, C.; Schulz, A.; Coxon, F.P.; Villa, A.; Helfrich, M.H. Osteopetrosis: Genetics, treatment and new insights into osteoclast function. Nat. Rev. Endocrinol. 2013, 9, 522–536. [Google Scholar] [CrossRef]
- Henriksen, K.; Bollerslev, J.; Everts, V.; Karsdal, M.A. Osteoclast Activity and Subtypes as a Function of Physiology and Pathology—Implications for Future Treatments of Osteoporosis. Endocr. Rev. 2011, 32, 31–63. [Google Scholar] [CrossRef] [Green Version]
- Frattini, A.; Pangrazio, A.; Susani, L.; Sobacchi, C.; Mirolo, M.; Abinun, M.; Andolina, M.; Flanagan, A.; Horwitz, E.M.; Mihci, E.; et al. Chloride Channel ClCN7 Mutations Are Responsible for Severe Recessive, Dominant, and Intermediate Osteopetrosis. J. Bone Miner. Res. 2003, 18, 1740–1747. [Google Scholar] [CrossRef]
- Balemans, W.; Van Wesenbeeck, L.; Van Hul, W. A Clinical and Molecular Overview of the Human Osteopetroses. Calcif. Tissue Int. 2005, 77, 263–274. [Google Scholar] [CrossRef]
- Cleiren, E.; Bénichou, O.; Van Hul, E.; Gram, J.; Bollerslev, J.; Singer, F.R.; Beaverson, K.; Aledo, A.; Whyte, M.P.; Yoneyama, T.; et al. Albers-Schonberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene. Hum. Mol. Genet. 2001, 10, 2861–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benichou, O.D.; Bénichou, B.; Copin, H.; De Vernejoul, M.C.; Van Hul, W. Further Evidence for Genetic Heterogeneity Within Type II Autosomal Dominant Osteopetrosis. J. Bone Miner. Res. 2000, 15, 1900–1904. [Google Scholar] [CrossRef]
- Bollerslev, J.; Andersen, P. Radiological, biochemical and hereditary evidence of two types of autosomal dominant osteopetrosis. Bone 1988, 9, 7–13. [Google Scholar] [CrossRef]
- Maurizi, A.; Capulli, M.; Curle, A.; Patel, R.; Ucci, A.; Côrtes, J.A.; Oxford, H.; Lamandé, S.R.; Bateman, J.F.; Rucci, N.; et al. Extra-skeletal manifestations in mice affected by Clcn7-dependent autosomal dominant osteopetrosis type 2 clinical and therapeutic implications. Bone Res. 2019, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Kida, Y.; Uchida, S.; Miyazaki, H.; Sasaki, S.; Marumo, F. Localization of mouse CLC-6 and CLC-7 mRNA and their functional complementation of yeast CLC gene mutant. Histochem. Cell Biol. 2001, 115, 189–194. [Google Scholar] [CrossRef]
- Hiroyama, Y.; Miike, T.; Sugino, S.; Taku, K. Creatine kinase brain isoenzyme in infantile osteopetrosis. Pediatr. Neurol. 1987, 3, 54–57. [Google Scholar] [CrossRef]
- Rössler, U.; Hennig, A.F.; Stelzer, N.; Bose, S.; Kopp, J.; Søe, K.; Cyganek, L.; Zifarelli, G.; Ali, S.; von der Hagen, M.; et al. Efficient generation of osteoclasts from human induced pluripotent stem cells and functional investigations of lethal CLCN7 -related osteopetrosis. J. Bone Miner. Res. 2021, 36, 1621–1635. [Google Scholar] [CrossRef]
- Teinert, J.; Behne, R.; Wimmer, M.; Ebrahimi-Fakhari, D. Novel insights into the clinical and molecular spectrum of congenital disorders of autophagy. J. Inherit. Metab. Dis. 2019, 43, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Bhatia, D.; Choi, M.E. Autophagy: A Lysosome-Dependent Process with Implications in Cellular Redox Homeostasis and Human Disease. Antioxid. Redox Signal. 2019, 30, 138–159. [Google Scholar] [CrossRef]
- Pata, M.; Vacher, J. Ostm1 Bifunctional Roles in Osteoclast Maturation: Insights From a Mouse Model Mimicking a Human OSTM1 Mutation. J. Bone Miner. Res. 2018, 33, 888–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neutzsky-Wulff, A.; Sims, N.; Supanchart, C.; Kornak, U.; Felsenberg, D.; Poulton, I.; Martin, T.; Karsdal, M.; Henriksen, K. Severe developmental bone phenotype in ClC-7 deficient mice. Dev. Biol. 2010, 344, 1001–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobacchi, C.; Villa, A.; Schulz, A.; Kornak, U. CLCN7-related Osteopetrosis. In GeneReviews(R); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 2022. [Google Scholar]
- Pangrazio, A.; Poliani, P.L.; Megarbane, A.; Lefranc, G.; Lanino, E.; Di Rocco, M.; Rucci, F.; Lucchini, F.; Ravanini, M.; Facchetti, F.; et al. Mutations in OSTM1 (Grey Lethal) Define a Particularly Severe Form of Autosomal Recessive Osteopetrosis With Neural Involvement. J. Bone Miner. Res. 2006, 21, 1098–1105. [Google Scholar] [CrossRef]
- Gerritsen, E.J.; Vossen, J.M.; Van Loo, I.H.; Hermans, J.; Helfrich, M.H.; Griscelli, C.; Fischer, A. Autosomal recessive osteopetrosis: Variability of findings at diagnosis and during the natural course. Pediatrics 1994, 93, 247–253. [Google Scholar] [CrossRef]
- Mazzolari, E.; Forino, C.; Razza, A.; Porta, F.; Villa, A.; Notarangelo, L.D. A single-center experience in 20 patients with infantile malignant osteopetrosis. Am. J. Hematol. 2009, 84, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Teti, A.; Econs, M.J. Osteopetroses, emphasizing potential approaches to treatment. Bone 2017, 102, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Campos-Xavier, A.B.; Casanova, J.-L.; Doumaz, Y.; Feingold, J.; Munnich, A.; Cormier-Daire, V. Intrafamilial phenotypic variability of osteopetrosis due to chloride channel 7 (CLCN7) mutations. Am. J. Med Genet. Part A 2005, 133A, 216–218. [Google Scholar] [CrossRef]
- Pang, Q.; Chi, Y.; Zhao, Z.; Xing, X.; Li, M.; Wang, O.; Jiang, Y.; Liao, R.; Sun, Y.; Dong, J.; et al. Novel mutations of CLCN7 cause autosomal dominant osteopetrosis type II (ADO-II) and intermediate autosomal recessive osteopetrosis (IARO) in Chinese patients. Osteoporos. Int. 2015, 27, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Pangrazio, A.; Pusch, M.; Caldana, E.; Frattini, A.; Lanino, E.; Tamhankar, P.M.; Phadke, S.; Lopez, A.G.M.; Orchard, P.; Mihci, E.; et al. Molecular and clinical heterogeneity in CLCN7-dependent osteopetrosis: Report of 20 novel mutations. Hum. Mutat. 2010, 31, E1071–E1080. [Google Scholar] [CrossRef]
- Letizia, C.; Taranta, A.; Migliaccio, S.; Caliumi, C.; Diacinti, D.; Delfini, E.; D’Erasmo, E.; Iacobini, M.; Roggini, M.; Albagha, O.M.E.; et al. Type II Benign Osteopetrosis (Albers-Schönberg Disease) Caused by a Novel Mutation in CLCN7 Presenting with Unusual Clinical Manifestations. Calcif. Tissue Int. 2003, 74, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Del Fattore, A.; Peruzzi, B.; Rucci, N.; Recchia, I.; Cappariello, A.; Longo, M.; Fortunati, D.; Ballanti, P.; Iacobini, M.; Luciani, M.; et al. Clinical, genetic, and cellular analysis of 49 osteopetrotic patients: Implications for diagnosis and treatment. J. Med Genet. 2005, 43, 315–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waguespack, S.G.; Hui, S.L.; DiMeglio, L.; Econs, M. Autosomal Dominant Osteopetrosis: Clinical Severity and Natural History of 94 Subjects with a Chloride Channel 7 Gene Mutation. J. Clin. Endocrinol. Metab. 2007, 92, 771–778. [Google Scholar] [CrossRef] [Green Version]
- Campos-Xavier, A.; Saraiva, J.M.; Ribeiro, L.M.; Munnich, A.; Cormier-Daire, V. Chloride channel 7 (CLCN7) gene mutations in intermediate autosomal recessive osteopetrosis. Hum. Genet. 2003, 112, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wang, W.; Mao, T.; Duan, X. Report of two Chinese patients suffering from CLCN7-related osteopetrosis and root dysplasia. J. Cranio-Maxillofac. Surg. 2012, 40, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Nicoli, E.-R.; Weston, M.R.; Hackbarth, M.; Becerril, A.; Larson, A.; Zein, W.M.; Baker, P.R.; Burke, J.D.; Dorward, H.; Davids, M.; et al. Lysosomal Storage and Albinism Due to Effects of a De Novo CLCN7 Variant on Lysosomal Acidification. Am. J. Hum. Genet. 2019, 104, 1127–1138. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Ren, D. Lysosomal Physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Gu, M.; Xu, H. Lysosomal Ion Channels as Decoders of Cellular Signals. Trends Biochem. Sci. 2019, 44, 110–124. [Google Scholar] [CrossRef]
- Ishida, Y.; Nayak, S.; Mindell, J.A.; Grabe, M. A model of lysosomal pH regulation. J. Gen. Physiol. 2013, 141, 705–720. [Google Scholar] [CrossRef] [Green Version]
- Astaburuaga, R.; Haro, O.D.Q.; Stauber, T.; Relόgio, A. A Mathematical Model of Lysosomal Ion Homeostasis Points to Differential Effects of Cl− Transport in Ca2+ Dynamics. Cells 2019, 8, 1263. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, B.E.; Huynh, K.K.; Brodovitch, A.; Jabs, S.; Stauber, T.; Jentsch, T.J.; Grinstein, S. A cation counterflux supports lysosomal acidification. J. Cell Biol. 2010, 189, 1171–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dyke, R.W. Acidification of rat liver lysosomes: Quantitation and comparison with endosomes. Am. J. Physiol. Content 1993, 265, 901–917. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, Y. Potassium ion dependent proton efflux and depolarization from spleen lysosomes. Biochem. Biophys. Res. Commun. 1988, 156, 211–216. [Google Scholar] [CrossRef]
- Marcoline, F.; Ishida, Y.; Mindell, J.; Nayak, S.; Grabe, M. A mathematical model of osteoclast acidification during bone resorption. Bone 2016, 93, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Alam, I.; Gray, A.K.; Chu, K.; Ichikawa, S.; Mohammad, K.S.; Capannolo, M.; Capulli, M.; Maurizi, A.; Muraca, M.; Teti, A.; et al. Generation of the first autosomal dominant osteopetrosis type II (ADO2) disease models. Bone 2014, 59, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Caetano-Lopes, J.; Lessard, S.; Hann, S.; Espinoza, K.; Kang, K.; Lim, K.; Horan, D.; Noonan, H.; Hu, D.; Baron, R.; et al. Clcn7F318L/+ as a new mouse model of Albers-Schönberg disease. Bone 2017, 105, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.; Koller, D.L.; Snyder, R.; Fishburn, T.; Lai, D.; Waguespack, S.G.; Foroud, T.; Econs, M.J. Analysis of variation in expression of autosomal dominant osteopetrosis type 2: Searching for modifier genes. Bone 2005, 37, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Steward, C.G. Neurological aspects of osteopetrosis. Neuropathol. Appl. Neurobiol. 2003, 29, 87–97. [Google Scholar] [CrossRef]
- Pressey, S.N.; O’Donnell, K.J.; Stauber, T.; Fuhrmann, J.C.; Tyynelä, J.; Jentsch, T.J.; Cooper, J.D. Distinct neuropathologic phenotypes after disrupting the chloride transport proteins ClC-6 or ClC-7/Ostm1. J. Neuropathol. Exp. Neurol. 2010, 69, 1228–1246. [Google Scholar] [CrossRef] [Green Version]
- Ohmi, K.; Greenberg, D.S.; Rajavel, K.S.; Ryazantsev, S.; Li, H.H.; Neufeld, E.F. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl. Acad. Sci. USA 2003, 100, 1902–1907. [Google Scholar] [CrossRef] [Green Version]
- Wada, R.; Tifft, C.J.; Proia, R.L. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc. Natl. Acad. Sci. USA 2000, 97, 10954–10959. [Google Scholar] [CrossRef] [Green Version]
- Kornak, U.; Schulz, A.; Friedrich, W.; Uhlhaas, S.; Kremens, B.; Voit, T.; Hasan, C.; Bode, U.; Jentsch, T.J.; Kubisch, C. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum. Mol. Genet. 2000, 9, 2059–2063. [Google Scholar] [CrossRef] [Green Version]
- Frattini, A.; Orchard, P.J.; Sobacchi, C.; Giliani, S.; Abinun, M.; Mattsson, J.P.; Keeling, D.J.; Andersson, A.-K.; Wallbrandt, P.; Zecca, L.; et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat. Genet. 2000, 25, 343–346. [Google Scholar] [CrossRef]
- Wartosch, L.; Stauber, T. A role for chloride transport in lysosomal protein degradation. Autophagy 2010, 6, 158–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinetti, A.; Rocchetta, F.; Costantino, E.; Frattini, A.; Caldana, E.; Rucci, F.; Bettiga, A.; Poliani, P.L.; Chigorno, V.; Sonnino, S. Brain lipid composition in grey-lethal mutant mouse characterized by severe malignant osteopetrosis. Glycoconj. J. 2008, 26, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Poet, M.; Kornak, U.; Schweizer, M.; Zdebik, A.A.; Scheel, O.; Hoelter, S.; Wurst, W.; Schmitt, A.; Fuhrmann, J.; Planells-Cases, R.; et al. Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc. Natl. Acad. Sci. USA 2006, 103, 13854–13859. [Google Scholar] [CrossRef] [Green Version]
- Stauber, T.; Weinert, S.; Jentsch, T.J. Cell Biology and Physiology of CLC Chloride Channels and Transporters. Compr. Physiol. 2012, 2, 1701–1744. [Google Scholar] [CrossRef]
- Polovitskaya, M.M.; Barbini, C.; Martinelli, D.; Harms, F.L.; Cole, F.S.; Calligari, P.; Bocchinfuso, G.; Stella, L.; Ciolfi, A.; Niceta, M.; et al. A Recurrent Gain-of-Function Mutation in CLCN6, Encoding the ClC-6 Cl−/H+-Exchanger, Causes Early-Onset Neurodegeneration. Am. J. Hum. Genet. 2020, 107, 1062–1077. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Cao, X.; Yin, F.; Wu, T.; Stauber, T.; Peng, J. West Syndrome Caused By a Chloride/Proton Exchange-Uncoupling CLCN6 Mutation Related to Autophagic-Lysosomal Dysfunction. Mol. Neurobiol. 2021, 58, 2990–2999. [Google Scholar] [CrossRef]
- Di Zanni, E.; Palagano, E.; Lagostena, L.; Strina, D.; Rehman, A.; Abinun, M.; De Somer, L.; Martire, B.; Brown, J.; Kariminejad, A.; et al. Pathobiologic Mechanisms of Neurodegeneration in Osteopetrosis Derived From Structural and Functional Analysis of 14 ClC -7 Mutants. J. Bone Miner. Res. 2021, 36, 531–545. [Google Scholar] [CrossRef]
- Sartelet, A.; Stauber, T.; Coppieters, W.; Ludwig, C.F.; Fasquelle, C.; Druet, T.; Zhang, Z.; Ahariz, N.; Cambisano, N.; Jentsch, T.J.; et al. A missense mutation accelerating the gating of the lysosomal Cl-/H+-exchanger ClC-7/Ostm1 causes osteopetrosis with gingival hamartomas in cattle. Dis. Model Mech. 2014, 7, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, C.F.; Ullrich, F.; Leisle, L.; Stauber, T.; Jentsch, T.J. Common gating of both CLC transporter subunits underlies voltage-dependent activation of the 2Cl-/1H+ exchanger ClC-7/Ostm1. J. Biol. Chem. 2013, 288, 28611–28619. [Google Scholar] [CrossRef] [Green Version]
- Bykova, E.A.; Zhang, X.-D.; Chen, T.-Y.; Zheng, J. Large movement in the C terminus of CLC-0 chloride channel during slow gating. Nat. Struct. Mol. Biol. 2006, 13, 1115–1119. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, H.; He, J.-W.; Gu, J.-M.; Hu, W.-W.; Hu, Y.-Q.; Li, M.; Liu, Y.-J.; Fu, W.-Z.; Yue, H.; et al. The virulence gene and clinical phenotypes of osteopetrosis in the Chinese population: Six novel mutations of the CLCN7 gene in twelve osteopetrosis families. J. Bone Miner. Metab. 2011, 30, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Barvencik, F.; Kurth, I.; Koehne, T.; Stauber, T.; Zustin, J.; Tsiakas, K.; Ludwig, C.F.; Beil, F.T.; Pestka, J.M.; Hahn, M.; et al. CLCN7andTCIRG1Mutations Differentially Affect Bone Matrix Mineralization in Osteopetrotic Individuals. J. Bone Miner. Res. 2013, 29, 982–991. [Google Scholar] [CrossRef]
- Waguespack, S.G.; Koller, D.L.; White, K.E.; Fishburn, T.; Carn, G.; Buckwalter, K.A.; Johnson, M.; Kocisko, M.; Evans, W.E.; Foroud, T.; et al. Chloride Channel 7 (ClCN7) Gene Mutations and Autosomal Dominant Osteopetrosis, Type II. J. Bone Miner. Res. 2003, 18, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, K.; Gram, J.; Schaller, S.; Dahl, B.H.; Dziegiel, M.H.; Bollerslev, J.; Karsdal, M.A. Characterization of Osteoclasts from Patients Harboring a G215R Mutation in ClC-7 Causing Autosomal Dominant Osteopetrosis Type II. Am. J. Pathol. 2004, 164, 1537–1545. [Google Scholar] [CrossRef] [Green Version]
- Schulz, P.; Werner, J.; Stauber, T.; Henriksen, K.; Fendler, K. The G215R mutation in the Cl-/H+-antiporter ClC-7 found in ADO II osteopetrosis does not abolish function but causes a severe trafficking defect. PLoS ONE 2010, 5, e12585. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wei, Z.; He, J.; Wang, C.; Zhang, Z. Novel mutations of CLCN7 cause autosomal dominant osteopetrosis type II (ADOII) and intermediate autosomal recessive osteopetrosis (ARO) in seven Chinese families. Postgrad. Med. 2017, 129, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.-W.; Tong, S.-F.; Wong, K.; Luo, Y.F.; Tang, H.-Y.; Ha, S.-Y.; Chan, M.H.-M. DNA-based diagnosis of malignant osteopetrosis by whole-genome scan using a single-nucleotide polymorphism microarray: Standardization of molecular investigations of genetic diseases due to consanguinity. J. Hum. Genet. 2007, 52, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Bonapace, G.; Moricca, M.T.; Talarico, V.; Graziano, F.; Pensabene, L.; Miniero, R. Identification of two novel mutations on CLCN7 gene in a patient with malignant ostopetrosis. Ital. J. Pediatr. 2014, 40, 90. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Shao, C.; Zheng, Y.; He, J.-W.; Fu, W.-Z.; Wang, C.; Zhang, Z.-L. Two novel mutations of CLCN7 gene in Chinese families with autosomal dominant osteopetrosis (type II). J. Bone Miner. Metab. 2015, 34, 440–446. [Google Scholar] [CrossRef]
- Phadke, S.R.; Fischer, B.; Gupta, N.; Ranganath, P.; Kabra, M.; Kornak, U. Novel mutations in Indian patients with autosomal recessive infantile malignant osteopetrosis. Indian J. Med Res. 2010, 131, 508–514. [Google Scholar]
- Zeng, B.; Li, R.; Hu, Y.; Hu, B.; Zhao, Q.; Liu, H.; Yuan, P.; Wang, Y. A novel mutation and a known mutation in the CLCN7 gene associated with relatively stable infantile malignant osteopetrosis in a Chinese patient. Gene 2016, 576, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Rashid, B.M.; Rashid, N.G.; Schulz, A.; Lahr, G.; Nore, B.F. A novel missense mutation in the CLCN7 gene linked to benign autosomal dominant osteopetrosis: A case series. J. Med Case Rep. 2013, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, N.; Kohmoto, T.; Naruto, T.; Masuda, K.; Komori, T.; Imoto, I. Novel CLCN7 compound heterozygous mutations in intermediate autosomal recessive osteopetrosis. Hum. Genome Var. 2017, 4, 17036. [Google Scholar] [CrossRef] [PubMed]
- Besbas, N.; Draaken, M.; Ludwig, M.; Deren, O.; Orhan, D.; Bilginer, Y.; Ozaltin, F. A novel CLCN7 mutation resulting in a most severe form of autosomal recessive osteopetrosis. Eur. J. Nucl. Med. Mol. Imaging 2009, 168, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Pusch, M. Myotonia caused by mutations in the muscle chloride channel geneCLCN1. Hum. Mutat. 2002, 19, 423–434. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Amino Acid Location | Disease | Current Amplitude | Current Activation | Lysosomal Localization | |
|---|---|---|---|---|---|---|
| L90P (splice variant) | N-terminus | ARO | normal | normal | normal | [102] |
| Y99C | N-terminus | ADO II | [74] | |||
| R126H | Helix B | ARO (neurodegen.) | reduced | accelerated | strongly reduced | [102] |
| W127G | Helix B | ADO II | [106] | |||
| L132P (L227del) | Helix B | ARO | [72] | |||
| D145fs | Helix B | ARO | [52] | |||
| D145G | Helix B | ADO II | accelerated | normal | [107] | |
| W179x | Helix C | ADO II | [52] | |||
| G203D | Loop helix C-D | IARO | [76] | |||
| L213F | Helix D | ADO II | normal | accelerated | [11,108] | |
| N214S (R767P) | Helix D | ARO | [72] | |||
| G215R | Helix D | ADO II and ADO II + renal tubular acidosis | ER retention | [18,52,54,109,110] | ||
| L224R (K691fs) | Helix E | IARO | [71] | |||
| G240E (W127G) | Helix E | ARO | [111] | |||
| G240R (A299E) | Helix E | ARO (neurodegen.) | strongly reduced | reduced | [11,102] | |
| G240R (R526W) | Helix E | ARO | [52] | |||
| G240R (L651P) | Helix E | ARO | [72] | |||
| P249L | Helix F | ADO II | [54,102] | |||
| P249R (S744F) | Helix F | ARO | [52] | |||
| I261F | Helix F | ARO | [112] | |||
| R271x | Loop helix F-G | ARO | [72] | |||
| R280C (splice variant) | Loop helix F-G | ARO | [113] | |||
| R286Q | Helix G | ADO II | normal | accelerated | [11,52] | |
| R286W | Helix G | ADO II | [71,108] | |||
| V289L | Helix G | ADO II | [114] | |||
| S290F | Helix G | ADO II | [106] | |||
| S290Y | Helix G | ADO II | [71] | |||
| G292E (R403Q) | Helix G | ARO (neurodeg.) | [60] | |||
| V297M | Helix G | ARO | strongly reduced | normal (increased overall expression) | [52,115] | |
| A299E | Helix G | ARO (neurodegen.) | strongly reduced | strongly reduced | [102,116] | |
| A299V | Helix G | ADO II/ARO (neurodegen.) | strongly reduced | strongly reduced | [102] | |
| E313K | Helix H | ADO II | [106] | |||
| A316G | Loop helix H-I | ADO II | [106] | |||
| F318L | Loop helix H-I | ADO II | reduced | normal | [52,72] | |
| F318S | Loop helix H-I | ADO II | [111] | |||
| W319R | Loop helix H-I | ADO II | [71] | |||
| L323P | Helix I | ADO II | normal | accelerated | normal | [102] |
| R326G | Helix I | ADO II | [71] | |||
| M332V (R767W) | Helix I | ARO | [52] | |||
| G347R | Helix I | ADO II | [71] | |||
| E374x (in frame insertion G306) | Loop helix I-J | ARO | [52] | |||
| P376L | Helix J | ARO | reduced | accelerated | strongly reduced | [102] |
| R403Q (G512R) | Helix J | ARO | [72] | |||
| R409W | Loop helix J-K | ADO II | [117] | |||
| V418M | Helix K | ADO II | [70] | |||
| V418M (R674Q) | Helix K | IARO | [70] | |||
| V418fs | Helix K | ARO | [72] | |||
| P470L | Loop helix K-L | IARO | [77] | |||
| P470Q | Loop helix K-L | IARO | [76] | |||
| S473N | Helix L | ADO II | [71] | |||
| L490F | Helix M | ADO | reduced | Normal (reduced overall expression) | [11,52] | |
| C502Y (V577M) | Helix M | IARO | [118] | |||
| A511T (G780W) | Loop helix M-N | ARO | [102] | |||
| G521R | Helix N | ARO (neurodegen.) | strongly reduced | reduced | [52] | |
| R526Q | Helix N | ARO | [72] | |||
| R526T | Helix N | ARO | [72] | |||
| R526W | Helix N | ARO | strongly reduced | reduced ER retention | [52] | |
| L549P | Helix O | ARO | [72] | |||
| Q555x (R762Q) | Helix O | ARO | [7] | |||
| R561Q | Loop Helix O-P | ARO | [119] | |||
| L564P | Helix P | ADO II | [71] | |||
| P582H | Helix Q | ARO (neurodegen.) | reduced | [102] | ||
| A590T | Helix Q | ARO | normal | [102] | ||
| L614P (Del exon 17) | Loop helix R–CBS1 | ARO | [52] | |||
| P619L | Loop helix R–CBS1 | ARO | reduced | [52,115] | ||
| P634fs | CBS1 | ARO | [72] | |||
| L651P | CBS1 | ARO | strongly reduced | normal | [52] | |
| R674Q | CBS1 | ADO II | [70] | |||
| G677V | CBS1 | ADO II | [52,54] | |||
| K689E | Loop CBS1-CBS2 | ADO II | [54] | |||
| K691E | Loop CBS1-CBS2 | ADO II | reduced | slower | reduced | [102] |
| R712x (E730x) | Loop CBS1-CBS2 | ARO | [72] | |||
| Y715C | Loop CBS1-CBS2 | Lysosomal storage + albinism | [78] | |||
| G741R | Loop CBS1-CBS2 | ADO II | [106] | |||
| R743W | Loop CBS1-CBS2 | ADO II | [106] | |||
| S744F | Loop CBS1-CBS2 | ARO | normal | [11,52] | ||
| Y746Q | CBS2 | ADO bovine | accelerated | normal | [103] | |
| S753W | CBS2 | ADO II | [111] | |||
| F758L | CBS2 | ADO II | [72] | |||
| R762L | CBS2 | ADO II | accelerated | [11,108] | ||
| R762Q | CBS2 | ARO | accelerated | [7,11] | ||
| R762W (splicing variant) | CBS2 | ADO II | [72] | |||
| L766P | CBS2 | ARO | [54] | |||
| R767P | CBS2 | ARO | strongly reduced | normal (reduced overall expression) | [11] | |
| R767Q | CBS2 | ARO | normal | accelerated | [11,52] | |
| R767W | CBS2 | ADO II | Strongly reduced | Normal (reduced overall expression) | [11,54] | |
| R767W (M332V) | CBS2 | ARO | [52] | |||
| G780R (splice variant) | CBS2 | ARO (neurodegen.) | [102] | |||
| A788D | CBS2 | ADO II | normal | [11,73] | ||
| R791C | CBS2 | ARO | normal | accelerated | strongly reduced | [102] |
| G796fs | CBS2 | ADO II | accelerated | [11,54] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zifarelli, G. The Role of the Lysosomal Cl−/H+ Antiporter ClC-7 in Osteopetrosis and Neurodegeneration. Cells 2022, 11, 366. https://doi.org/10.3390/cells11030366
Zifarelli G. The Role of the Lysosomal Cl−/H+ Antiporter ClC-7 in Osteopetrosis and Neurodegeneration. Cells. 2022; 11(3):366. https://doi.org/10.3390/cells11030366
Chicago/Turabian StyleZifarelli, Giovanni. 2022. "The Role of the Lysosomal Cl−/H+ Antiporter ClC-7 in Osteopetrosis and Neurodegeneration" Cells 11, no. 3: 366. https://doi.org/10.3390/cells11030366
APA StyleZifarelli, G. (2022). The Role of the Lysosomal Cl−/H+ Antiporter ClC-7 in Osteopetrosis and Neurodegeneration. Cells, 11(3), 366. https://doi.org/10.3390/cells11030366