
Discriminating Epithelial to Mesenchymal Transition Phenotypes in Circulating Tumor Cells Isolated from Advanced Gastrointestinal Cancer Patients

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. RUBYchip™ Microfluidic Device Design and Fabrication
2.2. Cell Culture
2.3. Spiking Experiments and Whole Blood Processing
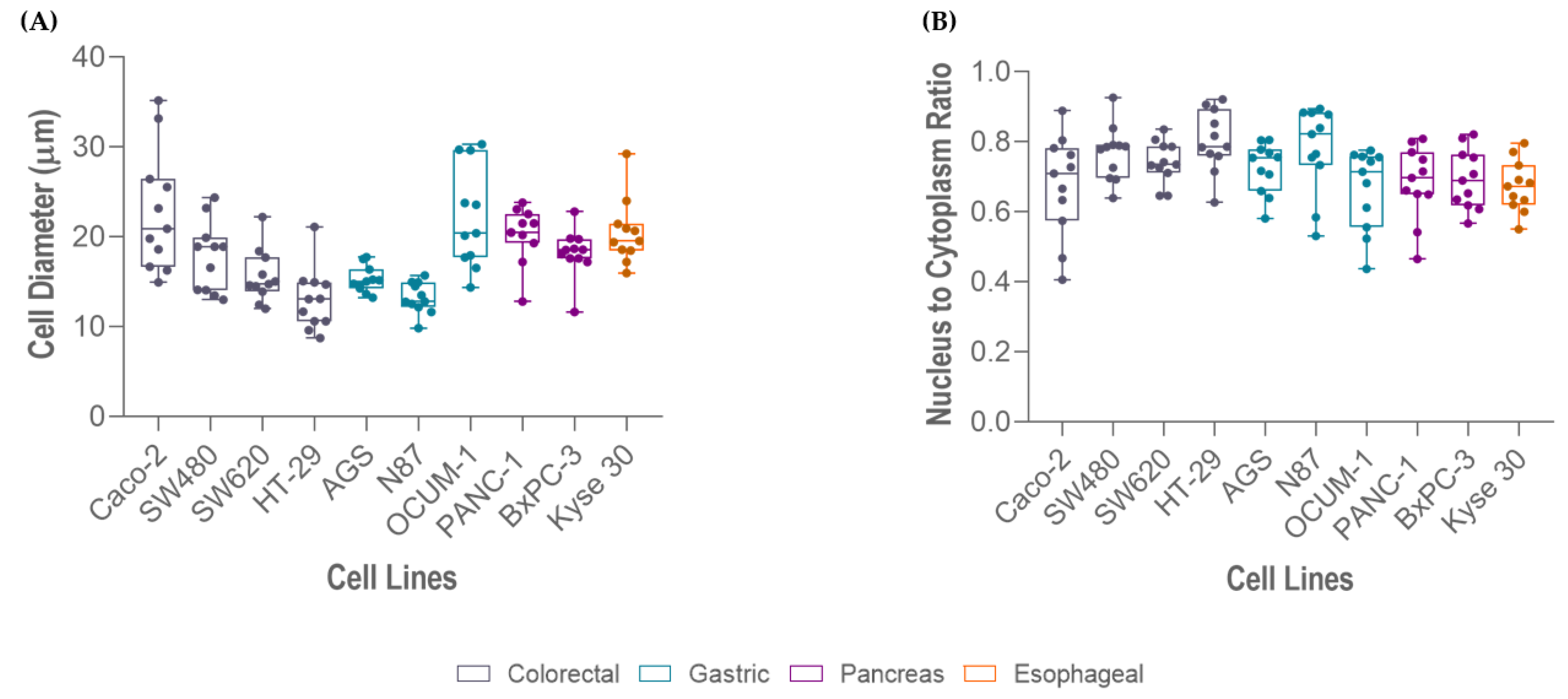
2.4. Cell Size and Nucleus-to-Cytoplasm Ratio
2.5. Immunocytochemistry Studies
2.6. Gastrointestinal Cancer Patient Samples
2.7. CTC Isolation and Characterization in Clinical Samples
3. Results
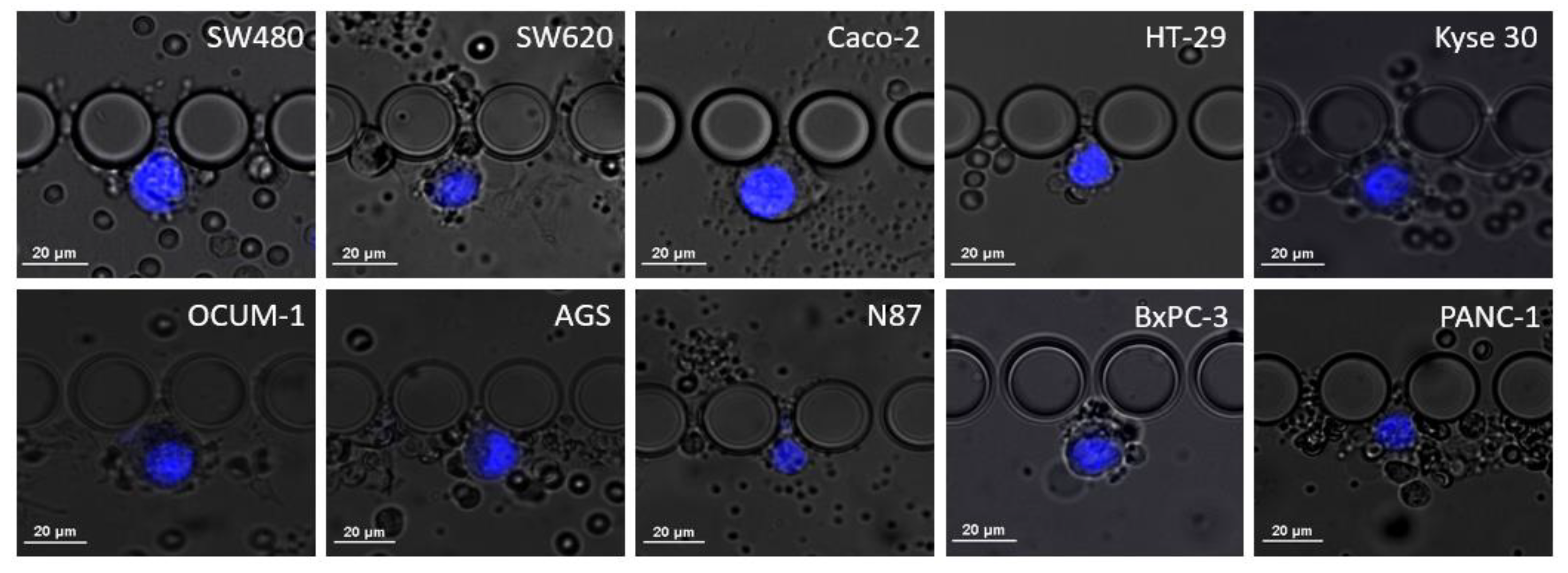
3.1. RUBYchip™ Performance Assessment Using Human Gastrointestinal Cell Lines
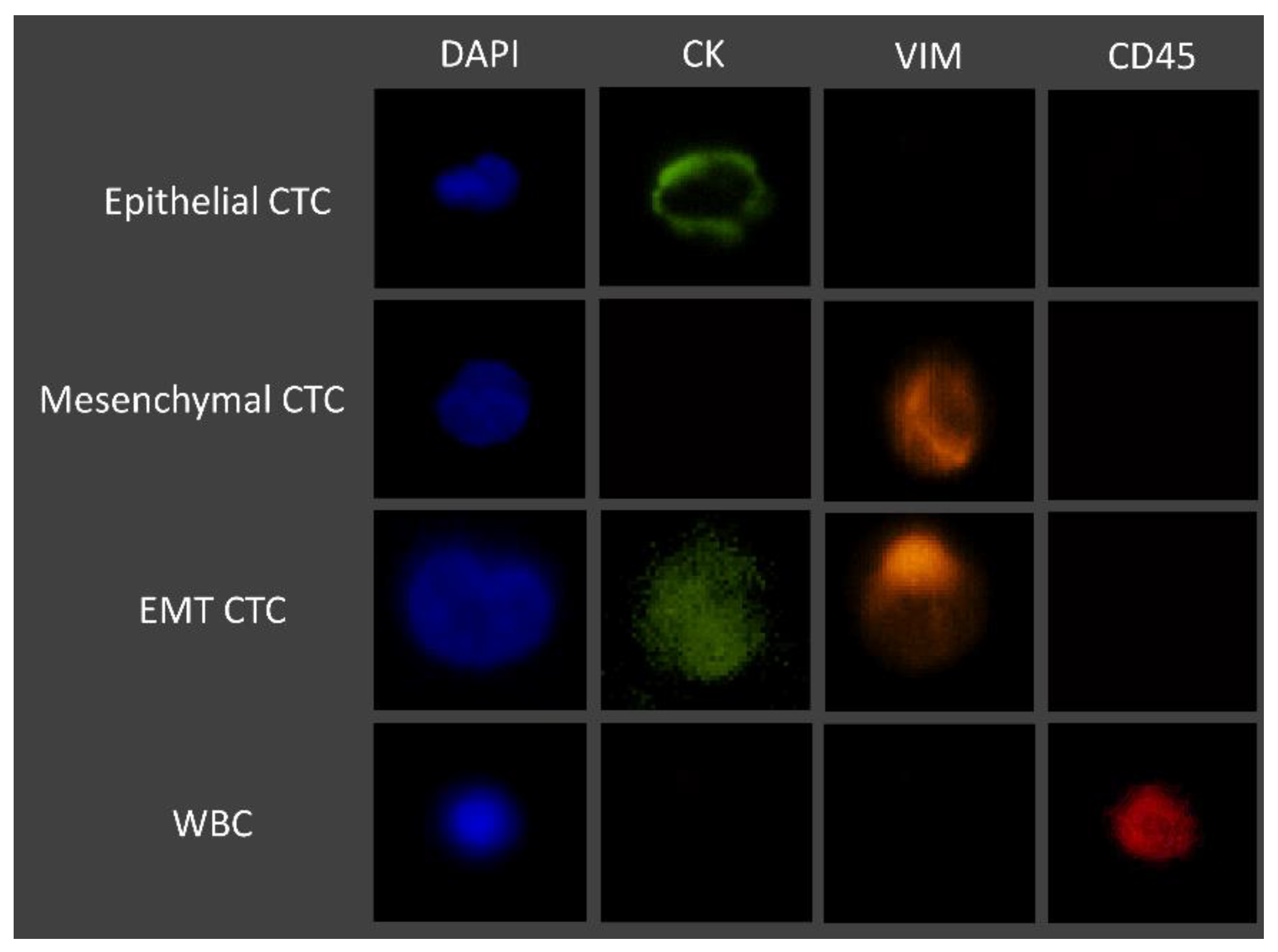
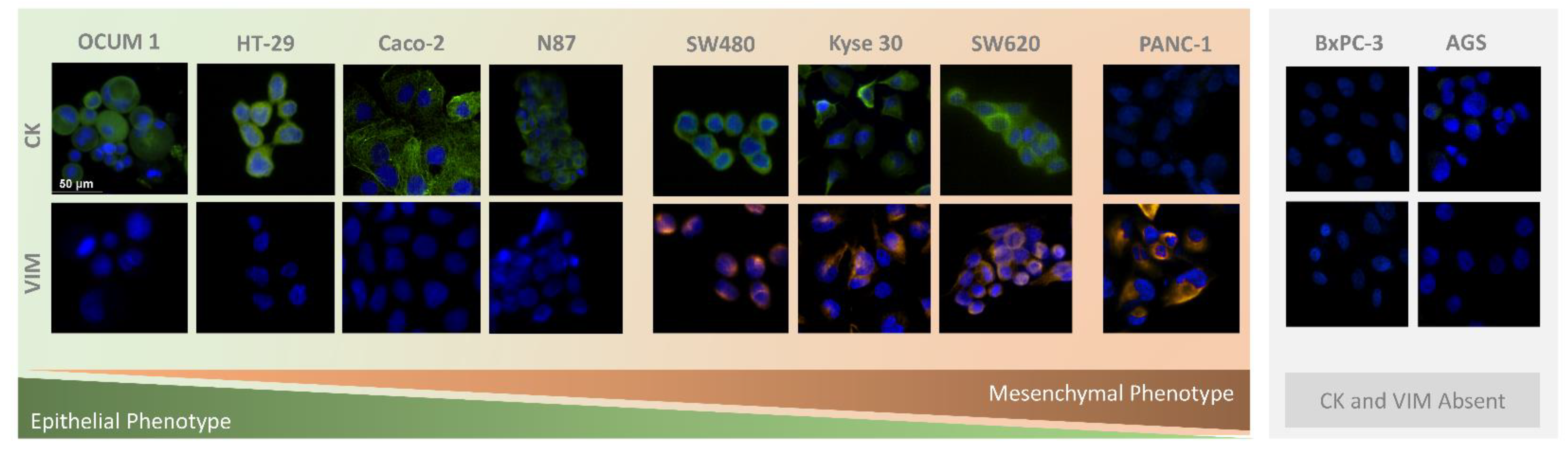
3.2. Immunocytochemistry Studies
3.3. CTC Enumeration in Gastrointestinal Cancer Patients’ Samples Using the RUBYchip™
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iwasaki, J.; Nihira, S.-I. Anti-angiogenic Therapy Against Gastrointestinal Tract Cancers. Jpn. J. Clin. Oncol. 2009, 39, 543–551. [Google Scholar] [CrossRef]
- Cervantes, A.; Roselló, S.; Roda, D.; Rodríguez-Braun, E. The treatment of advanced gastric cancer: Current strategies and future perspectives. Ann. Oncol. 2008, 19, v103–v107. [Google Scholar] [CrossRef]
- Capdevila, J.; Elez, E.; Peralta, S.; Macarulla, T.; Ramos, F.J.; Tabernero, J. Oxaliplatin-based chemotherapy in the management of colorectal cancer. Expert Rev. Anticancer. Ther. 2008, 8, 1223–1236. [Google Scholar] [CrossRef]
- Yang, C.; Chen, F.; Wang, S.; Xiong, B. Circulating Tumor Cells in Gastrointestinal Cancers: Current Status and Future Perspectives. Front. Oncol. 2019, 9, 1427. [Google Scholar] [CrossRef]
- Lee, M.W.; Kim, G.H.; Jeon, H.K.; Park, S.J. Clinical Application of Circulating Tumor Cells in Gastric Cancer. Gut Liver 2019, 13, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Konczalla, L.; Wöstemeier, A.; Kemper, M.; Karstens, K.-F.; Izbicki, J.; Reeh, M. Clinical Significance of Circulating Tumor Cells in Gastrointestinal Carcinomas. Diagnostics 2020, 10, 192. [Google Scholar] [CrossRef]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating tumor cells: Biology and clinical significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef] [PubMed]
- Cayrefourcq, L.; Thomas, F.; Mazard, T.; Assenat, E.; Assou, S.; Alix-Panabières, C. Selective treatment pressure in colon cancer drives the molecular profile of resistant circulating tumor cell clones. Mol. Cancer 2021, 20, 30. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zheng, G.; Cheng, B.; Chen, F.; Wang, Z.; Chen, Y.; Wang, Y.; Xiong, B. Circulating Tumor Cells (CTCs) Detected by RT-PCR and Its Prognostic Role in Gastric Cancer: A Meta-Analysis of Published Literature. PLoS ONE 2014, 9, e99259. [Google Scholar] [CrossRef]
- Sastre, J.; Maestro, M.L.; Puente, J.; Veganzones, S.; Alfonso, R.; Rafael, S.R.; Garcia-Saenz, J.A.; Vidaurreta, M.; Martín, M.; Arroyo, M.; et al. Circulating tumor cells in colorectal cancer: Correlation with clinical and pathological variables. Ann. Oncol. 2008, 19, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Pimienta, M.; Edderkaoui, M.; Wang, R.; Pandol, S.J. The Potential for Circulating Tumor Cells in Pancreatic Cancer Management. Front. Physiol. 2017, 8, 381. [Google Scholar] [CrossRef]
- Schulze, K.; Gasch, C.; Staufer, K.; Nashan, B.; Lohse, A.W.; Pantel, K.; Riethdorf, S.; Wege, H. Presence of EpCAM-positive circulating tumor cells as biomarker for systemic disease strongly correlates to survival in patients with hepatocellular carcinoma. Int. J. Cancer 2013, 133, 2165–2171. [Google Scholar] [CrossRef]
- Genna, A.; Vanwynsberghe, A.M.; Villard, A.V.; Pottier, C.; Ancel, J.; Polette, M.; Gilles, C. EMT-Associated Heterogeneity in Circulating Tumor Cells: Sticky Friends on the Road to Metastasis. Cancers 2020, 12, 1632. [Google Scholar] [CrossRef] [PubMed]
- Neelakantan, D.; Drasin, D.J.; Ford, H.L. Intratumoral heterogeneity: Clonal cooperation in epithelial-to-mesenchymal transition and metastasis. Cell Adhes. Migr. 2014, 9, 265–276. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsuji, T.; Hu, G.F.; Ibaragi, S. EMT and cell cooperativity in metastasis. Cancer Res. 2009, 69, 7135–7139. [Google Scholar] [CrossRef]
- Zhao, R.; Cai, Z.; Li, S.; Cheng, Y.; Gao, H.; Liu, F.; Wu, S.; Liu, S.; Dong, Y.; Zheng, L.; et al. Expression and clinical relevance of epithelial and mesenchymal markers in circulating tumor cells from colorectal cancer. Oncotarget 2016, 8, 9293–9302. [Google Scholar] [CrossRef]
- Han, D.; Chen, K.; Che, J.; Hang, J.; Li, H. Detection of Epithelial-Mesenchymal Transition Status of Circulating Tumor Cells in Patients with Esophageal Squamous Carcinoma. BioMed Res. Int. 2018, 2018, 7610154. [Google Scholar] [CrossRef]
- Zhao, X.-H.; Wang, Z.-R.; Chen, C.-L.; Di, L.; Bi, Z.-F.; Li, Z.-H.; Liu, Y.-M. Molecular detection of epithelial-mesenchymal transition markers in circulating tumor cells from pancreatic cancer patients: Potential role in clinical practice. World J. Gastroenterol. 2019, 25, 138–150. [Google Scholar] [CrossRef]
- Ning, Y.; Zhang, W.; Hanna, D.L.; Yang, D.; Okazaki, S.; Berger, M.D.; Miyamoto, Y.; Suenaga, M.; Schirripa, M.; El-Khoueiry, A.; et al. Clinical relevance of EMT and stem-like gene expression in circulating tumor cells of metastatic colorectal cancer patients. Pharm. J. 2018, 18, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, Q.; Li, H.; Wu, Y.; Feng, J.; Yan, Y. Mesenchymal circulating tumor cells (CTCs) and OCT4 mRNA expression in CTCs for prognosis prediction in patients with non-small-cell lung cancer. Clin. Transl. Oncol. 2017, 19, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112.e14. [Google Scholar] [CrossRef]
- Poulet, G.; Massias, J.; Taly, V. Liquid Biopsy: General Concepts. Acta Cytol. 2019, 63, 449–455. [Google Scholar] [CrossRef]
- Lei, K.F. A Review on Microdevices for Isolating Circulating Tumor Cells. Micromachines 2020, 11, 531. [Google Scholar] [CrossRef] [PubMed]
- Andree, K.C.; Van Dalum, G.; Terstappen, L.W.M.M. Challenges in circulating tumor cell detection by the CellSearch system. Mol. Oncol. 2015, 10, 395–407. [Google Scholar] [CrossRef]
- Wicha, M.S.; Hayes, D.F. Circulating Tumor Cells: Not All Detected Cells Are Bad and Not All Bad Cells Are Detected. J. Clin. Oncol. 2011, 29, 1508–1511. [Google Scholar] [CrossRef]
- Esmaeilsabzali, H.; Beischlag, T.V.; Cox, M.E.; Parameswaran, A.M.; Park, E.J. Detection and isolation of circulating tumor cells: Principles and methods. Biotechnol. Adv. 2013, 31, 1063–1084. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Skelley, A.M.; Merdek, K.D.; Sprott, K.M.; Jiang, C.; Pierceall, W.E.; Lin, J.; Stocum, M.; Carney, W.P.; Smirnov, D.A. Microfluidics and Circulating Tumor Cells. J. Mol. Diagn. 2013, 15, 149–157. [Google Scholar] [CrossRef]
- Myung, J.H.; Hong, S. Microfluidic devices to enrich and isolate circulating tumor cells. Lab. Chip 2015, 15, 4500–4511. [Google Scholar] [CrossRef]
- Ribeiro-Samy, S.; Oliveira, M.; Pereira-Veiga, T.; Muinelo-Romay, L.; Carvalho, S.; Gaspar, J.; Freitas, P.P.; López-López, R.; Costa, C.; Diéguez, L. Fast and efficient microfluidic cell filter for isolation of circulating tumor cells from unprocessed whole blood of colorectal cancer patients. Sci. Rep. 2019, 9, 8032. [Google Scholar] [CrossRef]
- Lima, L.; Neves, M.; Oliveira, M.I.; Dieguez, L.; Freitas, R.; Azevedo, R.; Gaiteiro, C.; Soares, J.; Ferreira, D.; Peixoto, A.; et al. Sialyl-Tn identifies muscle-invasive bladder cancer basal and luminal subtypes facing decreased survival, being expressed by circulating tumor cells and metastases. Urol. Oncol. Semin. Orig. Investig. 2017, 35, 675.e1–675.e8. [Google Scholar] [CrossRef]
- Neves, M.; Azevedo, R.; Lima, L.; Oliveira, M.I.; Peixoto, A.; Ferreira, D.; Soares, J.; Fernandes, E.; Gaiteiro, C.; Palmeira, C.; et al. Exploring sialyl-Tn expression in microfluidic-isolated circulating tumour cells: A novel biomarker and an analytical tool for precision oncology applications. New Biotechnol. 2018, 49, 77–87. [Google Scholar] [CrossRef]
- Lopes, C.; Piairo, P.; Chícharo, A.; Abalde-Cela, S.; Pires, L.R.; Corredeira, P.; Alves, P.; Muinelo-Romay, L.; Costa, L.; Diéguez, L. HER2 Expression in Circulating Tumour Cells Isolated from Metastatic Breast Cancer Patients Using a Size-Based Microfluidic Device. Cancers 2021, 13, 4446. [Google Scholar] [CrossRef] [PubMed]
- McInroy, L.; Määttä, A. Down-regulation of vimentin expression inhibits carcinoma cell migration and adhesion. Biochem. Biophys. Res. Commun. 2007, 360, 109–114. [Google Scholar] [CrossRef]
- Dai, S.; Wei, S.; Zhang, C.; Li, X.; Liu, Y.; Ma, M.; Lv, H.; Zhang, Z.; Zhao, L.; Shan, B. MTA2 promotes the metastasis of esophageal squamous cell carcinoma via EIF4E-Twist feedback loop. Cancer Sci. 2021, 112, 1060–1074. [Google Scholar] [CrossRef] [PubMed]
- Toiyama, Y.; Yasuda, H.; Saigusa, S.; Tanaka, K.; Inoue, Y.; Goel, A.; Kusunoki, M. Increased expression of Slug and Vimentin as novel predictive biomarkers for lymph node metastasis and poor prognosis in colorectal cancer. Carcinogenesis 2013, 34, 2548–2557. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Han, H.; Min, H.; Jin, J.; Yi, E.C.; Kim, Y. Comparative Proteomic Profiling of Pancreatic Ductal Adenocarcinoma Cell Lines. Mol. Cells 2014, 37, 888–898. [Google Scholar] [CrossRef]
- Barriere, G.; Fici, P.; Gallerani, G.; Fabbri, F.; Zoli, W.; Rigaud, M. Circulating tumor cells and epithelial, mesenchymal and stemness markers: Characterization of cell subpopulations. Ann. Transl. Med. 2014, 2, 109. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, A.A.S.; Hou, H.W.; Li, L.D.; Lim, C.T.; Han, J. Pinched flow coupled shear-modulated inertial microfluidics for high-throughput rare blood cell separation. Lab. Chip 2011, 11, 1870–1878. [Google Scholar] [CrossRef]
- Low, W.S.; Abas, W.A.B.W. Benchtop Technologies for Circulating Tumor Cells Separation Based on Biophysical Properties. BioMed Res. Int. 2015, 2015, 239362. [Google Scholar] [CrossRef] [PubMed]
- Matsusaka, S.; Chìn, K.; Ogura, M.; Suenaga, M.; Shinozaki, E.; Mishima, Y.; Terui, Y.; Mizunuma, N.; Hatake, K. Circulating tumor cells as a surrogate marker for determining response to chemotherapy in patients with advanced gastric cancer. Cancer Sci. 2010, 101, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Meropol, N.J.; Punt, C.J.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.A.; Mitchell, E.; et al. Relationship among circulating tumor cells, CEA and overall survival in patients with metastatic colorectal cancer. Ann. Oncol. 2013, 24, 420–428. [Google Scholar] [CrossRef]
- Sastre, J.; Vidaurreta, M.; Gómez, A.; Rivera, F.; Massutí, B.; López, M.R.; Abad, A.; Gallen, M.; Benavides, M.; Aranda, E.; et al. Prognostic Value of the Combination of Circulating Tumor Cells Plus KRAS in Patients With Metastatic Colorectal Cancer Treated With Chemotherapy Plus Bevacizumab. Clin. Color. Cancer 2013, 12, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Bork, U.; Rahbari, N.N.; Schölch, S.; Reissfelder, C.; Kahlert, C.; Büchler, M.W.; Weitz, J.; Koch, M. Circulating tumour cells and outcome in non-metastatic colorectal cancer: A prospective study. Br. J. Cancer 2015, 112, 1306–1313. [Google Scholar] [CrossRef]
- Van Dalum, G.; Stam, G.-J.; Scholten, L.F.; Mastboom, W.J.; Vermes, I.; Tibbe, A.G.; De Groot, M.R.; Terstappen, L.W. Importance of circulating tumor cells in newly diagnosed colorectal cancer. Int. J. Oncol. 2015, 46, 1361–1368. [Google Scholar] [CrossRef]
- Hou, J.; Zou, K.; Yang, C.; Leng, X.; Xu, Y. Clinicopathological and prognostic significance of circulating tumor cells in patients with esophageal cancer: A meta-analysis. OncoTargets Ther. 2018, 11, 8053–8061. [Google Scholar] [CrossRef]
- Han, L.; Chen, W.; Zhao, Q. Prognostic value of circulating tumor cells in patients with pancreatic cancer: A meta-analysis. Tumor Biol. 2013, 35, 2473–2480. [Google Scholar] [CrossRef]
- Khoja, L.; Backen, A.; Sloane, R.S.; Menasce, L.; Ryder, D.; Krebs, M.; Board, R.; Clack, G.; Hughes, A.; Blackhall, F.; et al. A pilot study to explore circulating tumour cells in pancreatic cancer as a novel biomarker. Br. J. Cancer 2011, 106, 508–516. [Google Scholar] [CrossRef]
- Bidard, F.C.; Huguet, F.; Louvet, C.; Mineur, L.; Bouché, O.; Chibaudel, B.; Artru, P.; Desseigne, F.; Bachet, J.-B.; Mathiot, C.; et al. Circulating tumor cells in locally advanced pancreatic adenocarcinoma: The ancillary CirCe 07 study to the LAP 07 trial. Ann. Oncol. 2013, 24, 2057–2061. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Circulating tumour cells and cell-free DNA in gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 73–74. [Google Scholar] [CrossRef]
- Veyrune, L.; Naumann, D.; Christou, N. Circulating Tumour Cells as Prognostic Biomarkers in Colorectal Cancer: A Systematic Review. Int. J. Mol. Sci. 2021, 22, 3437. [Google Scholar] [CrossRef]
- Huang, X.; Gao, P.; Sun, J.; Chen, X.; Song, Y.; Zhao, J.; Xu, H.; Wang, Z. Clinicopathological and prognostic significance of circulating tumor cells in patients with gastric cancer: A meta-analysis. Int. J. Cancer 2014, 136, 21–33. [Google Scholar] [CrossRef]
- Zhang, Q.; Shan, F.; Li, Z.; Gao, J.; Li, Y.; Shen, L.; Ji, J.; Lu, M. A prospective study on the changes and clinical significance of pre-operative and post-operative circulating tumor cells in resectable gastric cancer. J. Transl. Med. 2018, 16, 171. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gong, J.; Zhang, Q.; Lu, Z.; Gao, J.; Li, Y.; Cao, Y.; Shen, L. Dynamic monitoring of circulating tumour cells to evaluate therapeutic efficacy in advanced gastric cancer. Br. J. Cancer 2016, 114, 138–145. [Google Scholar] [CrossRef]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Nel, I.; Baba, H.A.; Ertle, J.; Weber, F.; Sitek, B.; Eisenacher, M.; Meyer, H.E.; Schlaak, J.F.; Hoffmann, A.-C. Individual Profiling of Circulating Tumor Cell Composition and Therapeutic Outcome in Patients with Hepatocellular Carcinoma. Transl. Oncol. 2013, 6, 420–428. [Google Scholar] [CrossRef]
- Satelli, A.; Mitra, A.; Brownlee, Z.; Xia, X.; Bellister, S.; Overman, M.J.; Kopetz, S.; Ellis, L.M.; Meng, Q.H.; Li, S. Epithelial–Mesenchymal Transitioned Circulating Tumor Cells Capture for Detecting Tumor Progression. Clin. Cancer Res. 2015, 21, 899–906. [Google Scholar] [CrossRef]
- Li, T.-T.; Liu, H.; Li, F.-P.; Hu, Y.-F.; Mou, T.-Y.; Lin, T.; Yu, J.; Zheng, L.; Li, G.-X. Evaluation of epithelial-mesenchymal transitioned circulating tumor cells in patients with resectable gastric cancer: Relevance to therapy response. World J. Gastroenterol. 2015, 21, 13259–13267. [Google Scholar] [CrossRef] [PubMed]
- Jie, X.-X.; Zhang, X.-Y.; Xu, C.-J. Epithelial-to-mesenchymal transition, circulating tumor cells and cancer metastasis: Mechanisms and clinical applications. Oncotarget 2017, 8, 81558–81571. [Google Scholar] [CrossRef]
- Li, Y.-M.; Xu, S.-C.; Li, J.; Han, K.-Q.; Pi, H.-F.; Zheng, L.; Zuo, G.-H.; Huang, X.-B.; Li, H.-Y.; Zhao, H.-Z.; et al. Epithelial–mesenchymal transition markers expressed in circulating tumor cells in hepatocellular carcinoma patients with different stages of disease. Cell Death Dis. 2013, 4, e831. [Google Scholar] [CrossRef] [PubMed]
- Kulemann, B.; Liss, A.S.; Warshaw, A.L.; Seifert, S.; Bronsert, P.; Glatz, T.; Pitman, M.B.; Hoeppner, J. KRAS mutations in pancreatic circulating tumor cells: A pilot study. Tumor Biol. 2015, 37, 7547–7554. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
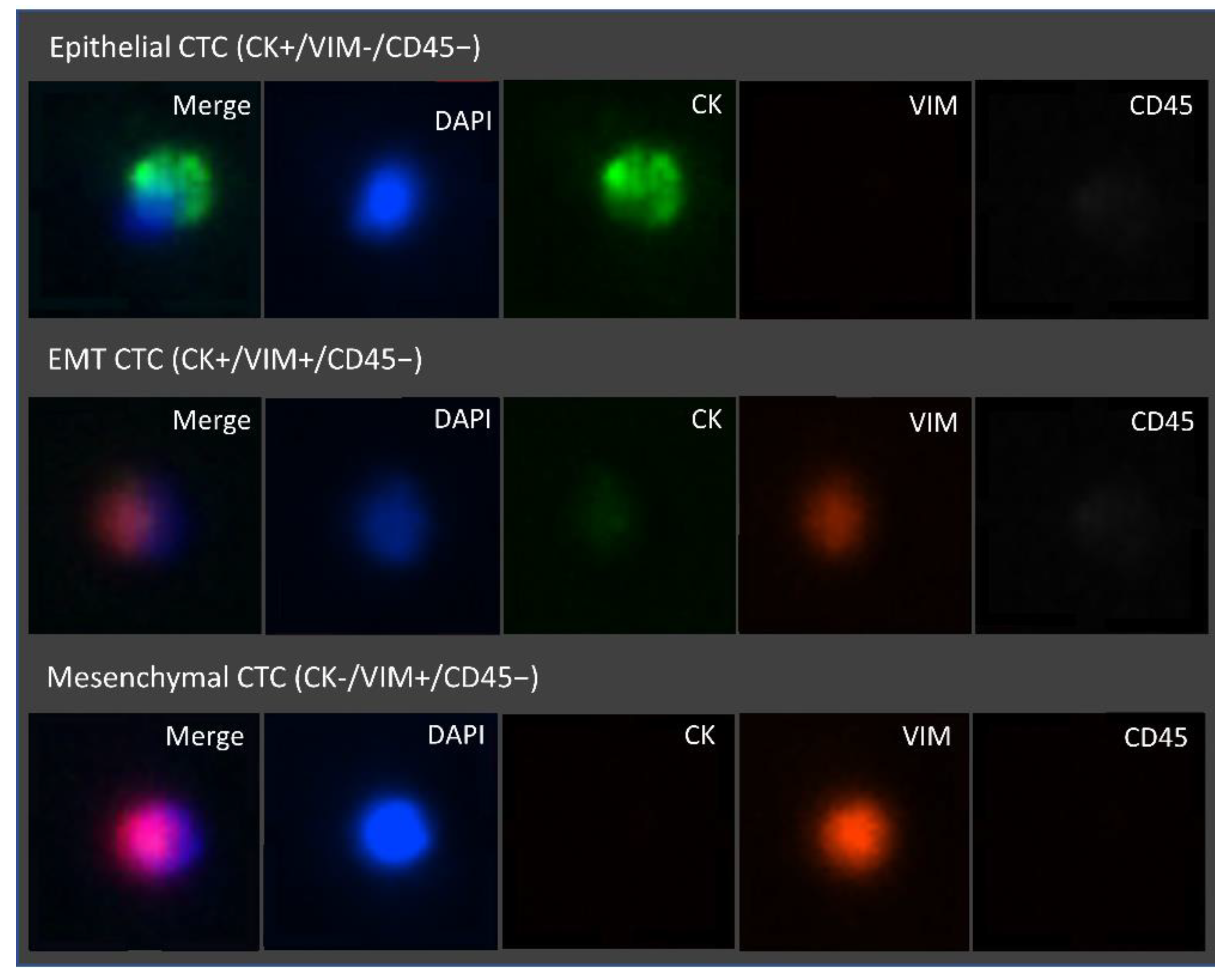{kind=link}
| Cell Line | Cell Size Range (µm) | Average Cell Size (µm) | Average NC Ratio |
|---|---|---|---|
| Caco 2 | 14.94–35.17 | 22.80 | 0.68 |
| OCUM 1 | 14.38–30.31 | 22.20 | 0.67 |
| Kyse 30 | 15.99–20.96 | 20.62 | 0.67 |
| PANC-1 | 12.82–23.82 | 19.91 | 0.68 |
| BxPC-3 | 11.65–25.73 | 19.17 | 0.69 |
| SW480 | 13.01–24.39 | 18.01 | 0.77 |
| SW620 | 12.00–22.23 | 15.59 | 0.74 |
| AGS | 13.25–17.74 | 15.25 | 0.72 |
| N87 | 9.84–19.30 | 13.23 | 0.78 |
| HT-29 | 8.75–21.10 | 13.03 | 0.80 |
| Patient Clinical Data | N |
|---|---|
| Cohort | |
| Number of patients | 11 |
| Tumor type | |
| Colorectal Cancer | 3 |
| Gastric Cancer | 3 |
| Esophageal Cancer | 3 |
| Pancreatic Cancer | 2 |
| Sex | |
| Female | 2 |
| Male | 9 |
| Average age at sample collection | 58 |
| Disease Stage | |
| Tumor (T) | |
| T1 | 1 |
| T2 | 0 |
| T3 | 4 |
| T4 | 4 |
| Tx | 2 |
| Lymph node metastasis | |
| N0 | 0 |
| N1 | 2 |
| N+ | 8 |
| Nx | 1 |
| Distant metastasis (M) | |
| M0 | 5 |
| M1 | 4 |
| Mx | 2 |
| CTC positive patients | 11 |
| Patients | Tumor Type | Disease Stage | CTC Enumeration (Baseline) | ||||
|---|---|---|---|---|---|---|---|
| Epithelial | EMT | Mesenchymal | Total | ||||
| P1 | CRC | TxNxM1 | 2 | 0 | 0 | 2 |  |
| P2 | CRC | T4N1M1 | 1 | 0 | 4 | 5 |  |
| P3 | CRC | T3a/bN+Mx | 2 | 0 | 1 | 3 |  |
| P4 | GC | T4aN+M1 | 2 | 9 | 33 | 44 |  |
| P5 | GC | T3/4aN+M0 | 1 | 0 | 5 | 6 |  |
| P6 | GC | T3/4N+M0 | 0 | 0 | 5 | 5 |  |
| P7 | EC | T1N1M0 | 6 | 0 | 3 | 9 |  |
| P8 | EC | T3N+Mx | 0 | 0 | 1 | 1 |  |
| P9 | EC | T4bN+M0 | 0 | 0 | 2 | 2 |  |
| P10 | PC | TxN+M0 | 8 | 0 | 7 | 15 |  |
| P11 | PC | T4N+M1 | 5 | 1 | 6 | 12 |  |
 Epithelial CTC;
Epithelial CTC;  EMT CTC;
EMT CTC;  Mesechymal CTC.
Mesechymal CTC.Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carneiro, A.; Piairo, P.; Teixeira, A.; Ferreira, D.; Cotton, S.; Rodrigues, C.; Chícharo, A.; Abalde-Cela, S.; Santos, L.L.; Lima, L.; et al. Discriminating Epithelial to Mesenchymal Transition Phenotypes in Circulating Tumor Cells Isolated from Advanced Gastrointestinal Cancer Patients. Cells 2022, 11, 376. https://doi.org/10.3390/cells11030376
Carneiro A, Piairo P, Teixeira A, Ferreira D, Cotton S, Rodrigues C, Chícharo A, Abalde-Cela S, Santos LL, Lima L, et al. Discriminating Epithelial to Mesenchymal Transition Phenotypes in Circulating Tumor Cells Isolated from Advanced Gastrointestinal Cancer Patients. Cells. 2022; 11(3):376. https://doi.org/10.3390/cells11030376
Chicago/Turabian StyleCarneiro, Adriana, Paulina Piairo, Alexandra Teixeira, Dylan Ferreira, Sofia Cotton, Carolina Rodrigues, Alexandre Chícharo, Sara Abalde-Cela, Lúcio Lara Santos, Luís Lima, and et al. 2022. "Discriminating Epithelial to Mesenchymal Transition Phenotypes in Circulating Tumor Cells Isolated from Advanced Gastrointestinal Cancer Patients" Cells 11, no. 3: 376. https://doi.org/10.3390/cells11030376
APA StyleCarneiro, A., Piairo, P., Teixeira, A., Ferreira, D., Cotton, S., Rodrigues, C., Chícharo, A., Abalde-Cela, S., Santos, L. L., Lima, L., & Diéguez, L. (2022). Discriminating Epithelial to Mesenchymal Transition Phenotypes in Circulating Tumor Cells Isolated from Advanced Gastrointestinal Cancer Patients. Cells, 11(3), 376. https://doi.org/10.3390/cells11030376