
Effect of FKBP12-Derived Intracellular Peptides on Rapamycin-Induced FKBP–FRB Interaction and Autophagy

 , , ,
, , ,  , , and
, , and
Abstract
:
1. Introduction
2. Experimental Procedures
2.1. Cell Culture
2.2. Protein Complementation Assay to Investigate the Effect of Peptides on PPI
2.3. Peptide Synthesis
2.4. Intrinsic Fluorescence Emission to Evaluate Rapamycin-VFD7-cpp Complex Formation
2.5. Structural Models for the VFD7-cpp and Derivatives
2.6. MD Simulations
2.7. InPeps Extraction and Quantification
2.8. Measurement of Cellular Viability after Hypoxia-Reoxygenation in Cell Culture
2.9. Statistical Analysis
3. Results
3.1. Intrinsic Fluorescence Emission Analyses of the Direct Interaction of VFD7-cpp and Rapamycin
3.2. Effect of VFD7-cpp and Derivatives on Rapamycin-Induced FKBP–FRB Interaction
3.3. Structural Models for the VFD7-cpp and Derivatives
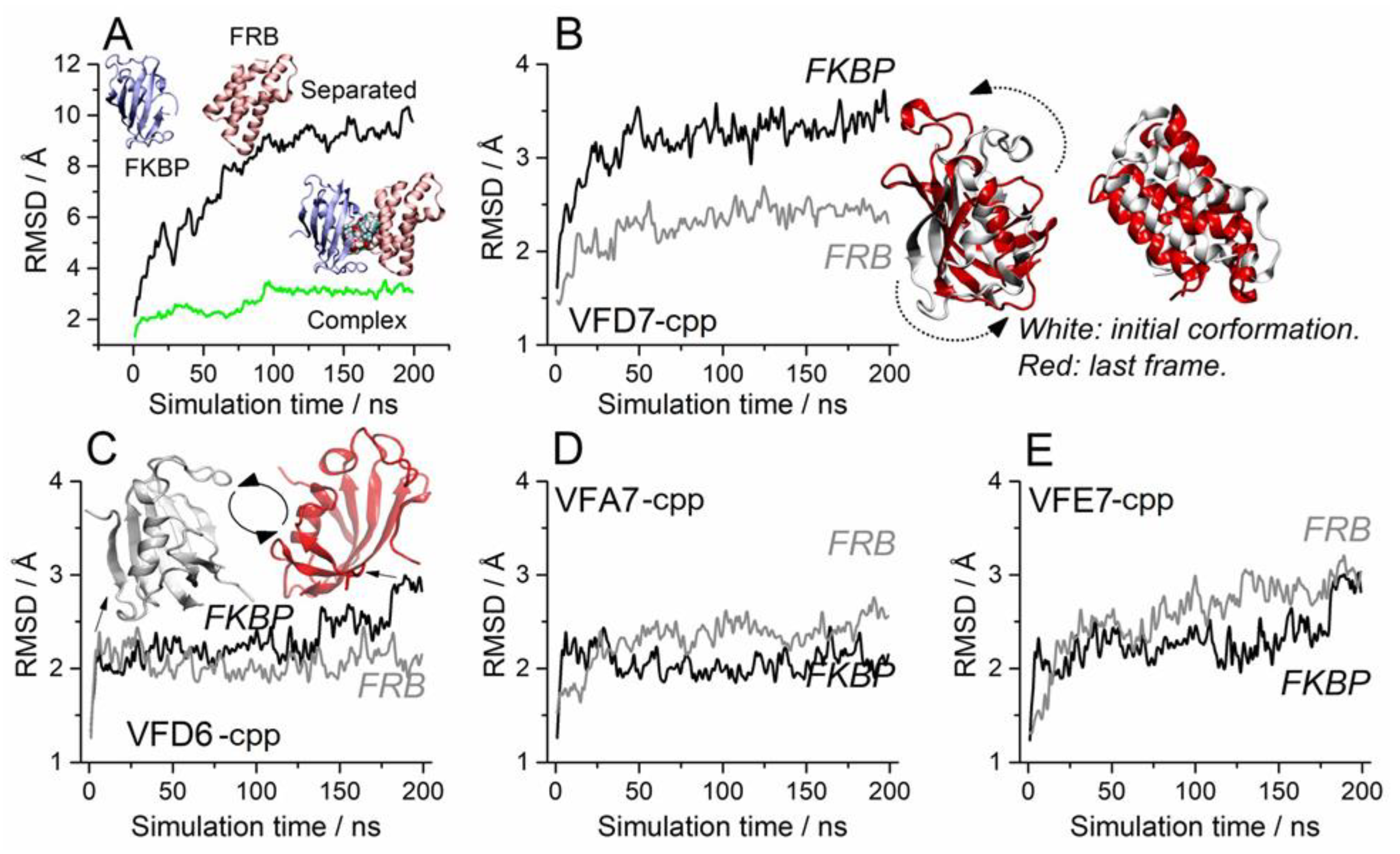
3.4. Molecular Dynamics to Evaluate the Effect of the VFD7-cpp and Related Peptides on Rapamycin-Induced FKBP and FRB Interaction
3.5. Impact on the FKBP Conformation Induced by the VFD7-cpp Peptide
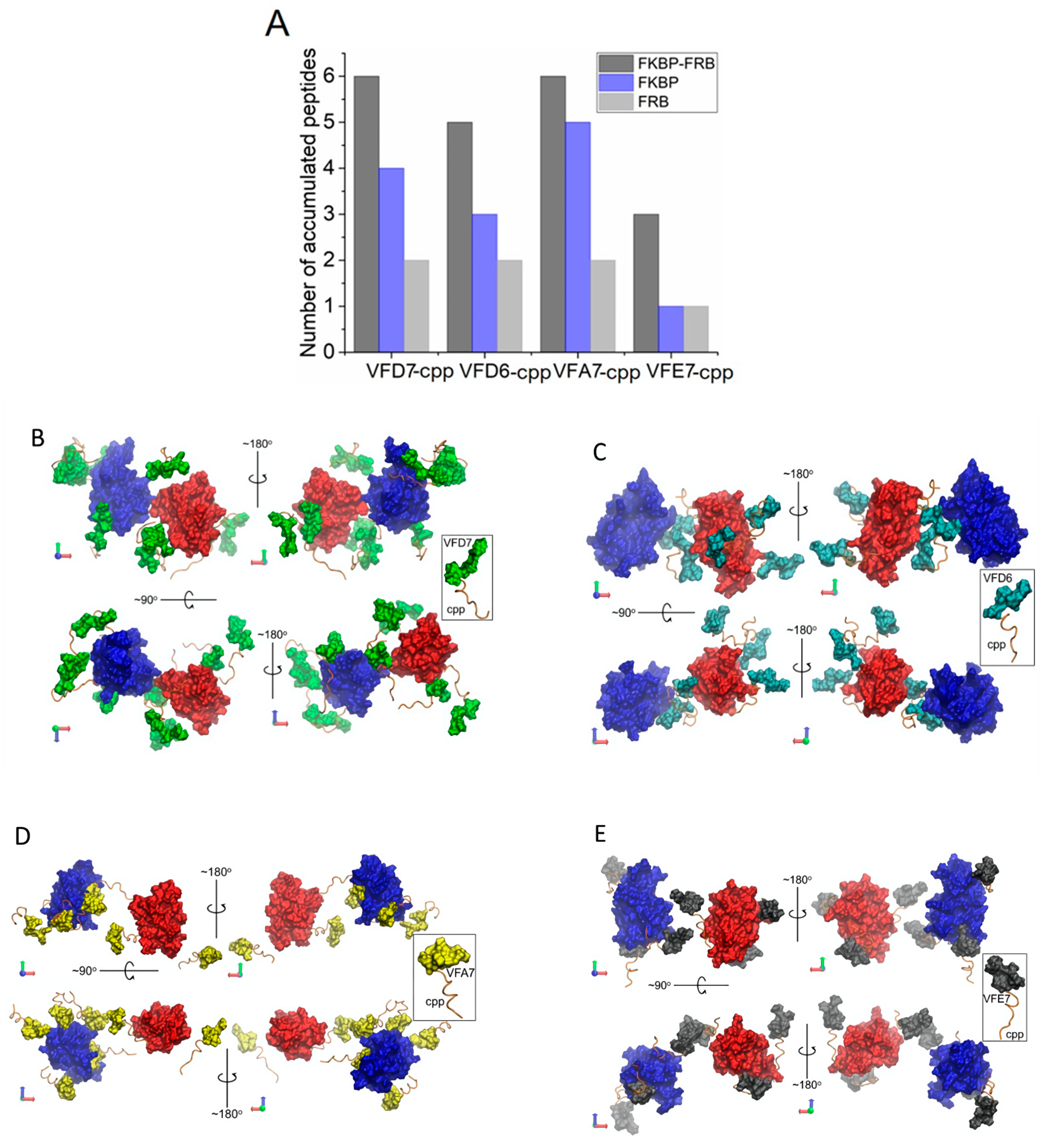
3.6. Surface Interactions between FKBP and FRB and the Effects of VFD7-cpp and Derived Peptides
3.7. InPeps Exist Associated as well as Unassociated/Free from Macromolecular Components of the Cell
3.8. Possible Pharmacological Effects of VFD7-cpp and Derived Peptides on Autophagy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vidal, M.; Cusick, M.E.; Barabasi, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Lu, W.; Liu, C.; Fang, J.; Hou, Y.; Handy, D.E.; Wang, R.; Zhao, Y.; Yang, Y.; Huang, J.; et al. A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat. Commun. 2019, 10, 3476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahni, N.; Yi, S.; Zhong, Q.; Jailkhani, N.; Charloteaux, B.; Cusick, M.E.; Vidal, M. Edgotype: A fundamental link between genotype and phenotype. Curr. Opin. Genet. Dev. 2013, 23, 649–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawson, T.; Nash, P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000, 14, 1027–1047. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein–protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef]
- Qiu, Y.; Li, X.; He, X.; Pu, J.; Zhang, J.; Lu, S. Computational methods-guided design of modulators targeting protein-protein interactions (PPIs). Eur. J. Med. Chem. 2020, 207, 112764. [Google Scholar] [CrossRef]
- De Araujo, C.B.; Heimann, A.S.; Remer, R.A.; Russo, L.C.; Colquhoun, A.; Forti, F.L.; Ferro, E.S. Intracellular Peptides in Cell Biology and Pharmacology. Biomolecules 2019, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Reckziegel, P.; Festuccia, W.T.; Britto, L.R.G.; Jang, K.L.L.; Romao, C.M.; Heimann, J.C.; Fogaca, M.V.; Rodrigues, N.S.; Silva, N.R.; Guimaraes, F.S.; et al. A novel peptide that improves metabolic parameters without adverse central nervous system effects. Sci. Rep. 2017, 7, 14781. [Google Scholar] [CrossRef] [Green Version]
- Wysocki, V.H.; Resing, K.A.; Zhang, Q.; Cheng, G. Mass spectrometry of peptides and proteins. Methods 2005, 35, 211–222. [Google Scholar] [CrossRef]
- Dasgupta, S.; Yang, C.; Castro, L.M.; Tashima, A.K.; Ferro, E.S.; Moir, R.D.; Willis, I.M.; Fricker, L.D. Analysis of the Yeast Peptidome and Comparison with the Human Peptidome. PLoS ONE 2016, 11, e0163312. [Google Scholar] [CrossRef]
- Gelman, J.S.; Sironi, J.; Castro, L.M.; Ferro, E.S.; Fricker, L.D. Peptidomic analysis of human cell lines. J. Proteome Res. 2011, 10, 1583–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaya, G.R.; Parada, C.A.; Eichler, R.A.; Yamaki, V.N.; Navon, A.; Heimann, A.S.; Figueiredo, E.G.; Ferro, E.S. Peptidomic profiling of cerebrospinal fluid from patients with intracranial saccular aneurysms. J. Proteom. 2021, 240, 104188. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Lahav-Baratz, S.; Ciechanover, A. Two distinct ubiquitin-dependent mechanisms are involved in NF-kappaB p105 proteolysis. Biochem. Biophys. Res. Commun. 2006, 345, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Haecker, H.; Karin, M.; Ciechanover, A. Mechanism of processing of the NF-kappa B2 p100 precursor: Identification of the specific polyubiquitin chain-anchoring lysine residue and analysis of the role of NEDD8-modification on the SCF(beta-TrCP) ubiquitin ligase. Oncogene 2004, 23, 2540–2547. [Google Scholar] [CrossRef] [Green Version]
- Bochtler, M.; Ditzel, L.; Groll, M.; Hartmann, C.; Huber, R. The proteasome. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 295–317. [Google Scholar] [CrossRef]
- Hughes, A.L.; Nei, M. Evolutionary relationships of the classes of major histocompatibility complex genes. Immunogenetics 1993, 37, 337–346. [Google Scholar] [CrossRef]
- Kasahara, M.; McKinney, E.C.; Flajnik, M.F.; Ishibashi, T. The evolutionary origin of the major histocompatibility complex: Polymorphism of class II alpha chain genes in the cartilaginous fish. Eur. J. Immunol. 1993, 23, 2160–2165. [Google Scholar] [CrossRef]
- Ferro, E.S.; Hyslop, S.; Camargo, A.C. Intracellullar peptides as putative natural regulators of protein interactions. J. Neurochem. 2004, 91, 769–777. [Google Scholar] [CrossRef]
- Fricker, L.D. Proteasome Inhibitor Drugs. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 457–476. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, S.; Fishman, M.; Mahallati, H.; Castro, L.; Tashima, A.; Ferro, E.; Fricker, L. Reduced Levels of Proteasome Products in a Mouse Striatal Cell Model of Huntington’s Disease. PLoS ONE 2015, 10, e0145333. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, S.; Castro, L.M.; Dulman, R.; Yang, C.; Schmidt, M.; Ferro, E.S.; Fricker, L.D. Proteasome inhibitors alter levels of intracellular peptides in HEK293T and SH-SY5Y cells. PLoS ONE 2014, 9, e103604. [Google Scholar] [CrossRef] [PubMed]
- Ferro, E.S.; Rioli, V.; Castro, L.M.; Fricker, L.D. Intracellular peptides: From discovery to function. EuPA Open Proteom. 2014, 3, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Gelman, J.S.; Sironi, J.; Berezniuk, I.; Dasgupta, S.; Castro, L.M.; Gozzo, F.C.; Ferro, E.S.; Fricker, L.D. Alterations of the Intracellular Peptidome in Response to the Proteasome Inhibitor Bortezomib. PLoS ONE 2013, 8, e53263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fricker, L.D.; Gelman, J.S.; Castro, L.M.; Gozzo, F.C.; Ferro, E.S. Peptidomic analysis of HEK293T cells: Effect of the proteasome inhibitor epoxomicin on intracellular peptides. J. Proteome Res. 2012, 11, 1981–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berezniuk, I.; Sironi, J.; Callaway, M.B.; Castro, L.M.; Hirata, I.Y.; Ferro, E.S.; Fricker, L.D. CCP1/Nna1 functions in protein turnover in mouse brain: Implications for cell death in Purkinje cell degeneration mice. Faseb J. 2010, 24, 1813–1823. [Google Scholar] [CrossRef] [Green Version]
- Fricker, L.D. Analysis of mouse brain peptides using mass spectrometry-based peptidomics: Implications for novel functions ranging from non-classical neuropeptides to microproteins. Mol. Biosyst. 2010, 6, 1355–1365. [Google Scholar] [CrossRef]
- Café-Mendes, C.C.C.; Ferro, E.S.S.; Torrão, A.S.S.; Crunfli, F.; Rioli, V.; Schmitt, A.; Falkai, P.; Britto, L.R.R.; Turck, C.W.W.; Martins-de-Souza, D. Peptidomic analysis of the anterior temporal lobe and corpus callosum from schizophrenia patients. J. Proteom. 2017, 151, 97–105. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Wang, F.; You, L.; Xu, P.; Cao, Y.; Chen, L.; Wen, J.; Guo, X.; Cui, X.; et al. Identification of intracellular peptides associated with thermogenesis in human brown adipocytes. J. Cell Physiol. 2019, 234, 7104–7114. [Google Scholar] [CrossRef]
- Gelman, J.S.; Dasgupta, S.; Berezniuk, I.; Fricker, L.D. Analysis of peptides secreted from cultured mouse brain tissue. Biochim. Biophys. Acta 2013, 1834, 2408–2417. [Google Scholar] [CrossRef] [Green Version]
- Gelman, J.S.; Fricker, L.D. Hemopressin and other bioactive peptides from cytosolic proteins: Are these non-classical neuropeptides? AAPS J. 2010, 12, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Heimann, A.S.; Dale, C.S.; Guimarães, F.S.; Reis, R.A.M.; Navon, A.; Shmuelov, M.A.; Rioli, V.; Gomes, I.; Devi, L.L.; Ferro, E.S. Hemopressin as a breakthrough for the cannabinoid field. Neuropharmacology 2021, 183, 108406. [Google Scholar] [CrossRef] [PubMed]
- Heimann, A.S.; Gomes, I.; Dale, C.S.; Pagano, R.L.; Gupta, A.; de Souza, L.L.; Luchessi, A.D.; Castro, L.M.; Giorgi, R.; Rioli, V.; et al. Hemopressin is an inverse agonist of CB1 cannabinoid receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 20588–20593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, M.F.; Cunha, F.M.; Berti, D.A.; Heimann, A.S.; Klitzke, C.F.; Rioli, V.; Oliveira, V.; Ferro, E.S. Substrate phosphorylation affects degradation and interaction to endopeptidase 24.15, neurolysin, and angiotensin-converting enzyme. Biochem. Biophys. Res. Commun. 2006, 339, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Heimann, A.S.; Favarato, M.H.; Gozzo, F.C.; Rioli, V.; Carreno, F.R.; Eberlin, M.N.; Ferro, E.S.; Krege, J.H.; Krieger, J.E. ACE gene titration in mice uncovers a new mechanism for ACE on the control of body weight. Physiol. Genom. 2005, 20, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, L.C.; Asega, A.F.; Castro, L.M.; Negraes, P.D.; Cruz, L.; Gozzo, F.C.; Ulrich, H.; Camargo, A.C.; Rioli, V.; Ferro, E.S. Natural intracellular peptides can modulate the interactions of mouse brain proteins and thimet oligopeptidase with 14-3-3epsilon and calmodulin. Proteomics 2012, 12, 2641–2655. [Google Scholar] [CrossRef] [PubMed]
- Cunha, F.M.; Berti, D.A.; Ferreira, Z.S.; Klitzke, C.F.; Markus, R.P.; Ferro, E.S. Intracellular peptides as natural regulators of cell signaling. J. Biol. Chem. 2008, 283, 24448–24459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewehr, M.C.F.; Teixeira, A.A.S.; Santos, B.A.C.; Biondo, L.A.; Gozzo, F.C.; Cordibello, A.M.; Eichler, R.A.S.; Reckziegel, P.; Da Silva, R.N.O.; Dos Santos, N.B.; et al. The Relevance of Thimet Oligopeptidase in the Regulation of Energy Metabolism and Diet-Induced Obesity. Biomolecules 2020, 10, 321. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Wang, X.; Proud, C.G. mTOR inhibitors in cancer therapy. F1000Research 2016, 5, 2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calne, R.; Lim, S.; Samaan, A.; Collier, D.S.J.; Pollard, S.; White, D.; Thiru, S. Rapamycin for immunosuppression in organ allografting. Lancet 1989, 334, 227. [Google Scholar] [CrossRef]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.A.; Pacold, M.E.; Cervantes, C.L.; Lim, D.; Lou, H.J.; Ottina, K.; Gray, N.S.; Turk, B.E.; Yaffe, M.B.; Sabatini, D.M. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 2013, 341, 1236566. [Google Scholar] [CrossRef] [Green Version]
- Thoreen, C.C.; Chantranupong, L.; Keys, H.R.; Wang, T.; Gray, N.S.; Sabatini, D.M. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012, 485, 109–113. [Google Scholar] [CrossRef]
- Lamming, D.W. Inhibition of the Mechanistic Target of Rapamycin (mTOR)-Rapamycin and Beyond. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Takahara, T.; Amemiya, Y.; Sugiyama, R.; Maki, M.; Shibata, H. Amino acid-dependent control of mTORC1 signaling: A variety of regulatory modes. J. Biomed. Sci. 2020, 27, 87. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Anti-aging: Senolytics or gerostatics (unconventional view). Oncotarget 2021, 12, 1821–1835. [Google Scholar] [CrossRef]
- Perl, A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat. Rev. Rheumatol. 2016, 12, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahé, E.; Morelon, E.; Lechaton, S.; Sang, K.-H.L.Q.; Mansouri, R.; Ducasse, M.-F.; Mamzer-Bruneel, M.-F.; de Prost, Y.; Kreis, H.; Bodemer, C. Cutaneous Adverse Events in Renal Transplant Recipients Receiving Sirolimus-Based Therapy1. Transplantation 2005, 79, 476–482. [Google Scholar] [PubMed] [Green Version]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langel, Ü. Cell-Penetrating Peptides and Transportan. Pharmaceutics 2021, 13, 987. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchardt, F.; Ruttekolk, I.R.; Verdurmen, W.P.; Lortat-Jacob, H.; Bürck, J.; Hufnagel, H.; Fischer, R.; Van den Heuvel, M.; Löwik, D.W.; Vuister, G.W. A cell-penetrating peptide derived from human lactoferrin with conformation-dependent uptake efficiency. J. Biol. Chem. 2009, 284, 36099–36108. [Google Scholar] [CrossRef] [Green Version]
- Shoari, A.; Tooyserkani, R.; Tahmasebi, M.; Löwik, D. Delivery of Various Cargos into Cancer Cells and Tissues via Cell-Penetrating Peptides: A Review of the Last Decade. Pharmaceutics 2021, 13, 1391. [Google Scholar] [CrossRef]
- Dixon, A.S.; Schwinn, M.K.; Hall, M.P.; Zimmerman, K.; Otto, P.; Lubben, T.H.; Butler, B.L.; Binkowski, B.F.; Machleidt, T.; Kirkland, T.A.; et al. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 2016, 11, 400–408. [Google Scholar] [CrossRef]
- Hall, M.P.; Unch, J.; Binkowski, B.F.; Valley, M.P.; Butler, B.L.; Wood, M.G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; Machleidt, T.; et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7, 1848–1857. [Google Scholar] [CrossRef]
- Ziegler, A.; Nervi, P.; Dürrenberger, M.; Seelig, J. The cationic cell-penetrating peptide CPPTAT derived from the HIV-1 protein TAT is rapidly transported into living fibroblasts: Optical, biophysical, and metabolic evidence. Biochemistry 2005, 44, 138–148. [Google Scholar] [CrossRef]
- Zaro, J.L.; Vekich, J.E.; Tran, T.; Shen, W.C. Nuclear localization of cell-penetrating peptides is dependent on endocytosis rather than cytosolic delivery in CHO cells. Mol. Pharm. 2009, 6, 337–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Araujo, C.B.; Russo, L.C.; Castro, L.M.; Forti, F.L.; do Monte, E.R.; Rioli, V.; Gozzo, F.C.; Colquhoun, A.; Ferro, E.S. A novel intracellular peptide derived from g1/s cyclin d2 induces cell death. J. Biol. Chem. 2014, 289, 16711–16726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philippe, G.J.B.; Craik, D.J.; Henriques, S.T. Converting peptides into drugs targeting intracellular protein–protein interactions. Drug Discov. Today 2021, 26, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Buyanova, M.; Pei, D. Targeting intracellular protein-protein interactions with macrocyclic peptides. Trends Pharmacol. Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Berti, D.A.; Morano, C.; Russo, L.C.; Castro, L.M.; Cunha, F.M.; Zhang, X.; Sironi, J.; Klitzke, C.F.; Ferro, E.S.; Fricker, L.D. Analysis of intracellular substrates and products of thimet oligopeptidase in human embryonic kidney 293 cells. J. Biol. Chem. 2009, 284, 14105–14116. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.; White, P. Fmoc Solid Phase Peptide Synthesis: A Practical Approach; OUP Oxford: Oxford, UK, 1999; Volume 222. [Google Scholar]
- Jaradat, D.M.M. Thirteen decades of peptide synthesis: Key developments in solid phase peptide synthesis and amide bond formation utilized in peptide ligation. Amino Acids 2018, 50, 39–68. [Google Scholar] [CrossRef]
- Stein, A.; Aloy, P. Contextual specificity in peptide-mediated protein interactions. PLoS ONE 2008, 3, e2524. [Google Scholar] [CrossRef] [Green Version]
- London, N.; Movshovitz-Attias, D.; Schueler-Furman, O. The structural basis of peptide-protein binding strategies. Structure 2010, 18, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Meszaros, B.; Erdos, G.; Dosztanyi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein disorder prediction: Implications for structural proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neron, B.; Menager, H.; Maufrais, C.; Joly, N.; Maupetit, J.; Letort, S.; Carrere, S.; Tuffery, P.; Letondal, C. Mobyle: A new full web bioinformatics framework. Bioinformatics 2009, 25, 3005–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Lee, H.; Lee, H.K.; Lee, S.W.; Ha, S.C.; Kwon, T.; Seo, J.K.; Lee, C.; Rhee, H.W. Proximity-Directed Labeling Reveals a New Rapamycin-Induced Heterodimer of FKBP25 and FRB in Live Cells. ACS Cent. Sci. 2016, 2, 506–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, L.; Andrade, R.; Birgin, E.G.; Martinez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.T.; Singh, R.P.; Klauda, J.B.; Hodoscek, M.; Brooks, B.R.; Woodcock, H.L., 3rd. CHARMMing: A new, flexible web portal for CHARMM. J. Chem. Inf. Model. 2008, 48, 1920–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, L. MDAnalysis; Version 17.224; Institute of Chemistry, University of Campinas: Campinas, SP, Brazil, 2017. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Udenfriend, S.; Stein, S.; Böhlen, P.; Dairman, W.; Leimgruber, W.; Weigele, M. Fluorescamine: A reagent for assay of amino acids, peptides, proteins, and primary amines in the picomole range. Science 1972, 178, 871–872. [Google Scholar] [CrossRef]
- Castro, L.M.; Berti, D.A.; Russo, L.C.; Coelho, V.; Gozzo, F.C.; Oliveira, V.; Ferro, E.S. Similar intracellular peptide profile of TAP1/beta2 microglobulin double-knockout mice and C57BL/6 wild-type mice as revealed by peptidomic analysis. AAPS J. 2010, 12, 608–616. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zhou, X.-J.; Zhang, H. Exploring the Role of Autophagy-Related Gene 5 (ATG5) Yields Important Insights Into Autophagy in Autoimmune/Autoinflammatory Diseases. Front. Immunol. 2018, 9, 2334. [Google Scholar] [CrossRef] [PubMed]
- Yogalingam, G.; Hwang, S.; Ferreira, J.C.B.; Mochly-Rosen, D. Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH) Phosphorylation by Protein Kinase Cδ (PKCδ) Inhibits Mitochondria Elimination by Lysosomal-like Structures following Ischemia and Reoxygenation-induced Injury *. J. Biol. Chem. 2013, 288, 18947–18960. [Google Scholar] [CrossRef] [Green Version]
- Rai, Y.; Pathak, R.; Kumari, N.; Sah, D.K.; Pandey, S.; Kalra, N.; Soni, R.; Dwarakanath, B.S.; Bhatt, A.N. Mitochondrial biogenesis and metabolic hyperactivation limits the application of MTT assay in the estimation of radiation induced growth inhibition. Sci. Rep. 2018, 8, 1531. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Chen, J.; Schreiber, S.L.; Clardy, J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 1996, 273, 239–242. [Google Scholar] [CrossRef]
- Reits, E.; Griekspoor, A.; Neijssen, J.; Groothuis, T.; Jalink, K.; van Veelen, P.; Janssen, H.; Calafat, J.; Drijfhout, J.W.; Neefjes, J. Peptide diffusion, protection, and degradation in nuclear and cytoplasmic compartments before antigen presentation by MHC class I. Immunity 2003, 18, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Paz, P.; Brouwenstijn, N.; Perry, R.; Shastri, N. Discrete proteolytic intermediates in the MHC class I antigen processing pathway and MHC I–dependent peptide trimming in the ER. Immunity 1999, 11, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, C.M.M.; Correa, C.N.; Iwai, L.K.; Ferro, E.S.; Castro, L.M. Characterization of Intracellular Peptides from Zebrafish (Danio rerio) Brain. Zebrafish 2019, 16, 240–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1–PGC-1α transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Daneshgar, N.; Rabinovitch, P.S.; Dai, D.F. TOR Signaling Pathway in Cardiac Aging and Heart Failure. Biomolecules 2021, 11, 168. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Simon, H.-U. Apoptosis regulation by autophagy gene 5. Crit. Rev. Oncol./Hematol. 2007, 63, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Jeong, H.; Yu, S.-W. Autophagy as a decisive process for cell death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential roles of Atg5 and FADD in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005, 280, 20722–20729. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Tan, S.-H.; Nicolas, V.; Bauvy, C.; Yang, N.-D.; Zhang, J.; Xue, Y.; Codogno, P.; Shen, H.-M. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013, 23, 508–523. [Google Scholar] [CrossRef] [Green Version]
- Var, S.R.; Shetty, A.V.; Grande, A.W.; Low, W.C.; Cheeran, M.C. Microglia and Macrophages in Neuroprotection, Neurogenesis, and Emerging Therapies for Stroke. Cells 2021, 10, 3555. [Google Scholar] [CrossRef]
- Varble, N.; Tutino, V.M.; Yu, J.; Sonig, A.; Siddiqui, A.H.; Davies, J.M.; Meng, H. Shared and distinct rupture discriminants of small and large intracranial aneurysms. Stroke 2018, 49, 856–864. [Google Scholar] [CrossRef]
- Johnson, S.C.; Kaeberlein, M. Rapamycin in aging and disease: Maximizing efficacy while minimizing side effects. Oncotarget 2016, 7, 44876–44878. [Google Scholar] [CrossRef] [Green Version]
- Salmon, A.B. About-face on the metabolic side effects of rapamycin. Oncotarget 2015, 6, 2585–2586. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T.; Scott, J.D. Signaling through scaffold, anchoring, and adaptor proteins. Science 1997, 278, 2075–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fesenko, I.; Azarkina, R.; Kirov, I.; Kniazev, A.; Filippova, A.; Grafskaia, E.; Lazarev, V.; Zgoda, V.; Butenko, I.; Bukato, O.; et al. Phytohormone treatment induces generation of cryptic peptides with antimicrobial activity in the Moss Physcomitrella patens. BMC Plant Biol. 2019, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Filippova, A.; Lyapina, I.; Kirov, I.; Zgoda, V.; Belogurov, A.; Kudriaeva, A.; Ivanov, V.; Fesenko, I. Salicylic acid influences the protease activity and posttranslation modifications of the secreted peptides in the moss Physcomitrella patens. J. Pept. Sci. 2019, 25, e3138. [Google Scholar] [CrossRef] [PubMed]
- Fesenko, I.; Khazigaleeva, R.; Govorun, V.; Ivanov, V. Analysis of Endogenous Peptide Pools of Physcomitrella patens Moss. Methods Mol. Biol. 2018, 1719, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Fesenko, I.A.; Arapidi, G.P.; Skripnikov, A.Y.; Alexeev, D.G.; Kostryukova, E.S.; Manolov, A.I.; Altukhov, I.A.; Khazigaleeva, R.A.; Seredina, A.V.; Kovalchuk, S.I.; et al. Specific pools of endogenous peptides are present in gametophore, protonema, and protoplast cells of the moss Physcomitrella patens. BMC Plant. Biol. 2015, 15, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navon, A.; Goldberg, A.L. Proteins are unfolded on the surface of the ATPase ring before transport into the proteasome. Mol. Cell 2001, 8, 1339–1349. [Google Scholar] [CrossRef]
- Rock, K.L.; York, I.A.; Goldberg, A.L. Post-proteasomal antigen processing for major histocompatibility complex class I presentation. Nat. Immunol. 2004, 5, 670–677. [Google Scholar] [CrossRef]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef]
- Gelman, J.S.; Sironi, J.; Castro, L.M.; Ferro, E.S.; Fricker, L.D. Hemopressins and other hemoglobin-derived peptides in mouse brain: Comparison between brain, blood, and heart peptidome and regulation in Cpefat/fat mice. J. Neurochem. 2010, 113, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Berezniuk, I.; Sironi, J.J.; Wardman, J.; Pasek, R.C.; Berbari, N.F.; Yoder, B.K.; Fricker, L.D. Quantitative peptidomics of Purkinje cell degeneration mice. PLoS ONE 2013, 8, e60981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiametti, L.O.; Correa, C.N.; Castro, L.M. Peptide Profile of Zebrafish Brain in a 6-OHDA-Induced Parkinson Model. Zebrafish 2021, 18, 55–65. [Google Scholar] [CrossRef]
- Monte, E.R.; Rossato, C.; Llanos, R.P.; Russo, L.C.; de Castro, L.M.; Gozzo, F.C.; de Araujo, C.B.; Peron, J.P.; Sant’Anna, O.A.; Ferro, E.S.; et al. Interferon-gamma activity is potentiated by an intracellular peptide derived from the human 19S ATPase regulatory subunit 4 of the proteasome. J. Proteom. 2017, 151, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, D.M.L.P.; Castro, L.M.; Rosa Neto, J.C.; Seelaender, M.; Neves, R.X.; Oliveira, V.; Forti, F.L.; Iwai, L.K.; Gozzo, F.C.; Todiras, M.; et al. Neurolysin knockout mice generation and initial phenotype characterization. J. Biol. Chem. 2014, 289, 15426–15440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, L.M.; Cavalcanti, D.M.; Araujo, C.B.; Rioli, V.; Icimoto, M.Y.; Gozzo, F.C.; Juliano, M.; Juliano, L.; Oliveira, V.; Ferro, E.S. Peptidomic analysis of the neurolysin-knockout mouse brain. J. Proteom. 2014, 111, 238–248. [Google Scholar] [CrossRef]
- Santos, N.B.D.; Franco, R.D.; Camarini, R.; Munhoz, C.D.; Eichler, R.A.S.; Gewehr, M.C.F.; Reckziegel, P.; Llanos, R.P.; Dale, C.S.; Silva, V.; et al. Thimet Oligopeptidase (EC 3.4.24.15) Key Functions Suggested by Knockout Mice Phenotype Characterization. Biomolecules 2019, 9, 382. [Google Scholar] [CrossRef] [Green Version]
- Berti, D.A.; Russo, L.C.; Castro, L.M.; Cruz, L.; Gozzo, F.C.; Heimann, J.C.; Lima, F.B.; Oliveira, A.C.; Andreotti, S.; Prada, P.O. Identification of intracellular peptides in rat adipose tissue: Insights into insulin resistance. Proteomics 2012, 12, 2668–2681. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H. Regulation of Autophagy by mTOR Signaling Pathway. Adv. Exp. Med. Biol. 2019, 1206, 67–83. [Google Scholar] [CrossRef]
- Wu, K.; Seylani, A.; Wu, J.; Wu, X.; Bleck, C.K.E.; Sack, M.N. BLOC1S1/GCN5L1/BORCS1 is a critical mediator for the initiation of autolysosomal tubulation. Autophagy 2021, 17, 3707–3724. [Google Scholar] [CrossRef]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef]
- Gangloff, Y.-G.; Mueller, M.; Dann, S.G.; Svoboda, P.; Sticker, M.; Spetz, J.-F.; Um, S.H.; Brown, E.J.; Cereghini, S.; Thomas, G. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol. Cell. Biol. 2004, 24, 9508–9516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eltschinger, S.; Loewith, R. TOR complexes and the maintenance of cellular homeostasis. Trends Cell Biol. 2016, 26, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Dancey, J. mTOR signaling and drug development in cancer. Nat. Rev. Clin. Oncol. 2010, 7, 209. [Google Scholar] [CrossRef]
- Walters, H.E.; Cox, L.S. mTORC inhibitors as broad-spectrum therapeutics for age-related diseases. Int. J. Mol. Sci. 2018, 19, 2325. [Google Scholar] [CrossRef] [Green Version]
- Bischof, E.; Siow, R.C.; Zhavoronkov, A.; Kaeberlein, M. The potential of rapalogs to enhance resilience against SARS-CoV-2 infection and reduce the severity of COVID-19. Lancet Healthy Longev. 2021, 2, e105–e111. [Google Scholar] [CrossRef]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef]
- Horvath, S.; Lu, A.T.; Cohen, H.; Raj, K. Rapamycin retards epigenetic ageing of keratinocytes independently of its effects on replicative senescence, proliferation and differentiation. Aging 2019, 11, 3238–3249. [Google Scholar] [CrossRef]
- Wilkinson, J.E.; Burmeister, L.; Brooks, S.V.; Chan, C.C.; Friedline, S.; Harrison, D.E.; Hejtmancik, J.F.; Nadon, N.; Strong, R.; Wood, L.K. Rapamycin slows aging in mice. Aging Cell 2012, 11, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Jean, W.C.; Spellman, S.R.; Nussbaum, E.S.; Low, W.C. Reperfusion injury after focal cerebral ischemia: The role inflammation and the the rapeutic horizon. Neurosurgery 1998, 43, 1382–1396. [Google Scholar] [CrossRef]
- De la Torre, B.G.; Albericio, F. Peptide Therapeutics 2.0. Molecules 2020, 25, 2293. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2019. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2020, 25, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid Sequences | Symbol |
|---|---|
| VFDVELLKLE | VFD10 |
| VFDVELL | VFD7 |
| YGRKKKRRQRRR | cpp |
| VFDVELLKLEYGRKKKRRQRRR | VFD10-cpp |
| VFDVELLYGRKKKRRQRRR | VFD7-cpp |
| VFAVELLYGRKKKRRQRRR | VFA7-cpp |
| VFEVELLYGRKKKRRQRRR | VFE7-cpp |
| FDVELLYGRKKKRRQRRR | VFD6-cpp |
| DVELLYGRKKRRQRRR | VFD5-cpp |
| VELLYGRKKRRQRRR | VFD4-cpp |
| EVELLYGRKKKRRQRRR | EVE5-cpp |
| AVELLYGRKKKRRQRRR | AVE5-cpp |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parada, C.A.; de Oliveira, I.P.; Gewehr, M.C.F.; Machado-Neto, J.A.; Lima, K.; Eichler, R.A.S.; Lopes, L.R.; Bechara, L.R.G.; Ferreira, J.C.B.; Festuccia, W.T.; et al. Effect of FKBP12-Derived Intracellular Peptides on Rapamycin-Induced FKBP–FRB Interaction and Autophagy. Cells 2022, 11, 385. https://doi.org/10.3390/cells11030385
Parada CA, de Oliveira IP, Gewehr MCF, Machado-Neto JA, Lima K, Eichler RAS, Lopes LR, Bechara LRG, Ferreira JCB, Festuccia WT, et al. Effect of FKBP12-Derived Intracellular Peptides on Rapamycin-Induced FKBP–FRB Interaction and Autophagy. Cells. 2022; 11(3):385. https://doi.org/10.3390/cells11030385
Chicago/Turabian StyleParada, Carolina A., Ivan Pires de Oliveira, Mayara C. F. Gewehr, João Agostinho Machado-Neto, Keli Lima, Rosangela A. S. Eichler, Lucia R. Lopes, Luiz R. G. Bechara, Julio C. B. Ferreira, William T. Festuccia, and et al. 2022. "Effect of FKBP12-Derived Intracellular Peptides on Rapamycin-Induced FKBP–FRB Interaction and Autophagy" Cells 11, no. 3: 385. https://doi.org/10.3390/cells11030385
APA StyleParada, C. A., de Oliveira, I. P., Gewehr, M. C. F., Machado-Neto, J. A., Lima, K., Eichler, R. A. S., Lopes, L. R., Bechara, L. R. G., Ferreira, J. C. B., Festuccia, W. T., Censoni, L., Tersariol, I. L. S., & Ferro, E. S. (2022). Effect of FKBP12-Derived Intracellular Peptides on Rapamycin-Induced FKBP–FRB Interaction and Autophagy. Cells, 11(3), 385. https://doi.org/10.3390/cells11030385