
S-Adenosylmethionine: From the Discovery of Its Inhibition of Tumorigenesis to Its Use as a Therapeutic Agent





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
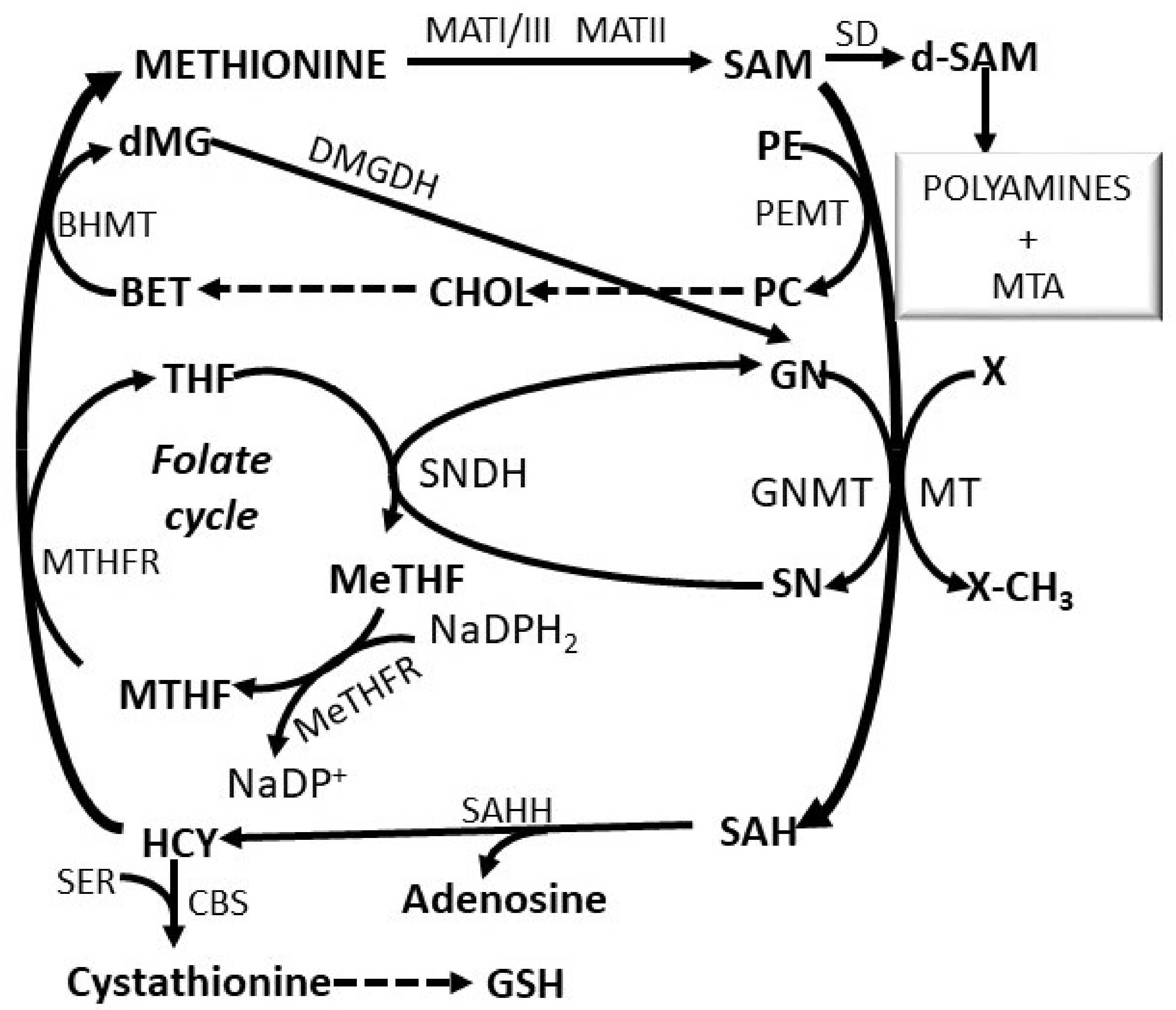
2. The Methionine Adenosyltransferase Switch
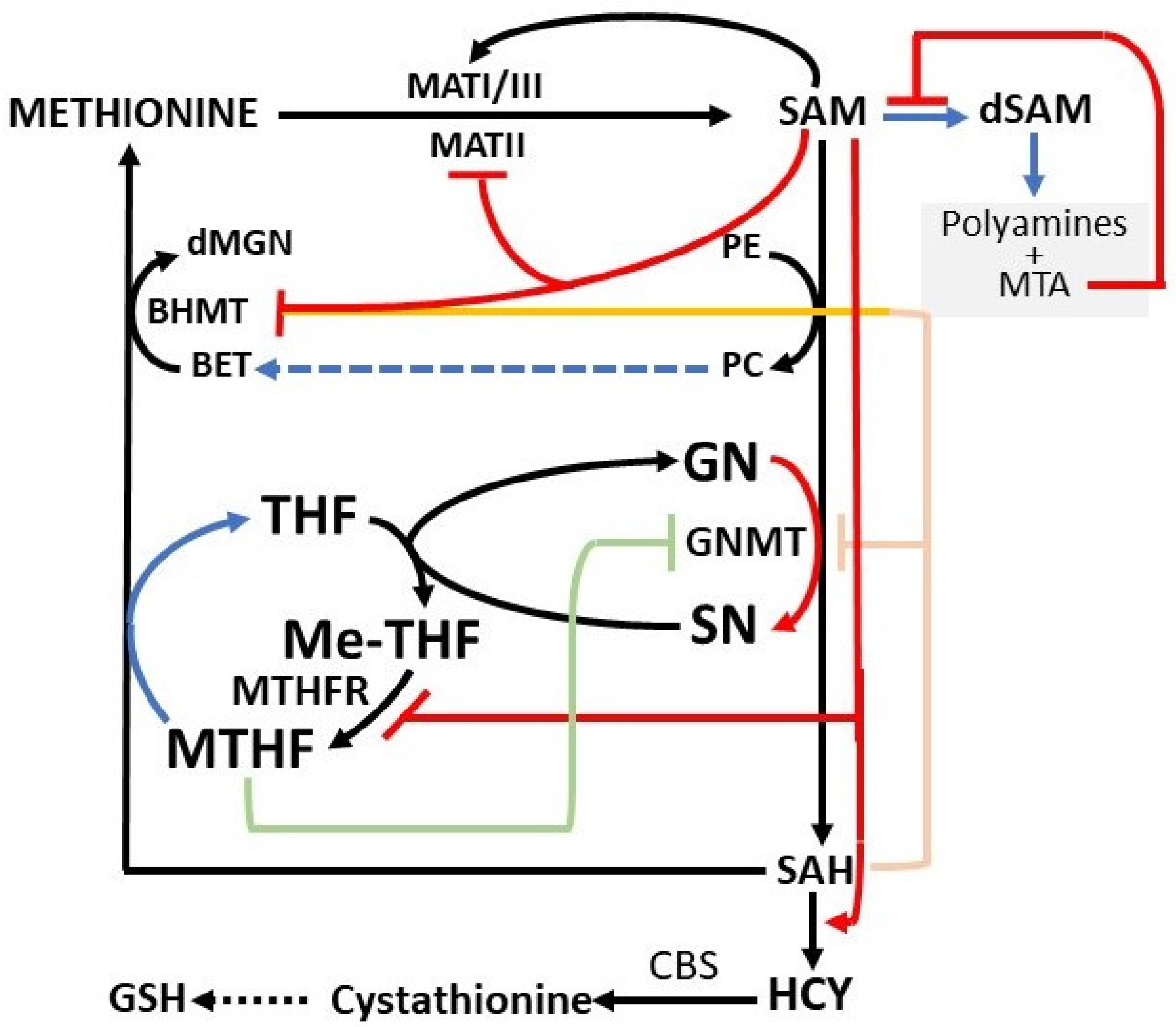
3. Regulatory Mechanisms of the Methionine Cycle
4. The Deregulation of Methionine Metabolism in Preneoplastic and Neoplastic Liver
5. SAM Inhibitory Effects
SAM Metabolism and Epigenetic Regulation
6. SAM and Human Disease
6.1. Alcoholic Liver Disease
6.2. Non-Alcoholic Fatty Liver Disease
6.3. SAM and Intra-Hepatic Cholestasis
6.4. SAM and Viral Hepatitis
6.5. SAM ant Tumor Therapy
6.6. SAM and Genetic Predisposition to Hepatocarcinogenesis
7. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
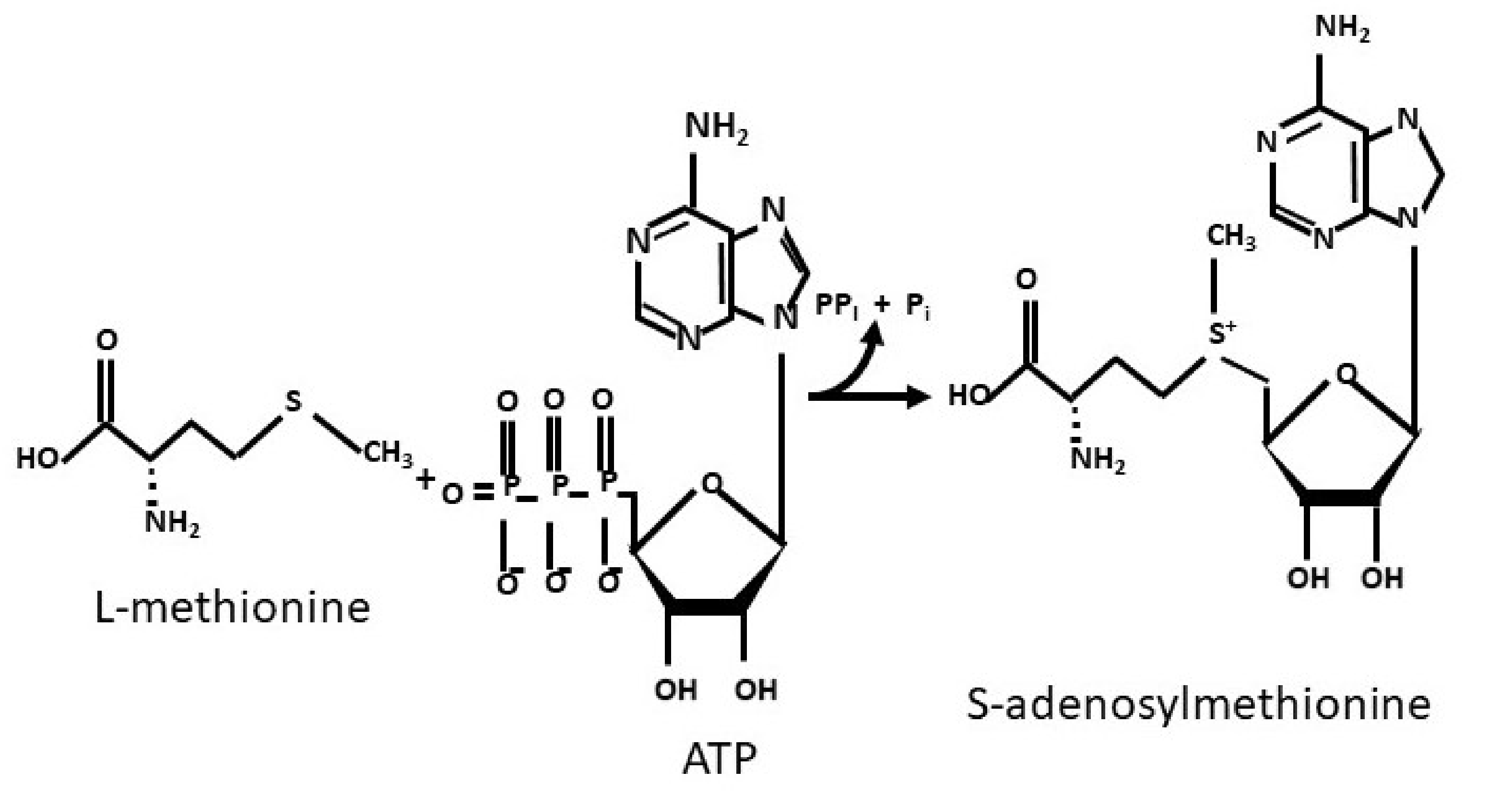
- Cantoni, G.L. Activation of methionine for transmethylation. J. Biol. Chem. 1951, 189, 745–750. [Google Scholar] [CrossRef]
- Feo, F.; Garcea, R.; Pascale, R.M.; Pirisi, L.; Daino, L.; Donaera, A. The variations of S-adenosyl-L-methionine content modu-late hepatocyte growth during phenobarbital promotion of diethylnitrosamine-induced rat liver carcinogenesis. Toxicol. Pathol. 1987, 5, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcea, R.; Pascale, R.M.; Daino, L.; Frassetto, S.; Cozzolino, P.; Ruggiu, M.E.; Vannini, M.G.; Gaspa, L.; Feo, F. Variations of ornithine decarboxylase activity and S-adenosyl-L-methionine and 5′-methylthioadenosine contents during the development of diethylnitrosamine-induced liver hyperplastic nodules and hepatocellular carcinomas. Carcinogenesis 1987, 8, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Ramani, K.; Mato, J.M.; Lu, S.C. Role of methionine adenosyltransferase genes in hepatocarcinogenesis. Cancers 2011, 3, 1480–1497. [Google Scholar] [CrossRef] [Green Version]
- Mato, J.M.; Lu, S.C. Role of S-adenosyl-l-methionine in liver health and injury. Hepatology 2007, 45, 1306–1312. [Google Scholar] [CrossRef]
- Miyazaki, J.H.; Yang, S.F. Metabolism of 5-methylthioribose to methionine. Plant. Physiol. 1987, 84, 277–281. [Google Scholar] [CrossRef] [Green Version]
- Bremer, J.; Greenberg, D.M. Methyl-transferring enzyme system of microsomes in the biosynthesis of lecithin (phosphatidyl-choline). Biochim. Biophys. Acta 1961, 46, 205–216. [Google Scholar] [CrossRef]
- Feo, F.; Pririsi, L.; Garcea, R.; Daino, L.; Pascale, R.M. The role of phosphatidylethanolamine methylation in the synthesis of phosphatidylcholine in acute ethanol intoxication. Res. Commun. Subst. Abuse 1982, 3, 499–502. [Google Scholar]
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B12, folate, and the methionine remethylation cycle-biochemistry, path-ways, and regulation. J. Inherit. Metab. Dis. 2019, 42, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, J.D.; Martin, J.J. Homocysteine. Int. J. Biochem. Cell Biol. 2000, 32, 385–389. [Google Scholar] [CrossRef]
- Ebara, S. Nutritional role of folate. Congenit. Anom. 2017, 57, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N. Role of methionine on epigenetic in animals. Anim. Nutr. 2018, 4, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Mladenović, D.; Radosavljević, T.; Hrnčić, D.; Rasic-Markovic, A.; Stanojlović, O. The effects of dietary methionine restriction on the function and metabolic reprogramming in the liver and brain—Implications for longevity. Rev. Neurosci. 2019, 30, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Mato, J.M.; Corrales, F.J.; Lu, S.C.; Avila, M.A. S-adenosylmethionine: A control switch that regulates liver function. FASEB J. 2002, 16, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avila, A.; Berasain, C.; Torres, L.; Martín-Duce, A.; Corrales, F.J.; Yang, H.; Prieto, J.; Lu, S.C.; Caballería, J.; Rodés, J.; et al. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepato-cellular carcinoma. J. Hepatol. 2000, 33, 907–914. [Google Scholar] [CrossRef] [Green Version]
- Frau, M.; Feo, F.; Pascale, R.M. Pleiotropic effects of methionine adenosyltransferases deregulation as determinants of liver cancer progression and prognosis. J. Hepatol. 2013, 59, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Feo, F.; Pascale, R.M.; Garcea, R.; Daino, L.; Pirisi, L.; Frassetto, S.; Ruggiu, M.E.; Di Padova, C.; Stramentinoli, G. Effect of the variations of S-adenosyl-L-methionine liver content on fat accumulation and ethanol metabolism in ethanol-intoxicated rats. Toxicol. Appl. Pharmacol. 1986, 83, 331–341. [Google Scholar] [CrossRef]
- Zhang, F.; Gu, J.X.; Zou, X.P.; Zhuge, Y.Z. Protective effects of S-adenosylmethionine against CCl4− and ethanol-induced ex-perimental hepatic fibrosis. Mol. Biol. 2016, 5, 284–290. [Google Scholar]
- Pascale, R.M.; Daino, L.; Garcea, R.; Frassetto, S.; Ruggiu, M.E.; Vannini, M.G.; Cozzolino, P.; Feo, F. Inhibition by ethanol of rat liver plasma membrane (Na+, K+)ATPase: Protective effect of S-adenosyl-L-methionine, L-methionine, and N-acetylcysteine. Toxicol. Appl. Pharmacol. 1989, 97, 216–229. [Google Scholar] [CrossRef]
- Feo, F.; Garcea, R.; Daino, L.; Pascale, R.M.; Pirisi, L.; Frassetto, S.; Ruggiu, M.E. Early stimulation of polyamine biosynthesis during promotion by phenobarbital of diethylnitrosamine-induced rat liver Carcinogenesis. The effects of variations of the S-adenosyl-L-methionine cellular pool. Carcinogenesis 1985, 6, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.D.; Martin, J. Inactivation of betaine-homocysteine methyltransferase by adenosylmethionine and adenosyle-thionine. Biochem. Biophys. Res. Commun. 1984, 118, 14–19. [Google Scholar] [CrossRef]
- Lu, S.C.; Alvarez, L.; Huang, Z.Z.; Chen, L.; An, W.; Corrales, F.J.; Avila, M.A.; Kanel, G.; Mato, J.M. Methionineadenosyl-transferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in prolifera-tion. Proc. Natl. Acad. Sci. USA 2001, 98, 5560–5565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, C.; Decha-Umphai, W.; Corbin, J. Phosphorylation modulates the activity of glycine N-methyltransferase, a folate binding protein. In vitro phosphorylation is inhibited by the natural folate ligand. J. Biol. Chem. 1989, 264, 9638–9642. [Google Scholar] [CrossRef]
- Reed, M.C.; Gamble, M.V.; Hall, M.N.; Nijhout, H.F. Mathematical analysis of the regulation of competing methyltransferas-es. BMC Syst. Biol. 2015, 9, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.C.; Lin, W.L.; Lin, Y.J.; Tang, F.Y.; Chen, Y.M. A novel role of the tumor suppressor GNMT in cellular defense against DNA damage. Int. J. Cancer 2014, 134, 799–810. [Google Scholar] [CrossRef]
- Wagner, C.; Briggs, W.T.; Cook, R.J. Inhibition of glycine N-methyltransferase activity by folate derivatives: Implications for regulation of methyl group metabolism. Biochem. Biophys. Res. Commun. 1985, 127, 746–752. [Google Scholar] [CrossRef]
- Murray, B.; Barbier-Torres, L.; Fan, W.; Mato, J.M.; Lu, S.C. Methionine adenosyltransferases in liver cancer. World J. Gastroenterol. 2019, 25, 4300–4319. [Google Scholar] [CrossRef]
- Pascale, R.M.; Peitta, G.; Simile, M.M.; Feo, F. Alterations of Methionine Metabolism as Potential Targets for the Prevention and Therapy of Hepatocellular Carcinoma. Medicina 2019, 55, 296. [Google Scholar] [CrossRef] [Green Version]
- Lozano-Rosas, M.G.; Chávez, E.; Velasco-Loyden, G.; Domínguez-López, M.; Martínez-Pérez, L.; Chagoya De Sánchez, V. Diminished S-adenosylmethionine biosynthesis and its metabolism in a model of hepatocellular carcinoma is recuperated by an adenosine derivative. Cancer Biol. Ther. 2020, 21, 81–94. [Google Scholar] [CrossRef]
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-Chantada, M.; Fernandez, D.; Embade, N.; Martínez-Lopez, N.; Varela-Rey, M.; Woodhoo, A.; Luka, Z.; Wagner, C.; Anglim, P.P.; Finnel, L.; et al. Hur/methylated-Hur and AUF1 regulate the expression of methionine adenosyltransferase during liver proliferation, differentiation and carcinogenesis. Gastroenterology 2012, 38, 1943–1953. [Google Scholar]
- Frau, M.; Tomasi, M.L.; Simile, M.M.; Demartis, M.I.; Salis, F.; Latte, G.; Calvisi, D.F.; Seddaiu, M.A.; Daino, L.; Feo, C.F.; et al. Role of transcriptional and posttranscriptional regulation of methionine adenosyltransferases in liver cancer progression. Hepatology 2012, 56, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasi, M.L.; Li, T.W.; Li, M.; Mato, J.M.; Lu, S.C. Inhibition of human methionine adenosyltransferase 1A transcription by coding region methylation. J. Cell. Physiol. 2012, 227, 1583–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Huang, Z.Z.; Wang, J.; Lu, S.C. The role of c-Myb and Sp1 in the up-regulation of methionine-adenosyltransferase 2A gene expression in human hepatocellular carcinoma. FASEB J. 2001, 15, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Sadda, M.R.; Yu, V.; Zeng, Y.; Lee, T.D.; Ou, X.; Chen, L.; Lu, S.C. Induction of human methionine adenosyltransferase 2A expression by tumor necrosis factor alpha. Role of NF-kappa B and AP-1. J. Biol. Chem. 2003, 278, 50887–50896. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, M.L.; Ryoo, M.; Ramani, K.; Tomasi, I.; Giordano, P.; Mato, J.M.; Lu, S.C. Methionine, adenosyltransferase 2 sumoy-lation positively regulate Bcl-2 expression in human colon and liver cancer cells. Oncotarget 2015, 6, 37706–37723. [Google Scholar] [CrossRef] [Green Version]
- LeGros, L.; Halim, A.B.; Chamberlin, M.; Geller, A.; Kotb, M. Regulation of the human MAT2B gene encoding the regulatory beta subunit of methionine adenosyltransferase, MAT II. J. Biol. Chem. 2001, 276, 24918–24924. [Google Scholar] [CrossRef] [Green Version]
- Xia, M.; Chen, Y.; Wang, L.C.; Zandi, E.; Yang, H.; Bemanian, S.; Martínez-Chantar, M.L.; Mato, J.M.; Lu, S.C. Novel function and intracellular localization of methionine adenosyltransferase 2beta splicing variants. J. Biol. Chem. 2010, 285, 20015–20021. [Google Scholar] [CrossRef] [Green Version]
- Ramani, K.; Yang, H.P.; Kuhlenkamp, J.; Tomasi, L.; Tsukamoto, H.; Mato, J.M.; Lu, S.C. Changes in methionine adenosyl-transferase and S-adenosylmethionine during hepatic stellate cell activation. Hepatology 2010, 51, 986–995. [Google Scholar]
- Yang, H.; Ara, A.I.; Magilnick, N.; Xia, M.; Ramani, K. Expression pattern, regulation and function of methionine adenosyl-transferase 2β alternative splicing variants in hepatoma cells. Gastroenterology 2008, 134, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Li, T.W.; Yang, H.; Moyer, M.P.; Mato, J.M.; Lu, S.C. Methionine, adenosyltransferase 2B-GIT1 complex serves as scaffold to regulate Ras/Rafi/mek1/2 activity in human liver and colon cancer cells. Am. J. Pathol. 2015, 185, 1135–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, B.H.; Neithardt, A.; Chu, D.B.; Shah, F.B.; Catt, K.J. Role of EGF receptor transactivation in phosphoinositide 3-kinase-dependent activation of MAP kinase by GPCRs. J. Cell. Physiol. 2006, 206, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Chantar, M.L.; Vázquez-Chantada, M.; Garnacho, M.; Varela-Rey, M.; Dotor, J.; Santamaria, M.; Martínez-Cruz, L.A.; Parada, L.A.; Lu, S.C.; Mato, J.M. S-adenosylmethionine regulates cytoplasmic HuR via AMP-activated kinase. Gastroenterology 2006, 131, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Santos, L.; Vazquez-Chantada, M.; Mato, J.M.; Martinez-Chantar, M.L. SAMe and HuR in liver physiology: Useful-ness of stem cells in hepatic differentiation research. Methods Mol. Biol. 2012, 826, 133–149. [Google Scholar]
- Peng, H.; Dara, L.; Li, T.W.; Zheng, Y.; Yang, H.; Tomasi, M.L.; Tomasi, I.; Giordano, P.; Mato, J.M.; Lu, S.C. MAT2B-GIT1 interplay activates MEK1/ERK 1 and 2 to induce growth in human liver and colon cancer. Hepatology 2013, 57, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.M.; Steitz, J.A. HuR and mRNA stability. Cell. Mol. Life Sci. 2001, 58, 266–277. [Google Scholar] [CrossRef]
- Wang, A.; Bao, Y.; Wu, Z.; Zhao, T.; Wang, D.; Shi, J.; Liu, B.; Sun, S.; Yang, F.; Wang, L.; et al. Long noncoding RNA EGFR-AS1 promotes cell growth and metastasis via affecting HuR mediated mRNA stability of EGFR in renal cancer. Cell Death Dis. 2019, 10, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papatheofani, V.; Levidou, G.; Sarantis, P.; Koustas, E.; Karamouzis, M.V.; Pergaris, A.; Kouraklis, G.; Theocharis, S. HuR Protein in Hepatocellular Carcinoma: Implications in Development, Prognosis and Treatment. Biomedicines 2021, 9, 119. [Google Scholar] [CrossRef]
- García-Román, R.; Salazar-González, D.; Rosas, S.; Arellanes-Robledo, J.; Beltrán-Ramírez, O.; Fattel-Fazenda, S.; Vil-la-Treviño, S. The differential NF-kB modulation by S-adenosyl-L-methionine, N-acetylcysteine and quercetin on the promo-tion stage of chemical hepatocarcinogenesis. Free Radic. Res. 2008, 42, 331–343. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Pinna, F.; Pellegrino, R.; Sanna, V.; Sini, M.; Daino, L.; Simile, M.M.; De Miglio, M.R.; Frau, M.; Tomasi, M.L.; et al. Ras-driven proliferation and apoptosis signaling during rat liver carcinogenesis is under genetic control. Int. J. Cancer 2008, 123, 2057–2064. [Google Scholar] [CrossRef] [PubMed]
- Frau, M.; Simile, M.M.; Tomasi, M.L.; Demartis, M.I.; Daino, L.; Brozzetti, S.; Feo, C.F.; Massarelli, G.; Solinas, G.; Feo, F.; et al. An expression signature of phenotypic resistance to hepatocellular carcinoma identified by cross-species gene expression analysis. Cell. Oncol. 2012, 35, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichhorn, P.J.; Creyghton, M.P.; Bernards, R. Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys. Acta 2009, 1795, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Martínez-López, N.; Varela-Rey, M.; Fernández-Ramos, D.; Woodhoo, A.; Vázquez-Chantada, M.; Embade, N.; Espi-nosa-Hevia, L.; Bustamante, F.J.; Parada, L.A.; Rodriguez, M.S.; et al. Activation of LKB1-Akt pathway independent of PI3 Kinase plays a critical role in the proliferation of hepatocellular carcinoma from NASH. Hepatology 2010, 52, 1621–1631. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Cho, M.E.; Li, T.W.; Peng, H.; Ko, K.S.; Mato, J.M.; Lu, S.C. MicroRNAs regulate methionine adenosyltransferase 1A expression in hepatocellular carcinoma. J. Clin. Investig. 2013, 123, 285–298. [Google Scholar] [CrossRef]
- Ez-santodLo, T.; Tsai, W.C.; Chen, S.T. MicroRNA-21–3p, a berberine-induced miRNA, directly down-regulates human methionine adenosyltransferases 2A and 2B and inhibits hepatoma cell growth. PLoS ONE 2013, 8, e75628. [Google Scholar]
- Simile, M.M.; Peitta, G.; Tomasi, K.L.; Brozzetti, S.; Feo, C.F.; Porcu, A.; Cigliano, A.; Calvisi, D.F.; Feo, F.; Pascale, R.M. Mi-croRNA-203 impacts on the growth, aggressiveness and prognosis of hepatocellular carcinoma by targeting MAT2A and MAT2B genes. Oncotarget 2019, 10, 2835–2854. [Google Scholar] [CrossRef] [Green Version]
- Stramentinoli, G.; Gualano, M.; Ideo, G. Protective role of S-adenosyl-L-methionine in liver injury induced by D-galactosamine in rats. Biochem. Pharmacol. 1978, 27, 1431–1433. [Google Scholar] [CrossRef]
- McMillan, J.M.; McMillan, D.C. S-adenosylmethionine but not glutathione protects against galactosamine-induced cytotoxi-city in rat hepatocyte cultures. Toxicology 2006, 222, 175–184. [Google Scholar] [CrossRef]
- Stramentinoli, G.; Pezzoli, C.; Galli-Kienle, M. Protective role of S-adenosyl-L-methionine against acetominophen-induced mortality and hepato-toxicity in mice. Biochem. Pharmacol. 1979, 28, 1567–1571. [Google Scholar] [CrossRef]
- Gong, Z.; Yan, S.; Zhang, P.; Huang, Y.; Wang, L. Effects of S-adenosylmethionine on liver methionine metabolism and steatosis with ethanol-induced liver injury in rats. Hepatol. Int. 2008, 2, 346–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Ruiz, C.; Morales, A.; Colell, A.; Ballesta, A.; Rodes, J.; Kaplowitz, N.; Fernández-Checa, J.C. Feeding S-adenosyl-L-methionine attenuates both ethanol-induced depletion of mitochondrial glutathione and mitochondrial dys-function in periportal and perivenous rat hepatocytes. Hepatology 1995, 21, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; García-Ruiz, C.; Morales, A.; Ballesta, A.; Ookhtens, M.; Rodés, J.; Kaplowitz, N.; Fernández-Checa, J.C. Transport of reduced glutathione in hepatic mitochondria and mitoplasts from ethanol-treated rats: Effect of membrane physical properties and S-adenosyl-L-methionine. Hepatology 1997, 26, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Barak, A.J.; Beckenhauer, H.C.; Mailliard, M.E.; Kharbanda, K.K.; Tuma, D.J. Betaine lowers elevated s-adenosylhomocysteine levels in hepatocytes from ethanol-fed rats. J. Nutr. 2003, 133, 2845–2848. [Google Scholar] [CrossRef]
- Casini, A.; Banchetti, E.; Milani, S.; Maggioni Moratti, E.; Surrenti, C. S-adenosylmethionine inhibits collagen synthesis by human fibroblasts in vitro. Methods Find. Exp. Clin. Pharmacol. 1989, 11, 331–334. [Google Scholar]
- Kharbanda, K.K.; Rogers, D.D., 2nd; Mailliard, M.E.; Siford, G.L.; Barak, A.J.; Beckenhauer, H.C.; Sorrell, M.F.; Tuma, D.J. A comparison of the effects of betaine and S-adenosylmethionine on ethanol-induced changes in methionine metabolism and steatosis in rat hepatocytes. J. Nutr. 2005, 135, 519–524. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Zhou, Z.; Chen, T.; Hill, D.; Kang, J.; Barve, S.; McClain, C. S-adenosyl-methionine (SAMe) protects against acute alcohol induced hepatotoxicity in mice. J. Nutr. Biochem. 2003, 14, 591–597. [Google Scholar] [CrossRef]
- Lieber, C.S.; Casini, A.; DeCarli, L.M.; Kim, C.I.; Lowe, N.; Sasaki, R.; Leo, M.A. S-adenosyl-L-methionine attenuates alco-hol-induced liver injury in the baboon. Hepatology 1990, 11, 165–172. [Google Scholar] [CrossRef]
- Lieber, C.S.; Leo, M.A.; Cao, Q.; Mak, K.M.; Ren, C.; Ponomarenko, A.; Wang, X.; Decarli, L.M. The Combination of S-adenosylmethionine and dilinoleoylpho-sphatidylcholine attenuates non-alcoholic steatohepatitis produced in rats by a high-fat diet. Nutr. Res. 2007, 27, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [Green Version]
- Hartz, C.S.; Schalinske, K.L. Phosphatidylethanolamine N-methyltransferase and regulation of homocysteine. Nutr. Rev. 2006, 64, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, F.; Smith, T.K. The Kennedy pathway—De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef]
- Pascale, R.; Pirisi, L.; Daino, L.; Zanetti, S.; Satta, A.; Bartoli, E.; Feo, F. Role of phosphatidylethanolamine methylation in the synthesis of phosphatidylcholine by hepatocytes isolated from choline-deficient rats. FEBS Lett. 1982, 145, 293–297. [Google Scholar] [CrossRef] [Green Version]
- Frau, M.; Feo, C.F.; Feo, F.; Pascale, R.M. New insights on the role of epigenetic alterations in hepatocellular carcinoma. J. Hepatocell. Carcinoma 2014, 1, 65–83. [Google Scholar] [PubMed] [Green Version]
- Garcea, R.; Daino, L.; Pascale, R.M.; Simile, M.M.; Puddu, M.; Frassetto, S.; Frassetto, S.; Cozzolino, P.; Seddaiu, M.A.; Gaspa, L.; et al. Inhibition of promotion and persistent nodule growth by S-adenosyl-L-methionine in rat liver carcinogenesis: Role of remodeling and apoptosis. Cancer Res. 1989, 49, 1850–1856. [Google Scholar] [PubMed]
- Calvisi, F.; Simile, M.M.; Ladu, S.; Pellegrino, R.; De Murtas, V.; Pinna, F.; Tomasi, M.L.; Frau, M.; Virdis, P.; De Miglio, M.R.; et al. Altered methionine metabolism and global DNA methylation in liver cancer: Relationship with genomic instability and prognosis. Int. J. Cancer 2007, 121, 2410–2420. [Google Scholar] [CrossRef] [PubMed]
- Simile, M.M.; Saviozzi, M.; De Miglio, M.R.; Muroni, M.R.; Nufris, A.; Pascale, R.; Malvaldi, G.; Feo, F. Persistent chemopreventive effect of S-adenosyl-L-methionine on the development of liver putative preneoplastic lesions induced by thiobenzamide in diethylnitrosamine-initiated rats. Carcinogenesis 1996, 17, 1533–1537. [Google Scholar] [CrossRef]
- Pascale, R.M.; Simile, M.M.; Satta, G.; Seddaiu, M.A.; Daino, L.; Pinna, G.; Vinci, M.A.; Gaspa, L.; Feo, F. Comparative effects of L-methionine, S-adenosyl-L-methionine and 5′-methylthioadenosine on the growth of preneoplastic lesions and DNA methylation in rat liver during the early stages of hepatocarcinogenesis. Anticancer Res. 1991, 11, 1617–1624. [Google Scholar]
- Pascale, R.M.; Marras, V.; Simile, M.M.; Daino, L.; Pinna, G.; Bennati, S.; Carta, M.; Seddaiu, M.A.; Massarelli, G.; Feo, F. Chemoprevention of rat liver carcinogenesis by S-adenosyl-L-methionine: A long-term study. Cancer Res. 1992, 52, 4979–4986. [Google Scholar]
- Gerbracht, U.; Eigenbrodt, E.; Simile, M.M.; Pascale, R.M.; Gaspa, L.; Daino, L.; Seddaiu, M.A.; De Miglio, M.R.; Nufris, A.; Feo, F. Effect of S-adenosyl-L-methionine on the development of preneoplastic foci and the activity of some carbohydrate metabolizing enzymes in the liver, during experimental hepatocarcinogenesis. Anticancer Res. 1993, 13, 1965–1972. [Google Scholar]
- Lu, S.C.; Ramani, K.; Ou, X.; Lin, M.; Yu, V.; Ko, K.; Park, R.; Bottiglieri, T.; Tsukamoto, H.; Kanel, G.; et al. S-adenosylmethionine in the chemoprevention and treatment of hepatocellular carcinoma in a rat model. Hepatology 2009, 50, 462–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.W.; Yang, H.; Peng, H.; Xia, M.; Mato, J.M.; Lu, S.C. Effects of S-adenosylmethionine and methylthioadenosine on in-flammation-induced colon cancer in mice. Carcinogenesis 2012, 33, 427–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Xia, M.; Lin, M.; Yang, H.; Kuhlenkamp, J.; Li, T.; Sodir, N.M.; Chen, Y.H.; Josef-Lenz, H.; Laird, P.W.; et al. Role of methionine adenosyltransferase 2A and S-adenosylmethionine in mitogen-induced growth of human colon cancer cells. Gastroenterology 2007, 133, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Garcea, R.; Daino, L.; Pascale, R.; Simile, M.M.; Puddu, M.; Ruggiu, M.E.; Seddaiu, M.A.; Satta, G.; Sequenza, M.J.; Feo, F. Pro-tooncogene methylation and expression in regenerating liver and preneoplastic liver nodules induced in the rat by diethyl-nitrosamine: Effect of variations of S-adenosylmethionine:S-adenosylhomocysteine ratio. Carcinogenesis 1989, 10, 1183–1192. [Google Scholar] [CrossRef]
- Pascale, R.M.; Simile, M.M.; De Miglio, M.R.; Nufris, A.; Daino, L.; Seddaiu, M.A.; Rao, P.M.; Rajalakshmi, S.; Sarma, D.S.; Feo, F. Chemoprevention by S-adenosyl-L-methionine of rat liver carcinogenesis initiated by 1,2-dimethylhydrazine and promoted by orotic acid. Carcinogenesis 1995, 16, 427–430. [Google Scholar] [CrossRef]
- Simile, M.M.; Banni, S.; Angioni, E.; Carta, G.; De Miglio, M.R.; Muroni, M.R.; Calvisi, D.F.; Carru, A.; Pascale, R.M.; Feo, F. 5′-Methylthioadenosine administration prevents lipid peroxidation and fibrogenesis induced in rat liver by car-bon-tetrachloride intoxication. J. Hepatol. 2001, 34, 386–394. [Google Scholar] [CrossRef]
- Li, J.; Ramani, K.; Sun, Z.; Zee, C.; Grant, E.G.; Yang, H.; Xia, M.; Oh, P.; Ko, K.; Mato, J.M.; et al. Forced expression of methionine adenosyltransferase 1A in human hepatoma cells suppresses in vivo tumorigenicity in mice. Am. J. Pathol. 2010, 176, 2456–2466. [Google Scholar] [CrossRef]
- Mato, J.M.; Alvarez, L.; Ortiz, P.; Mingorance, J.; Durán, C.; Pajares, M.A. S-adenosyl-L-methionine synthetase and methionine metabolism deficiencies in cirrhosis. Adv. Exp. Med. Biol. 1994, 368, 113–117. [Google Scholar]
- Sekowska, A.; Ashida, H.; Danchin, A. Revisiting the methionine salvage pathway and its paralogues. Microb. Biotechnol. 2019, 12, 77–97. [Google Scholar] [CrossRef]
- Pascale, R.M.; Simile, M.M.; Gaspa, L.; Daino, L.; Seddaiu, M.A.; Pinna, G.; Carta, M.; Zolo, P.; Feo, F. Alterations of ornithine decarboxylase gene during the progression of rat liver carcinogenesis. Carcinogenesis 1993, 14, 1077–1080. [Google Scholar] [CrossRef]
- Tomasi, M.L.; Ramani, K.; Lopitz-Otsoa, F.; Rodriguez, M.S.; Li, T.W.; Ko, K.; Yang, H.; Bardag-Gorce, F.; Iglesias-Ara, A.; Feo, F.; et al. S-adenosylmethionine regulates dual-specificity mitogen-activated protein kinase phosphatase 1 expression in mouse and human hepatocytes. Hepatology 2010, 51, 2152–2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvisi, D.F.; Pinna, F.; Meloni, F.; Ladu, S.; Pellegrino, R.; Sini, M.; Daino, L.; Simile, M.M.; De Miglio, M.R.; Virdis, P.; et al. Dual specificity phosphatase 1 ubiquitination in extracellular signal-regulated kinase mediated control of growth in human hepatocellular carcinoma. Cancer Res. 2008, 68, 4192–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvisi, D.F.; Pinna, F.; Ladu, S.; Pellegrino, R.; Simile, M.M.; Frau, M.; De Miglio, M.R.; Tomasi, M.L.; Sanna, V.; Muroni, M.R.; et al. Forkhead box M1B is a determinant of rat susceptibility to hepatocarcinogenesis and sustains ERK activity in human HCC. Gut 2009, 58, 679–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, L.; Mo, P.; Huang, W.; Zhang, L.; Wang, Y.; Zhu, H.; Tian, D.; Liu, J.; Chen, Z.; Zhang, Y.; et al. The TNF-a/ROS/HIF-1-induced upregulation of FoxMI expression promotes HCC proliferation and resistance to apoptosis. Carcinogenesis 2012, 3, 2250–2259. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Liu, L.; Zhao, Y.; Zhang, J.; Wang, D.; Chen, J.; He, Y.; Wu, J.; Zhang, Z.; Liu, Z. Hypoxia induces genomic DNA de-methylation through the activation of HIF-1α and transcriptional upregulation of MAT2A in hepatoma cells. Mol. Cancer Ther. 2011, 10, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Mazhar, S.; Taylor, S.E.; Sangodkar, J.; Narla, G. Different pathologic conditions leading to a decrease of the SAM cellular content are antagonized by the administration of exogenous SAM. Biochim. Biophys. Acta 2019, 1866, 51–63. [Google Scholar] [CrossRef]
- Zubiete-Franco, I.; García-Rodríguez, J.L.; Martínez-Uña, M.; Martínez-Lopez, N.; Woodhoo, A.; Juan, V.G.; Beraza, N.; Lage-Medina, S.; Andrade, F.; Fernandez, M.L.; et al. Methionine and S-adenosylmethionine levels are critical regulators of PP2A activity modulating lipophagy during steatosis. J. Hepatol. 2016, 64, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Millward, T.A.; Zolnierowicz, S.; Hemmings, B.A. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci. 1999, 24, 186–191. [Google Scholar] [CrossRef]
- Tomasi, M.L.; Iglesias-Ara, A.; Yang, H.; Ramani, K.; Feo, F.; Pascale, M.R.; Martínez-Chantar, M.L.; Mato, J.M.; Lu, S.C. S-adenosylmethionine regulates apurinic/apyrimidinic endonuclease 1 stability: Implication in hepatocarcinogenesis. Gastroenterology 2009, 136, 1025–1036. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Cheng, H.; Jiang, Q.; Li, H.; Wu, Z. APEX1 is a novel diagnostic and prognostic biomarker for hepatocellular carcinoma. Aging 2020, 12, 4573–4591. [Google Scholar] [CrossRef]
- Sadek, K.M.; Lebda, M.A.; Nasr, N.E.; Nasr, S.M.; El-Sayed, Y. Role of lncRNAs as prognostic markers of hepatic cancer and potential therapeutic targeting by S-adenosylmethionine via inhibiting PI3K/Akt signaling pathways. Environ. Sci. Pollut. Res. Int. 2018, 25, 20057–20070. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer. 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvisi, D.F.; Pinna, F.; Ladu, S.; Pellegrino, R.; Muroni, M.R.; Simile, M.M.; Frau, M.; Tomasi, M.L.; De Miglio, M.R.; Seddaiu, M.A.; et al. Aberrant iNOS signalling is under genetic control in rodent liver cancer and potentially prognostic for human disease. Carcinogenesis 2008, 29, 1639–1647. [Google Scholar] [CrossRef] [Green Version]
- Hezel, A.F.; Bardeesy, N. LKB1: Linking cell structure and tumor suppression. Oncogene 2008, 27, 6908–6919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-Chantada, M.; Ariz, U.; Varela-Rey, M.; Embade, N.; Martínez-Lopez, N.; Fernández-Ramos, D.; Gómez-Santos, L.; Lamas, S.; Lu, S.C.; Martínez-Chantar, M.L.; et al. Evidence for LKB1/AMP-activated protein kinase/endothelial nitric oxide synthase cascade regulated by hepatocyte growth factor, S-adenosylmethionine, and nitric oxide in hepatocyte proliferation. Hepatology 2009, 49, 608–617. [Google Scholar] [CrossRef]
- Corrales, F.; Giménez, A.; Alvarez, L.; Caballeria, J.; Pajares, M.A.; Andreu, H.; Parés, A.; Mato, J.M.; Rodés, J. S-adenosylmethionine treatment prevents carbon tetrachloride-induced S-adenosylmethionine synthetase inactivation and attenuates liver injury. Hepatology 1992, 16, 1022–1027. [Google Scholar] [CrossRef] [Green Version]
- Majano, P.L.; García-Monzón, C.; García-Trevijano, E.R.; Corrales, F.J.; Cámara, J.; Ortiz, P.; Mato, J.M.; Avila, M.A.; Moreno-Otero, R. S-Adenosylmethionine modulates inducible nitric oxide synthase gene expression in rat liver and isolated hepatocytes. J. Hepatol. 2001, 35, 692–699. [Google Scholar] [CrossRef]
- García-Trevijano, E.R.; Martínez-Chantar, M.L.; Latasa, M.U.; Mato, J.M.; Avila, M.A. NO sensitizes rat hepatocytes to proliferation by modifying S-adenosylmethionine levels. Gastroenterology 2002, 122, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Latasa, M.U.; Gil-Puig, C.; Fernández-Barrena, M.G.; Rodríguez-Ortigosa, C.M.; Banales, J.M.; Urtasun, R.; Goñi, S.; Méndez, M.; Arcelus, S.; Juanarena, N.; et al. Oral methylthioadenosine administration attenuates fibrosis and chronic liver disease progression in Mdr2−/− mice. PLoS ONE 2010, 5, e15690. [Google Scholar] [CrossRef]
- Zhao, R.X.; Xu, Z.X. Targeting the LKB1 tumor suppressor. Curr. Drug Targets 2014, 15, 32–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.W.; Wong, L.L.; Tse, E.Y.; Liu, H.F.; Leong, V.Y.; Lee, J.M.; Hardie, D.G.; Ng, I.O.; Ching, Y.P. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012, 72, 4394–4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvisi, D.F.; Frau, M.; Tomasi, M.L.; Feo, F.; Pascale, R.M. Deregulation of signaling pathways in prognostic subtypes of hepatocellular carcinoma: Novel insights from interspecies comparison. Biochim. Biophys. Acta 2012, 826, 215–237. [Google Scholar]
- Hevia, H.; Varela-Rey, M.; Corrales, F.J.; Berasain, C.; Martínez-Chantar, M.L.; Latasa, M.U.; Lu, S.C.; Mato, J.M.; García-Trevijano, E.R.; Avila, M.A. 5′-methylthioadenosine modulates the inflammatory response to endotoxin in mice and in rat hepatocytes. Hepatology 2004, 39, 1088–10998. [Google Scholar] [CrossRef] [PubMed]
- Andreu-Pérez, P.; Esteve-Puig, R.; de Torre-Minguela, C.; López-Fauqued, M.; Bech-Serra, J.J.; Tenbaum, S.; García-Trevijano, E.R.; Canals, F.; Merlino, G.; Avila, M.A.; et al. Protein arginine methyltransferase 5 regulates ERK1/2 signal transduction amplitude and cell fate through CRAF. Sci. Signal. 2011, 4, ra58. [Google Scholar] [CrossRef] [PubMed]
- Cebrian, A.; Pharoah, P.D.; Ahmed, S.; Ropero, S.; Fraga, M.F.; Smith, P.L.; Conroy, D.; Luben, R.; Perkins, B.; Easton, D.F.; et al. Genetic variants in epigenetic genes and breast cancer risk. Carcinogenesis 2006, 27, 1661–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wettergren, Y.; Odin, E.; Carlsson, G.; Gustavsson, B. MTHFR, MTR, and MTRR polymorphisms in relation to p16INK4A hypermethylation in mucosa of patients with colorectal cancer. Mol. Med. 2010, 16, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef]
- Mato, J.M.; Camara, J.; Fernandez de Paz, J.; Caballeria, L.; Coll, S.; Caballero, A.; García-Buey, L.; Beltrán, J.; Benita, V.; Caballería, J.; et al. S-adenosylmethionine in alcoholic liver cirrhosis: A randomized, placebo-controlled, double-blind, multi-center clinical trial. J. Hepatol. 1999, 30, 1081–1089. [Google Scholar] [CrossRef]
- Diaz Belmont, A.; Dominguez Henkel, R.; Uribe Ancira, F. Parenteral S-adenosylmethionine compared to placebos in the treatment of alcoholic liver diseases. An. Med. Interna. 1996, 13, 9–15. [Google Scholar]
- Stepuro, I.; Solodunov, A.A.; Solodunov, T.P.; Iaroshevich, N.A.; IuM, O. Distribution of pyridox-al-5-phosphate between proteins and low molecular weight components of plasma: Effect of ENG. Ukr. Biokhimicheskii Zhurnal 1988, 60, 34–41. [Google Scholar]
- Loguercio, C.; Nardi, G.; Argenzio, F.; Aurilio, C.; Petrone, E.; Grella, A.; Del Vecchio Blanco, C.; Coltorti, M. Effect of S-adenosyl-L-methionine administration on red blood cell cysteine and glutathione levels. Alcohol 1994, 29, 597–604. [Google Scholar]
- Vendemiale, G.; Altomare, E.; Trizio, T.; Le Grazie, C.; Di Padova, C.; Salerno, M.T.; Carrieri, V.; Albano, O. Effects of oral S-adenosyl-L-methionine on hepatic glutathione in patients with liver disease. Scand. J. Gastroenterol. 1989, 24, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Halsted, C.H.; Medici, V. Vitamin-dependent methionine metabolism and alcoholic liver disease. Adv. Nutr. 2011, 2, 421–427. [Google Scholar] [CrossRef]
- Lumeng, L. The role of acetaldehyde in mediating the deleterious effect of ethanol on pyridoxal 5′-phosphate metabolism. J. Clin. Investig. 1978, 62, 86–293. [Google Scholar] [CrossRef] [Green Version]
- Medici, V.; Virata, M.C.; Peerson, J.M.; Stabler, S.P.; French, S.W.; Gregory, J.F., 3rd; Albanese, A.; Bowlus, C.L.; Devaraj, S.; Panacek, E.A.; et al. S-adenosyl-L-methionine treatment for alcoholic liver disease: A double-blinded, randomized, placebo-controlled trial. Alcohol Clin. Exp. Res. 2011, 35, 1960–1965. [Google Scholar] [CrossRef] [Green Version]
- Rambaldi, A.; Gluud, C. S-adenosyl-L-methionine for alcoholic liver diseases. Cochrane Database Syst. Rev. 2016, 2, CD002235. [Google Scholar]
- Dietrich, P.; Hellerbrand, C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 637–653. [Google Scholar] [CrossRef]
- Sanyal, A.J. AGA technical review on nonalcoholic fatty liver disease. Gastroenterology 2002, 123, 705–1725. [Google Scholar] [CrossRef]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef]
- Dorna, W.; Lagente, V. Intestinally derived bacterial products stimulate development of nonalcoholic steatohepatitis. Pharmacol. Res. 2019, 141, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Higarza, S.G.; Arboleya, S.; Gueimonde, M.; Gómez-Lázaro, E.; Arias, J.L.; Arias, N. Neurobehavioral dysfunction in non-alcoholic steatohepatitis is associated with hyperammonemia, gut dysbiosis, and metabolic and functional brain re-gional deficits. PLoS ONE 2019, 14, e0223019. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Cooksley, W.G.; Hanson, R.; Searle, J.; Halliday, J.W.; Powell, L.W. The natural history of nonalcoholic steato-hepatitis: A follow-up study of forty-two patients for up to 21 years. Hepatology 1990, 11, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Suzuki, H.; Sakaguchi, S. Common pathogenic mechanism in development progression of liver injury caused by non-alcoholic or alcoholic steatohepatitis. J. Toxicol. Sci. 2007, 32, 453–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Concas, D.; Kudo, H.; Levene, A.; Pollard, J.; Charlton, P.; Thomas, H.C.; Thursz, M.R.; Goldin, R.D. Impact of pan-caspase inhibition in animal models of established steatosis and non-alcoholic steatohepatitis. J. Hepatol. 2010, 53, 542–550. [Google Scholar] [CrossRef]
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.Z.; Li, Y.T.; Wu, W.R.; Shi, D.; Fang, D.Q.; Yang, L.Y.; Bian, X.Y.; Wu, J.J.; Wang, Q.; Jiang, X.W.; et al. Dynamic alterations in the gut microbiota and metabolome during the development of methionine-choline-deficient diet-induced nonalcoholic steatohepatitis. World J. Gastroenterol. 2018, 24, 2468–2481. [Google Scholar] [CrossRef]
- Farrell, G.C.; Larter, C.Z.; Hou, J.Y.; Zhang, R.H.; Yeh, M.M.; Williams, J.; dela Pena, A.; Francisco, R.; Osvath, S.R.; Brooling, J.; et al. Apoptosis in experimental NASH is associated with p53 activation and TRAIL receptor expression. J. Gastroenterol. Hepatol. 2009, 24, 443–452. [Google Scholar] [CrossRef]
- Goldin, R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol. 2006, 87, 1–16. [Google Scholar]
- Wortham, M.; He, L.; Gyamfi, M.; Copple, B.L.; Wan, Y.J. The transition from fatty liver to NASH associates with SAMe de-pletion in db/db mice fed a methionine choline-deficient diet. Dig. Dis. Sci. 2008, 53, 2761–2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastre, J.; Serviddio, G.; Pereda, J.; Minana, J.B.; Arduini, A.; Vendemiale, G.; Poli, G.; Pallardo, F.V.; Vina, J. Mitochondrial function in liver disease. Front. Biosci. 2007, 12, 1200–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalhan, S.C.; Edmison, J.; Marczewski, S.; Dasarathy, S.; Gruca, L.L.; Bennett, C.; Duenas, C.; Lopez, R. Methionine and protein metabolism in non-alcoholic steatohepatitis: Evidence for lower rate of transmethylation of methionine. Clin. Sci. 2011, 121, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. NASH CRN. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 16751685. [Google Scholar] [CrossRef] [Green Version]
- Shiasi Arani, K.; Taghavi Ardakani, A.; Moazami Goudarzi, R.; Talari, H.R.; Hami, K.; Akbari, H.; Akbari, N. Effect of Vitamin E and Metformin on Fatty Liver Disease in Obese Children-Randomized Clinical Trial. Iran. J. Public Health 2014, 43, 1417–1423. [Google Scholar]
- Akcam, M.; Boyaci, A.; Pirgon, O.; Kaya, S.; Uysal, S.; Dundar, B.N. Therapeutic effect of metformin and vitamin E versus prescriptive diet in obese adolescents with fatty liver. Int. J. Vitam. Nutr. Res. 2011, 1, 398–406. [Google Scholar] [CrossRef]
- Bakir, M.B.; Salama, M.A.; Refaat, R.; Ali, M.A.; Khalifa, E.A.; Kamel, M.A. Evaluating the therapeutic potential of one-carbon donors in nonalcoholic fatty liver disease. Eur. J. Pharmacol. 2019, 847, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Cordero, P.; Gomez-Uriz, A.M.; Campion, J.; Milagro, F.I.; Martinez, J.A. Dietary supplementation with methyl donors re-duces fatty liver and modifies the fatty acid synthase DNA methylation profile in rats fed an obesogenic diet. Genes Nutr. 2013, 8, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Mahjoubin-Tehran, M.; De Vincentis, A.; Mikhailidis, D.P.; Atkin, S.L.; Mantzoros, C.S.; Jamialahmadi, T.; Sahebkar, A. Non-alcoholic fatty liver disease and steatohepatitis: State of the art on effective therapeutics based on the gold standard method for diagnosis. Mol. Metab. 2021, 50, 101049. [Google Scholar] [CrossRef]
- Shneider, B.L. Progressive intrahepatic cholestasis: Mechanisms, diagnosis and therapy. Pediatr. Transplant. 2004, 8, 609–612. [Google Scholar] [CrossRef]
- Copple, B.L.; Jaeschke, H.; Klaassen, C.D. Oxidative stress and the pathogenesis of cholestasis. Semin. Liver Dis. 2010, 30, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Correa, J.A.; De La Cruz, J.P.; Martin-Aurioles, E.; Lopez-Egea, M.A.; Ortiz, P.; Sanchez de la Cuesta, F. Effects of S-adenosyl-L-methionine on hepatic and renal oxidative stress in an experimental model of acute biliary obstruction in rats. Hepatology 1997, 26, 121–127. [Google Scholar] [PubMed]
- Datsko, V.A.; Fedoniuk, L.Y.; Ivankiv, Y.I.; Kurylo, K.I.; Volska, A.S.; Malanchuk, S.L.; Oleshchuk, O.M. Experimental cirrho-sis: Liver morphology and function. Wiad Lek 2020, 73, 947–952. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, L.; Victor, D.W.; Xin, Y.; Xuan, S. Ursodeoxycholic Acid and S-adenosylmethionine for the Treatment of Intra-hepatic Cholestasis of Pregnancy: A Meta-analysis. Hepat. Mon. 2016, 16, e38558. [Google Scholar] [CrossRef] [Green Version]
- Frezza, M.; Surrenti, C.; Manzillo, G.; Fiaccadori, F.; Bortolini, M.; Di Padova, C. Oral S-adenosylmethionine in the sympto-matic treatment of intrahepatic cholestasis. A double-blind, placebo-controlled study. Gastroenterology 1990, 99, 211–215. [Google Scholar] [CrossRef]
- Coltorti, M.; Bortolini, M.; Di Padova, C. A review of the studies on the clinical use of S-adenosylmethionine (SAMe) for the symptomatic treatment of intrahepatic cholestasis. Methods Find. Exp. Clin. Pharmacol. 1990, 12, 69–78. [Google Scholar]
- Fiorelli, G. S-adenosylmethionine in the treatment of intrahepatic cholestasis of chronic liver disease: A field trial. Curr. Ther. Res. Clin. Exp. 1999, 60, 335–348. [Google Scholar] [CrossRef]
- Duong, F.H.; Filipowicz, M.; Tripodi, M.; La Monica, N.; Heim, M.H. Hepatitis C virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology 2004, 126, 263–277. [Google Scholar] [CrossRef]
- Bernsmeier, C.; Duong, F.H.; Christen, V.; Pugnale, P.; Negro, F.; Terracciano, L.; Heim, M.H. Virus-induced over-expression of protein phosphatase 2A inhibits insulin signalling in chronic hepatitis C. J. Hepatol. 2008, 49, 429–440. [Google Scholar] [CrossRef]
- Filipowicz, M.; Bernsmeier, C.; Terracciano, L.; Duong, F.H.; Heim, M.H. S-adenosyl-methionine and betaine improve early virological response in chronic hepatitis C patients with previous nonresponse. PLoS ONE 2010, 5, e15492. [Google Scholar] [CrossRef]
- Castelli, G.; Pelosi, E.; Testa, U. Liver Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Cancers 2017, 9, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Yang, H.; Fan, W.; Tu, J.; Li, T.W.H.; Wang, J.; Shen, H.; Yang, J.; Xiong, T.; Steggerda, J.; et al. Mechanisms of MAFG Dysregulation in Cholestatic Liver Injury and Development of Liver Cancer. Gastroenterology 2018, 155, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.Q.; Li, H.C.; Teng, F.; Chang, Q.M.; Wu, X.B.; Feng, J.F.; Zhang, Z.P. Long noncoding RNA MAFG-AS1 facilitates the progression of hepatocellular carcinoma via targeting miR-3196/OTX1 axis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 12131–12143. [Google Scholar]
- Invernizzi, P.; Floreani, A.; Carbone, M.; Marzioni, M.; Craxi, A.; Muratori, L.; Vespasiani Gentilucci, U.; Gardini, I.; Gasbar-rini, A.; Kruger, P.; et al. Primary Biliary Cholangitis: Advances in management and treatment of the disease. Dig. Liver Dis. 2017, 49, 841–846. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.B.; Da, M.X.; Yao, J.B.; Duan, Y.X. S-Adenosylmethionine Inhibits Expression of Vascular Endothelial Growth Fac-tor-C Protein and Cellular Proliferation in Gastric Cancer. Sichuan Da Xue Xue Bao Yi Xue Ban 2015, 46, 384–388. [Google Scholar] [PubMed]
- Morgan, T.R.; Osann, K.; Bottiglieri, T.; Pimstone, N.; Hoefs, J.C.; Hu, K.Q.; Hassanein, T.; Boyer, T.D.; Kong, L.; Chen, W.P.; et al. Phase II randomized, controlled trial of S-Adenosylmethionine in reducing serum α-fetoprotein in patients with hepatitis C cirrhosis and elevated AFP. Cancer Prev. Res. 2015, 8, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Ilisso, C.P.; Delle Cave, D.; Mosca, L.; Pagano, M.; Coppola, A.; Mele, L.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine regulates apoptosis and autophagy in MCF-7 breast cancer cells through the modulation of specific microRNAs. Cancer Cell Int. 2018, 18, 197. [Google Scholar] [CrossRef]
- Mahmood, N.; Arakelian, A.; Cheishvili, D.; Szyf, M.; Rabbani, S.A. S-adenosylmethionine in combination with decitabine shows enhanced anti-cancer effects in repressing breast cancer growth and metastasis. J. Cell. Mol. Med. 2020, 24, 10322–10337. [Google Scholar] [CrossRef]
- Ilisso, C.P.; Castellano, M.; Zappavigna, S.; Lombardi, A.; Vitale, G. The methyl donor S-adenosylmethionine potentiates doxorubicin effects on apoptosis of hormone-dependent breast cancer cell lines. Endocrine 2015, 50, 212–222. [Google Scholar] [CrossRef]
- Mosca, L.; Vitiello, F.; Coppola, A.; Borzacchiello, L.; Ilisso, C.P.; Pagano, M.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. Ther-apeutic Potential of the Natural Compound S-Adenosylmethionine as a Chemoprotective Synergistic Agent in Breast, and Head and Neck Cancer Treatment: Current Status of Research. Int. J. Mol. Sci. 2020, 21, 8547. [Google Scholar] [CrossRef]
- Cave, D.D.; Desiderio, V.; Mosca, L.; Ilisso, C.P.; Mele, L.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine-mediated apoptosis is potentiated by autophagy inhibition induced by chloroquine in human breast cancer cells. J. Cell. Physiol. 2018, 233, 1370–1383. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, J.; Tang, J. miR-34a may regulate sensitivity of breast cancer cells to adriamycin via targeting Notch1. Zhonghua Zhong Liu Za Zhi 2014, 36, 892–896. [Google Scholar] [PubMed]
- Shukeir, N.; Pakneshan, P.; Chen, G.; Szyf, M.; Rabbani, S.A. Alteration of the methylation status of tumor-promoting genes decreases prostate cancer cell invasiveness and tumorigenesis in vitro and in vivo. Cancer Res. 2006, 166, 9202–9210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zsigrai, S.; Kalmár, A.; Nagy, Z.B.; Barták, B.K.; Valcz, G.; Szigeti, K.A.; Galamb, O.; Dankó, T.; Sebestyén, A.; Barna, G.; et al. S-Adenosylmethionine Treatment of Colorectal Cancer Cell Lines Alters DNA Methylation, DNA Repair and Tumor Pro-gression-Related Gene Expression. Cells 2020, 9, 1864. [Google Scholar] [CrossRef]
- Parashar, S.; Cheishvili, D.; Arakelian, A.; Hussain, Z.; Tanvir, I.; Khan, H.A.; Szyf, M.; Rabbani, S.A. S-adenosylmethionine blocks osteosarcoma cells proliferation and invasion in vitro and tumor metastasis in vivo: Therapeutic and diagnostic clinical applications. Cancer Med. 2015, 4, 732–744. [Google Scholar] [CrossRef]
- Pascale, R.M.; Simile, M.M.; Peitta, G.; Seddaiu, M.A.; Feo, F.; Calvisi, D.F. Experimental Models to Define the Genetic Pre-disposition to Liver Cancer. Cancers 2019, 11, 1450. [Google Scholar] [CrossRef] [Green Version]
- Manenti, G.; Binelli, G.; Gariboldi, M.; Canzian, F.; De Gregorio, L.; Falvella, F.S.; Dragani, T.A.; Pierotti, M.A. Multiple loci affect genetic predisposition to hepatocarcinogenesis in mice. Genomics 1994, 23, 118–124. [Google Scholar] [CrossRef]
- Lee, J.S.; Chu, I.S.; Mikaelyan, A.; Calvisi, D.F.; Heo, J.; Reddy, J.K.; Thorgeirsson, S.S. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat. Genet. 2004, 36, 1306–1311. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pascale, R.M.; Simile, M.M.; Calvisi, D.F.; Feo, C.F.; Feo, F. S-Adenosylmethionine: From the Discovery of Its Inhibition of Tumorigenesis to Its Use as a Therapeutic Agent. Cells 2022, 11, 409. https://doi.org/10.3390/cells11030409
Pascale RM, Simile MM, Calvisi DF, Feo CF, Feo F. S-Adenosylmethionine: From the Discovery of Its Inhibition of Tumorigenesis to Its Use as a Therapeutic Agent. Cells. 2022; 11(3):409. https://doi.org/10.3390/cells11030409
Chicago/Turabian StylePascale, Rosa M., Maria M. Simile, Diego F. Calvisi, Claudio F. Feo, and Francesco Feo. 2022. "S-Adenosylmethionine: From the Discovery of Its Inhibition of Tumorigenesis to Its Use as a Therapeutic Agent" Cells 11, no. 3: 409. https://doi.org/10.3390/cells11030409
APA StylePascale, R. M., Simile, M. M., Calvisi, D. F., Feo, C. F., & Feo, F. (2022). S-Adenosylmethionine: From the Discovery of Its Inhibition of Tumorigenesis to Its Use as a Therapeutic Agent. Cells, 11(3), 409. https://doi.org/10.3390/cells11030409