
Epiregulin as an Alternative Ligand for Leptin Receptor Alleviates Glucose Intolerance without Change in Obesity


 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Human Tissues
2.3. Animal Studies
- (1)
- Control Lepob mice group, injected with 0.1 mL sterile PBS (n = 7), and
- (2)
- EREG-treated group of Lepob mice (n = 7), injected intraperitoneally with PBS containing EREG (50 ng/g body weight (BW)). EREG was injected every other day for 26 days. Echo-MRI analysis was performed at the beginning and the end of the study. Blood was collected by cardiac puncture at the end of the study.
2.4. Glucose Tolerance Test (GTT)
2.5. EREG and Insulin Tolerance Tests (Single Exposure)
2.6. Body Composition Analysis
2.7. Adipocyte Culture
2.8. Glucose Uptake Assay
2.9. Transient Transfection and GLUT4 Translocation Measurement
2.10. Western Blot
2.11. Immunoprecipitation
2.12. Enzyme-Linked Immunosorbent Assay (ELISA)
2.13. Epiregulin Protein Modeling
2.14. Quartz Crystal Microbalance with Dissipation (QCM-D) Binding Assay
2.15. Statistical Analysis
3. Results
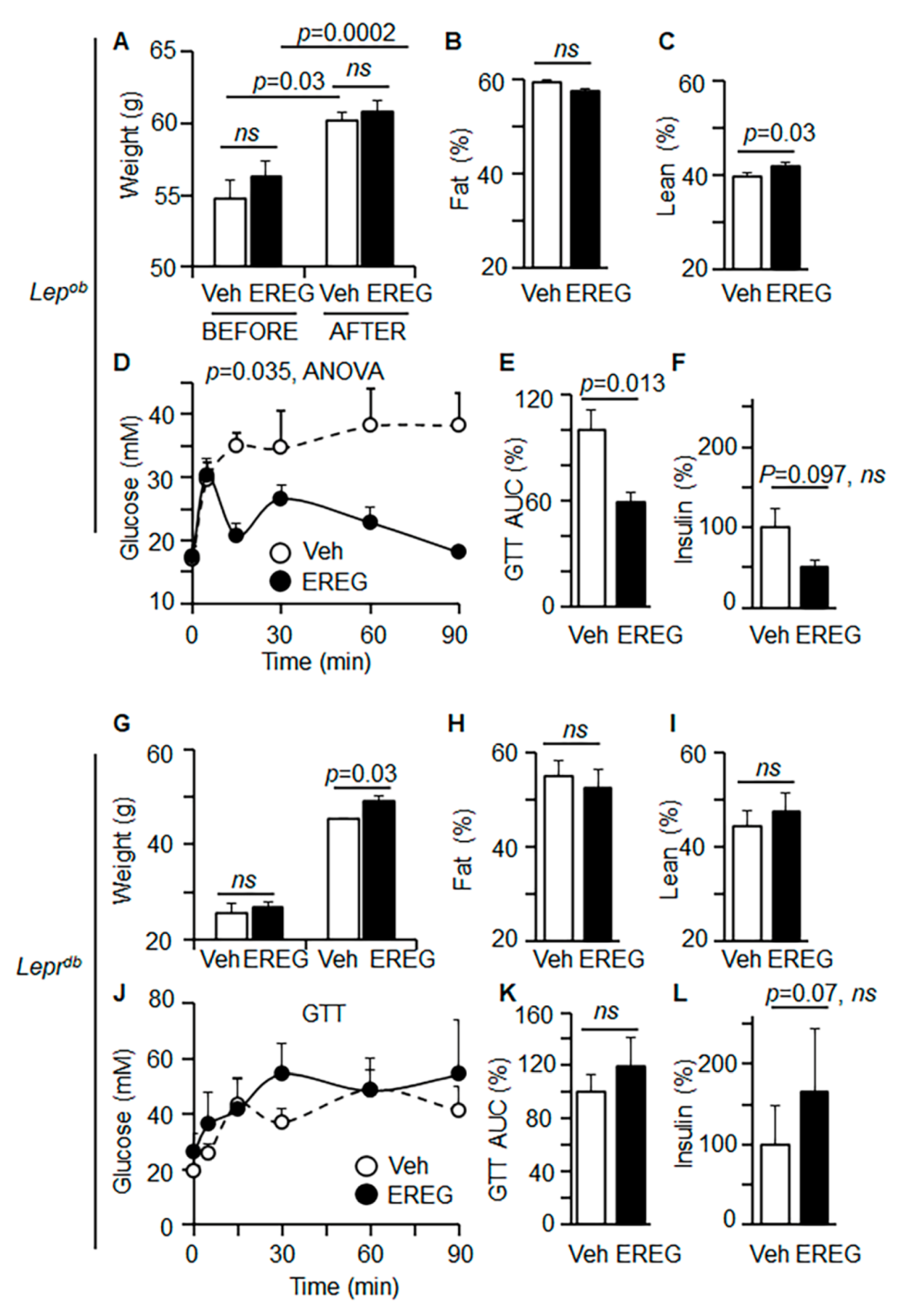
3.1. EREG Regulated Glucose Metabolism under Leptin Deficient Conditions but Required LepR
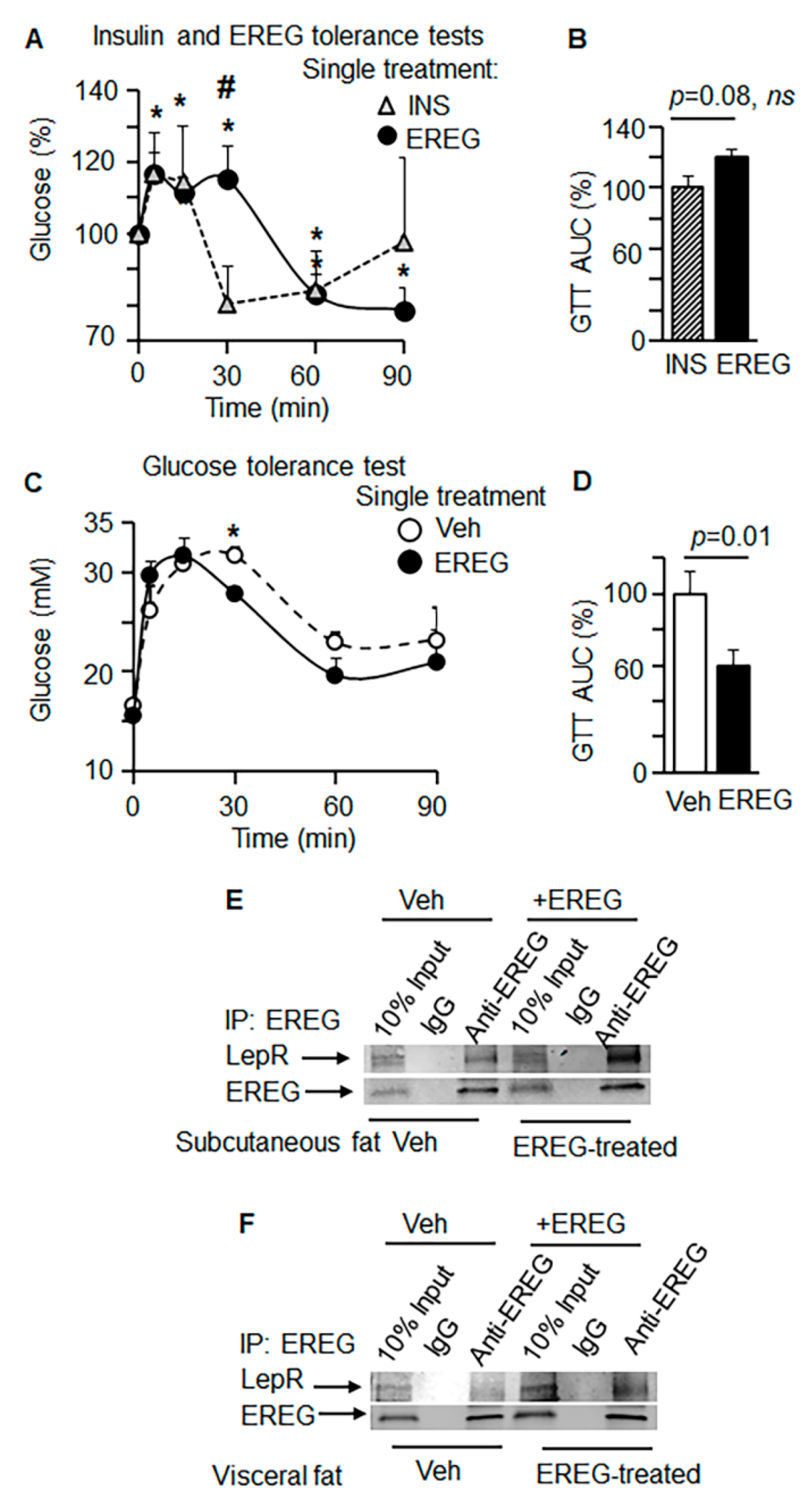
3.2. EREG Binds to LepR
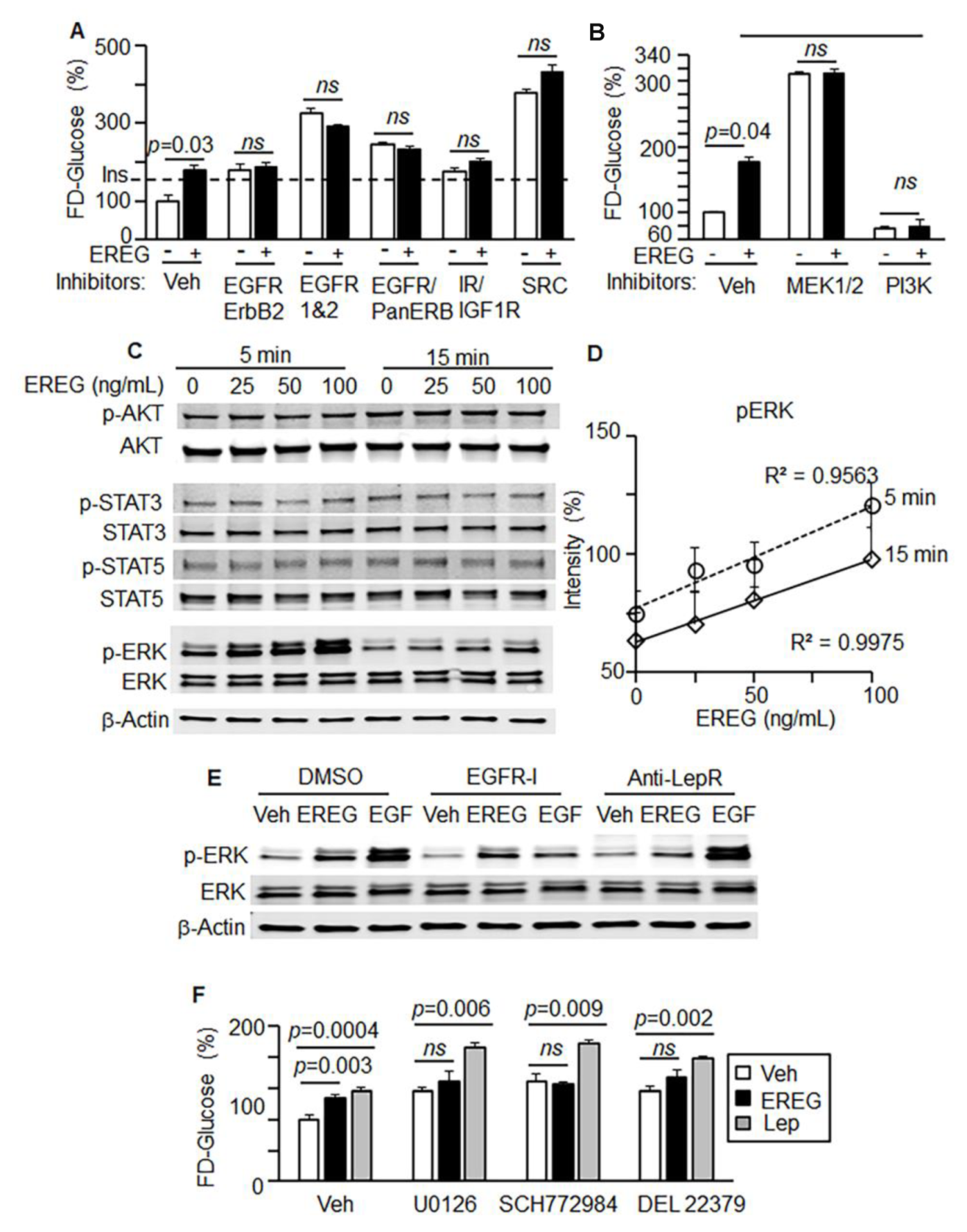
3.3. EREG Stimulation of Glucose Uptake In Vitro Depends on LepR
3.4. EREG Stimulated Glucose Uptake via the ERK/PIK3 Signaling Pathway
3.5. EREG Interact with LepR In Vitro
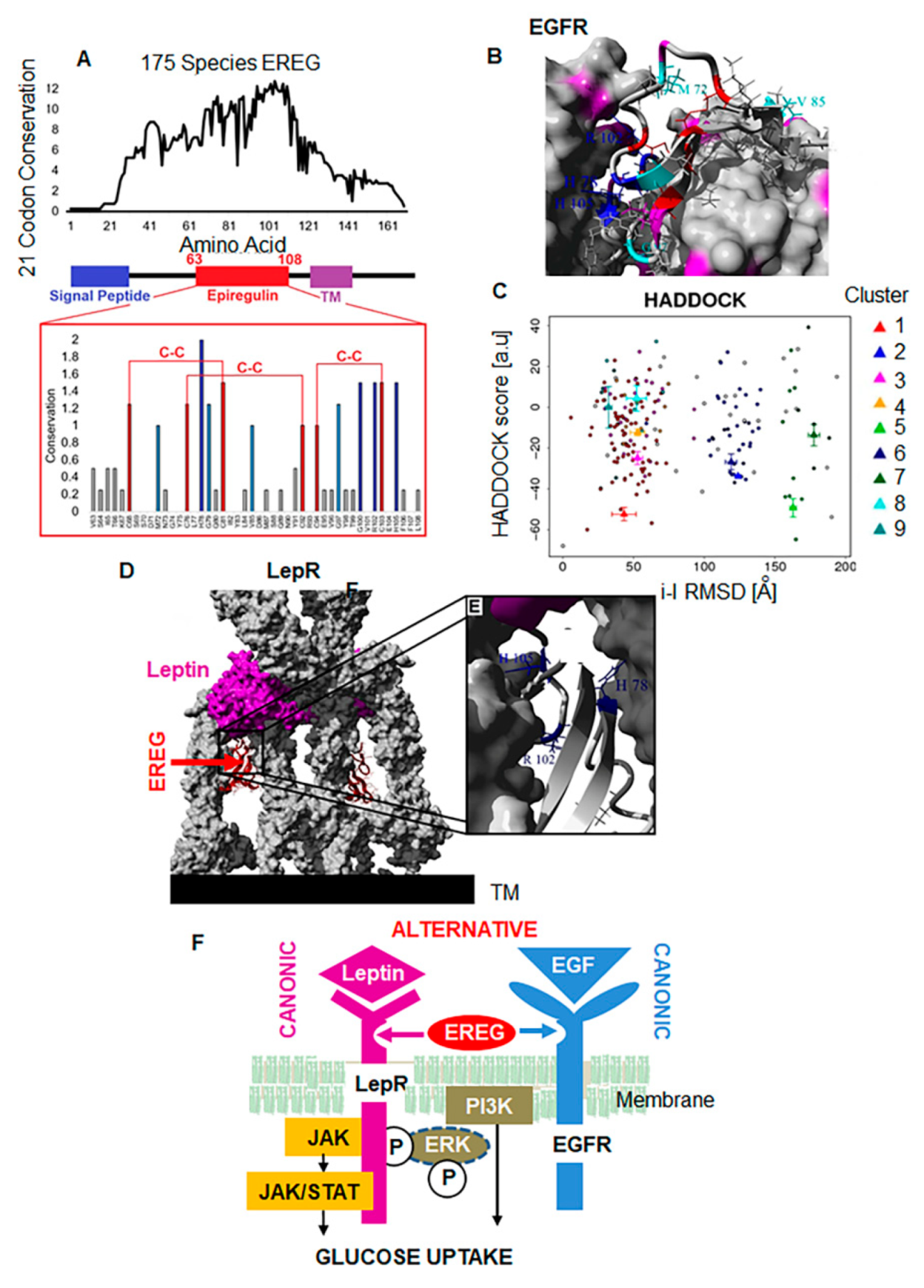
3.6. EREG Evolutionary Evolved as an Alternative Ligand for LepR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fujikawa, T.; Berglund, E.D.; Patel, V.R.; Ramadori, G.; Vianna, C.R.; Vong, L.; Thorel, F.; Chera, S.; Herrera, P.L.; Lowell, B.B.; et al. Leptin Engages a Hypothalamic Neurocircuitry to Permit Survival in the Absence of Insulin. Cell Metab. 2013, 18, 431–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.M. Leptin and the endocrine control of energy balance. Nat. Metab. 2019, 1, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G., Jr.; Leibel, R.L.; Seeley, R.J.; Schwartz, M.W. Obesity and leptin resistance: Distinguishing cause from effect. Trends Endocrinol. Metab. 2010, 21, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Charukeshi Chandrasekera, P.; Pippin, J.J. Leptin- and Leptin Receptor-Deficient Rodent Models: Relevance for Human Type 2 Diabetes. Curr. Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef] [Green Version]
- Londraville, R.L.; Prokop, J.; Duff, R.J.; Liu, Q.; Tuttle, M. On the Molecular Evolution of Leptin, Leptin Receptor, and Endospanin. Front. Endocrinol. 2017, 8, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peelman, F.; Zabeau, L.; Moharana, K.; Savvides, S.N.; Tavernier, J. 20 YEARS OF LEPTIN: Insights into signaling assemblies of the leptin receptor. J. Endocrinol. 2014, 223, T9–T23. [Google Scholar] [CrossRef]
- Procaccini, C.; Lourenco, E.V.; Matarese, G.; La Cava, A. Leptin Signaling: A Key Pathway in Immune Responses. Curr. Signal Transduct. Ther. 2009, 4, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Bjørbæk, C.; Uotani, S.; da Silva, B.; Flier, J.S. Divergent Signaling Capacities of the Long and Short Isoforms of the Leptin Receptor. J. Biol. Chem. 1997, 272, 32686–32695. [Google Scholar] [CrossRef] [Green Version]
- Bates, S.H.; Stearns, W.H.; Dundon, T.A.; Schubert, M.; Tso, A.W.K.; Wang, Y.; Banks, A.; Lavery, H.J.; Haq, A.K.; Maratos-Flier, E.; et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature 2003, 421, 856–859. [Google Scholar] [CrossRef]
- Lee, A.; Sun, Y.; Lin, T.; Song, N.-J.; Mason, M.L.; Leung, J.H.; Kowdley, D.; Wall, J.; Brunetti, A.; Fitzgerald, J.; et al. Amino acid-based compound activates atypical PKC and leptin receptor pathways to improve glycemia and anxiety like behavior in diabetic mice. Biomaterials 2020, 239, 119839. [Google Scholar] [CrossRef]
- Tsutsumi, A.; Motoshima, H.; Kondo, T.; Kawasaki, S.; Matsumura, T.; Hanatani, S.; Igata, M.; Ishii, N.; Kinoshita, H.; Kawashima, J.; et al. Caloric restriction decreases ER stress in liver and adipose tissue in ob/ob mice. Biochem. Biophys. Res. Commun. 2011, 404, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.E.; Jung, Y.; Min, S.; Nam, M.; Heo, R.W.; Jeon, B.T.; Song, D.H.; Yi, C.O.; Jeong, E.A.; Kim, H.; et al. Caloric restriction of db/db mice reverts hepatic steatosis and body weight with divergent hepatic metabolism. Sci. Rep. 2016, 6, 30111. [Google Scholar] [CrossRef] [PubMed]
- Wauman, J.; Zabeau, L.; Tavernier, J. The Leptin Receptor Complex: Heavier Than Expected? Front. Endocrinol. 2017, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabeau, L.; Jensen, C.J.; Seeuws, S.; Venken, K.; Verhee, A.; Catteeuw, D.; van Loo, G.; Chen, H.; Walder, K.; Hollis, J.; et al. Leptin’s metabolic and immune functions can be uncoupled at the ligand/receptor interaction level. Cell Mol. Life Sci. 2015, 72, 629–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabeau, L.; Wauman, J.; Dam, J.; van Lint, S.; Burg, E.; de Geest, J.; Rogge, E.; Silva, A.; Jockers, R.; Tavernier, J. A novel leptin receptor antagonist uncouples leptin’s metabolic and immune functions. Cell. Mol. Life Sci. 2019, 76, 1201–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed, D.M.; Bessman, N.J.; Kiyatkin, A.; Salazar-Cavazos, E.; Byrne, P.O.; Moore, J.O.; Valley, C.C.; Ferguson, K.M.; Leahy, D.J.; Lidke, D.S.; et al. EGFR Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics. Cell 2017, 171, 683–695.e618. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, H.; Komurasaki, T.; Uchida, D.; Takayama, Y.; Isobe, T.; Okuyama, T.; Hanada, K. Epiregulin: A novel epidermal growth factor with mitogenic activity for rat primary hepatocytes. J. Biol. Chem. 1995, 270, 7495–7500. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Nakamura, T.; Mizuguchi, M.; Miura, K.; Tada, M.; Aizawa, T.; Gomi, T.; Miyamoto, K.; Kawano, K. Solution structure of epiregulin and the effect of its C-terminal domain for receptor binding affinity. FEBS Lett. 2003, 553, 232–238. [Google Scholar] [CrossRef]
- Chen, J.; Zeng, F.; Forrester, S.J.; Eguchi, S.; Zhang, M.Z.; Harris, R.C. Expression and Function of the Epidermal Growth Factor Receptor in Physiology and Disease. Physiol. Rev. 2016, 96, 1025–1069. [Google Scholar] [CrossRef]
- Sunaga, N.; Kaira, K. Epiregulin as a therapeutic target in non-small-cell lung cancer. Lung Cancer (Auckl) 2015, 6, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Jing, C.; Jin, Y.H.; You, Z.; Qiong, Q.; Jun, Z. Prognostic value of amphiregulin and epiregulin mRNA expression in metastatic colorectal cancer patients. Oncotarget 2016, 7, 55890–55899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.Y.; Tseng, C.H.; Feng, P.H.; Sun, W.L.; Ho, S.C.; Lin, C.W.; van Hiep, N.; Luo, C.S.; Tseng, Y.H.; Chen, T.T.; et al. 3-Nitrobenzanthrone promotes malignant transformation in human lung epithelial cells through the epiregulin-signaling pathway. Cell Biol. Toxicol. 2021, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Atlas, H.P. Available online: https://www.proteinatlas.org/ENSG00000124882-EREG/tissue+cell+type (accessed on 1 January 2022).
- Yasmeen, R.; Shen, Q.; Lee, A.; Leung, J.H.; Kowdley, D.; DiSilvestro, D.J.; Xu, L.; Yang, K.; Maiseyeu, A.; Bal, N.C.; et al. Epiregulin induces leptin secretion and energy expenditure in high-fat diet-fed mice. J. Endocrinol. 2018, 239, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Yasmeen, R.; Marbourg, J.; Xu, L.; Yu, L.; Fadda, P.; Flechtner, A.; Lee, L.J.; Popovich, P.G.; Ziouzenkova, O. Induction of innervation by encapsulated adipocytes with engineered vitamin A metabolism. Transl. Res. 2018, 192, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Riese, D.J., 2nd; Cullum, R.L. Epiregulin: Roles in normal physiology and cancer. Semin Cell Dev. Biol. 2014, 28, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasmeen, R.; Reichert, B.; Deiuliis, J.; Yang, F.; Lynch, A.; Meyers, J.; Sharlach, M.; Shin, S.; Volz, K.S.; Green, K.B.; et al. Autocrine Function of Aldehyde Dehydrogenase 1 as a Determinant of Diet- and Sex-Specific Differences in Visceral Adiposity. Diabetes 2013, 62, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogan, J.S.; McKee, A.E.; Lodish, H.F. Insulin-responsive compartments containing GLUT4 in 3T3-L1 and CHO cells: Regulation by amino acid concentrations. Mol. Cell. Biol. 2001, 21, 4785–4806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prokop, J.W.; Lazar, J.; Crapitto, G.; Smith, D.C.; Worthey, E.A.; Jacob, H.J. Molecular modeling in the age of clinical genomics, the enterprise of the next generation. J. Mol. Model 2017, 23, 75. [Google Scholar] [CrossRef] [Green Version]
- van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Voinova, M.V.; Rodahl, M.; Jonson, M.; Kasemo, B. Viscoelastic acoustic response of layered polymer films at fluid-solid interfaces: Continuum mechanics approach. Phys. Scr. 1999, 59, 391–396. [Google Scholar] [CrossRef] [Green Version]
- Kuntz, E.; Broca, C.; Komurasaki, T.; Kaltenbacher, M.C.; Gross, R.; Pinget, M.; Damge, C. Effect of epiregulin on pancreatic beta cell growth and insulin secretion. Growth Factors 2005, 23, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Seron, K.; Corset, L.; Vasseur, F.; Boutin, P.; Gomez-Ambrosi, J.; Salvador, J.; Fruhbeck, G.; Froguel, P. Distinct impaired regulation of SOCS3 and long and short isoforms of the leptin receptor in visceral and subcutaneous fat of lean and obese women. Biochem. Biophys. Res. Commun. 2006, 348, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Viesti ACollares, R.; Salgado, W., Jr.; Pretti da Cunha Tirapelli, D.; dos Santos, J.S. The expression of LEP, LEPR, IGF1 and IL10 in obesity and the relationship with microRNAs. PLoS ONE 2014, 9, e93512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buettner, C.; Muse, E.D.; Cheng, A.; Chen, L.; Scherer, T.; Pocai, A.; Su, K.; Cheng, B.; Li, X.; Harvey-White, J.; et al. Leptin controls adipose tissue lipogenesis via central, STAT3-independent mechanisms. Nat. Med. 2008, 14, 667–675. [Google Scholar] [CrossRef]
- Roman, A.E.; Reis, D.; Romanatto, T.; Maimoni, D.; Ferreira, E.A.; Santos, G.A.; Torsoni, A.S.; Velloso, L.A.; Torsoni, M.A. Central leptin action improves skeletal muscle AKT, AMPK, and PGC1 alpha activation by hypothalamic PI3K-dependent mechanism. Mo.l Cell. Endocrinol. 2010, 314, 62–69. [Google Scholar] [CrossRef]
- Toyoda, H.; Komurasaki, T.; Ikeda, Y.; Yoshimoto, M.; Morimoto, S. Molecular cloning of mouse epiregulin, a novel epidermal growth factor-related protein, expressed in the early stage of development. FEBS Lett. 1995, 377, 403–407. [Google Scholar] [CrossRef] [Green Version]
- DiSilvestro, D.J.; Melgar-Bermudez, E.; Yasmeen, R.; Fadda, P.; Lee, L.J.; Kalyanasundaram, A.; Gilor, C.L.; Ziouzenkova, O. Leptin Production by Encapsulated Adipocytes Increases Brown Fat, Decreases Resistin, and Improves Glucose Intolerance in Obese Mice. PLoS ONE 2016, 11, e0153198. [Google Scholar] [CrossRef]
- Martin, L.J.; Smith, S.B.; Khoutorsky, A.; Magnussen, C.A.; Samoshkin, A.; Sorge, R.E.; Cho, C.; Yosefpour, N.; Sivaselvachandran, S.; Tohyama, S.; et al. Epiregulin and EGFR interactions are involved in pain processing. J. Clin. Investig. 2017, 127, 3353–3366. [Google Scholar] [CrossRef] [Green Version]
- Kavalakatt, S.; Khadir, A.; Madhu, D.; Koistinen, H.A.; Al-Mulla, F.; Tuomilehto, J.; Abubaker, J.; Tiss, A. Urocortin 3 overexpression reduces ER stress and heat shock response in 3T3-L1 adipocytes. Sci. Rep. 2021, 11, 15666. [Google Scholar] [CrossRef]
- Graham, T.E.; Kahn, B.B. Tissue-specific alterations of glucose transport and molecular mechanisms of intertissue communication in obesity and type 2 diabetes. Horm. Metab Res. 2007, 39, 717–721. [Google Scholar] [CrossRef] [Green Version]
- Actis Dato, V.; Sanchez, M.C.; Chiabrando, G.A. LRP1 mediates the IGF-1-induced GLUT1 expression on the cell surface and glucose uptake in Muller glial cells. Sci. Rep. 2021, 11, 4742. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.M.; Zhou, L.; Perez-Tilve, D.; Greer, T.; Grant, E.; Wancata, L.; Thomas, A.; Pfluger, P.T.; Basford, J.E.; Gilham, D.; et al. Adipocyte LDL receptor-related protein-1 expression modulates postprandial lipid transport and glucose homeostasis in mice. J. Clin. Investig. 2007, 117, 3271–3282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 2018, 15, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.; Mikolajczak, M.; Kersten, C.; Fleetwood-Walker, S. ErbB1-dependent signalling and vesicular trafficking in primary afferent nociceptors associated with hypersensitivity in neuropathic pain. Neurobiol. Dis. 2020, 142, 104961. [Google Scholar] [CrossRef]
- Xie, H.; Lin, L.; Tong, L.; Jiang, Y.; Zheng, M.; Chen, Z.; Jiang, X.; Zhang, X.; Ren, X.; Qu, W.; et al. AST1306, a novel irreversible inhibitor of the epidermal growth factor receptor 1 and 2, exhibits antitumor activity both in vitro and in vivo. PLoS ONE 2011, 6, e21487. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Kang, T.; Li, X.; Niu, C. Insulin-like growth factor binding proteins inhibit oocyte maturation of zebrafish. Gen. Comp. Endocrinol. 2019, 281, 83–90. [Google Scholar] [CrossRef]
- Ple, P.A.; Green, T.P.; Hennequin, L.F.; Curwen, J.; Fennell, M.; Allen, J.; der Brempt, C.L.; Costello, G. Discovery of a new class of anilinoquinazoline inhibitors with high affinity and specificity for the tyrosine kinase domain of c-Src. J. Med. Chem. 2004, 47, 871–887. [Google Scholar] [CrossRef]
- Ozcan, L.; Xu, X.; Deng, S.X.; Ghorpade, D.S.; Thomas, T.; Cremers, S.; Hubbard, B.; Serrano-Wu, M.H.; Gaestel, M.; Landry, D.W.; et al. Treatment of Obese Insulin-Resistant Mice With an Allosteric MAPKAPK2/3 Inhibitor Lowers Blood Glucose and Improves Insulin Sensitivity. Diabetes 2015, 64, 3396–3405. [Google Scholar] [CrossRef] [Green Version]
- Prada, P.O.; Ropelle, E.R.; Mourao, R.H.; de Souza, C.T.; Pauli, J.R.; Cintra, D.E.; Schenka, A.; Rocco, S.A.; Rittner, R.; Franchini, K.G.; et al. EGFR tyrosine kinase inhibitor (PD153035) improves glucose tolerance and insulin action in high-fat diet-fed mice. Diabetes 2009, 58, 2910–2919. [Google Scholar] [CrossRef] [Green Version]
- Van Epps-Fung, M.; Hardy, R.W.; Williford, J.; Gupta, K.; Wells, A. Epidermal growth factor induces glucose storage in transgenic 3T3-L1 adipocytes overexpressing epidermal growth factor receptors. Diabetes 1996, 45, 1619–1625. [Google Scholar] [CrossRef]
- Morton, G.J.; Gelling, R.W.; Niswender, K.D.; Morrison, C.D.; Rhodes, C.J.; Schwartz, M.W. Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2005, 2, 411–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, H.; Kane, S.; Sano, E.; Miinea, C.P.; Asara, J.M.; Lane, W.S.; Garner, C.W.; Lienhard, G.E. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J. Biol. Chem. 2003, 278, 14599–14602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Arias, E.B.; Qi, N.R.; Saunders, T.L.; Cartee, G.D. In vivo glucoregulation and tissue-specific glucose uptake in female Akt substrate 160 kDa knockout rats. PLoS ONE 2020, 15, e0223340. [Google Scholar] [CrossRef] [PubMed]
- Rodenhausen, K.B.; Kasputis, T.; Pannier, A.K.; Gerasimov, J.Y.; Lai, R.Y.; Solinsky, M.; Tiwald, T.E.; Wang, H.; Sarkar, A.; Hofmann, T.; et al. Combined optical and acoustical method for determination of thickness and porosity of transparent organic layers below the ultra-thin film limit. Rev. Sci. Instrum. 2011, 82, 103111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, E.J.; Jha, S.; Restaino, C.R.; Dayananth, P.; Zhu, H.; Cooper, A.; Carr, D.; Deng, Y.; Jin, W.; Black, S.; et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013, 3, 742–750. [Google Scholar] [CrossRef] [Green Version]
- Herrero, A.; Pinto, A.; Colon-Bolea, P.; Casar, B.; Jones, M.; Agudo-Ibanez, L.; Vidal, R.; Tenbaum, S.P.; Nuciforo, P.; Valdizan, E.M.; et al. Small Molecule Inhibition of ERK Dimerization Prevents Tumorigenesis by RAS-ERK Pathway Oncogenes. Cancer Cell 2015, 28, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Favata, M.F.; Horiuchi, K.Y.; Manos, E.J.; Daulerio, A.J.; Stradley, D.A.; Feeser, W.S.; van Dyk, D.E.; Pitts, W.J.; Earl, R.A.; Hobbs, F.; et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998, 273, 18623–18632. [Google Scholar] [CrossRef] [Green Version]
- Mercer, J.G.; Hoggard, N.; Williams, L.M.; Lawrence, C.B.; Hannah, L.T.; Trayhurn, P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. 1996, 387, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, A.A.; Carmo, J.M.d.; Freeman, J.N.; Tallam, L.S.; Hall, J.E. A functional melanocortin system may be required for chronic CNS-mediated antidiabetic and cardiovascular actions of leptin. Diabetes 2009, 58, 1749–1756. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, E.C.; Myers, M.G., Jr. Leptin receptor signaling and the regulation of mammalian physiology. Int. J. Obes. 2008, 32 (Suppl. S7), S8–S12. [Google Scholar] [CrossRef] [Green Version]
- Verma, V.; Khoury, S.; Parisien, M.; Cho, C.; Maixner, W.; Martin, L.J.; Diatchenko, L. The dichotomous role of epiregulin in pain. Pain 2020, 161, 1052–1064. [Google Scholar] [CrossRef] [PubMed]
- Duquenne, M.; Folgueira, C.; Bourouh, C.; Millet, M.; Silva, A.; Clasadonte, J.; Imbernon, M.; Fernandois, D.; Martinez-Corral, I.; Kusumakshi, S.; et al. Leptin brain entry via a tanycytic LepR-EGFR shuttle controls lipid metabolism and pancreas function. Nat. Metab. 2021, 3, 1071–1090. [Google Scholar] [CrossRef] [PubMed]
- Do Carmo, J.M.; da Silva, A.A.; Gava, F.N.; Moak, S.P.; Dai, X.; Hall, J.E. Impact of leptin deficiency compared with neuronal-specific leptin receptor deletion on cardiometabolic regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R552–R562. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhu, A.X. Targeting insulin-like growth factor axis in hepatocellular carcinoma. J. Hematol. Oncol. 2011, 4, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, N.-J.; Lee, A.; Yasmeen, R.; Shen, Q.; Yang, K.; Kumar, S.B.; Muhanna, D.; Arnipalli, S.; Noria, S.F.; Needleman, B.J.; et al. Epiregulin as an Alternative Ligand for Leptin Receptor Alleviates Glucose Intolerance without Change in Obesity. Cells 2022, 11, 425. https://doi.org/10.3390/cells11030425
Song N-J, Lee A, Yasmeen R, Shen Q, Yang K, Kumar SB, Muhanna D, Arnipalli S, Noria SF, Needleman BJ, et al. Epiregulin as an Alternative Ligand for Leptin Receptor Alleviates Glucose Intolerance without Change in Obesity. Cells. 2022; 11(3):425. https://doi.org/10.3390/cells11030425
Chicago/Turabian StyleSong, No-Joon, Aejin Lee, Rumana Yasmeen, Qiwen Shen, Kefeng Yang, Shashi Bhushan Kumar, Danah Muhanna, Shanvanth Arnipalli, Sabrena F. Noria, Bradley J. Needleman, and et al. 2022. "Epiregulin as an Alternative Ligand for Leptin Receptor Alleviates Glucose Intolerance without Change in Obesity" Cells 11, no. 3: 425. https://doi.org/10.3390/cells11030425