
Immunity and Nutrition: The Right Balance in Inflammatory Bowel Disease

 ,
,  ,
,  , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Immunity and IBD
3. Nutrition and IBD
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mak, W.Y.; Zhao, M.; Ng, S.C.; Burisch, J. The epidemiology of inflammatory bowel disease: East meets west. J. Gastroenterol. Hepatol. 2020, 35, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel diasease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Fiocchi, C. Ulcerative colitis. N. Engl. J. Med. 2011, 365, 1713–1725. [Google Scholar] [CrossRef] [Green Version]
- Molodecky, N.A.; Soon, S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 42, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cader, M.Z.; Kaser, A. Recent advances in inflammatory bowel disease: Mucosal immune cells in intestinal inflammatory. Gut 2013, 62, 1653–1664. [Google Scholar] [CrossRef]
- Harlan, W.R., III. Inflammatory Bowel Disease: Epidemiology, Evaluation, Treatment, and Health Maintenance. N. C. Med. J. 2016, 7, 198–201. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- Venkataraman, G.R.; Rivas, M.A. Rare and common variant discovery in complex disease: The IBD case study. Hum. Mol. Genet. 2019, 28, R162–R169. [Google Scholar] [CrossRef]
- Coleman, O.I.; Haller, D. ER Stress and the UPR in Shaping Intestinal Tissue Homeostasis and Immunity. Front. Immunol. 2019, 10, 2825. [Google Scholar] [CrossRef]
- Brewer, J.W.; Hendershot, L.M. Building an antibody factory: A job for the unfolded protein response. Nat. Immunol. 2005, 6, 23–29. [Google Scholar] [CrossRef]
- De Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, H.; Trier Moller, F.; Andersen, V.; Harbord, M. Heritability in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1428–1434. [Google Scholar] [CrossRef] [Green Version]
- Schreiner, P.; Martinho-Grueber, M.; Studerus, D.; Vavricka, S.R.; Tilg, H.; Biedermann, L. Nutrition in Inflammatory Bowel Disease. Digestion 2020, 101, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Wark, G.; Samocha-Bonet, D.; Ghaly, S.; Danta, M. The Role of Diet in the Pathogenesis and Management of Inflammatory Bowel Disease: A Review. Nutrients 2020, 13, 135. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Varum, F.; Bravo, R.; Furrer, E.; Bojic, D.; Basit, A.W. Inflammatory bowel disease: Exploring gut pathophysiology for novel therapeutic targets. Transl. Res. 2016, 176, 38–68. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Günther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananthakrishnan, A.N.; Khalili, H.; Pan, A.; Higuchi, L.M.; de Silva, P.; Richter, J.M.; Fuchs, C.S.; Chan, A.T. Association between depressive symptoms and incidence of Crohn’s disease and ulcerative colitis: Results from the Nurses’ Health Study. Clin. Gastroenterol. Hepatol. 2013, 11, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Petronis, A.; Petroniene, R. Epigenetics of inflammatory bowel disease. Gut 2000, 47, 302–306. [Google Scholar] [CrossRef] [Green Version]
- Rengarajan, S.; Vivio, E.E.; Parkes, M.; Peterson, D.A.; Roberson, E.D.O.; Newberry, R.D.; Ciorba, M.A.; Hsieh, C.S. Dynamic immunoglobulin responses to gut bacteria during inflammatory bowel disease. Gut Microbes 2020, 11, 405–420. [Google Scholar] [CrossRef]
- Jiang, W.; Su, J.; Zhang, X.; Cheng, X.; Zhou, J.; Shi, R.; Zhang, H. Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease. Inflamm. Res. 2014, 63, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, K.; Hwang, Y.; Nikolaev, A.; Atreya, R.; Dornhoff, H.; Steiner, S.; Lehr, H.A.; Wirtz, S.; Vieth, M.; Waisman, A.; et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat. Immunol. 2014, 15, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Nalleweg, N.; Chiriac, M.T.; Podstawa, E.; Lehmann, C.; Rau, T.T.; Atreya, R.; Krauss, E.; Hundorfean, G.; Fichtner-Feigl, S.; Hartmann, A.; et al. IL-9 and its receptor are predominantly involved in the pathogenesis of UC. Gut 2015, 64, 743–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartor, R.B. Mechanisms of disease: Pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Biancheri, P.; Allan, P.; Corazza, G.R.; Di Sabatino, A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 3–10. [Google Scholar] [CrossRef]
- Salim, S.Y.; Söderholm, J.D. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm. Bowel Dis. 2011, 17, 362–381. [Google Scholar] [CrossRef]
- Uehara, A.; Fujimoto, Y.; Fukase, K.; Takada, H. Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol. Immunol. 2007, 44, 3100–3111. [Google Scholar] [CrossRef]
- De Souz, H.S.P.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef]
- Neurath Markus, F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1beta and the autoinflammatory diseases. N. Engl. J. Med. 2009, 360, 2467–2470. [Google Scholar] [CrossRef]
- Pizarro, T.T.; Michie, M.H.; Bentz, M.; Woraratanadharm, J.; Smith, M.F., Jr.; Foley, E.; Moskaluk, C.A.; Bickston, S.J.; Cominelli, F. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: Expression and localization in intestinal mucosal cells. J. Immunol. 1999, 162, 6829–6835. [Google Scholar] [PubMed]
- Dinarello, C.A.; Novick, D.; Puren, A.J.; Fantuzzi, G.; Shapiro, L.; Mühl, H.; Yoon, D.Y.; Reznikov, L.L.; Kim, S.H.; Rubinstein, M. Overview of interleukin-18, more than an interferon-gamma inducing factor. J. Leukoc. Biol. 1998, 63, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Fiocchi, C. Cytokines and animal models: A combined path to inflammatory bowel disease pathogenesis. Gastroenterology 1993, 104, 1202–1205. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; de Haar, C.; Chen, M.; Deuring, J.; Gerrits, M.M.; Smits, R.; Xia, B.; Kuipers, E.J.; van der Woude, C.J. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut 2010, 59, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Munoz, F.; Dominguez-Lopez, A.; Yamamoto-Furusho, J.K. Role of cytokines in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 4280–4288. [Google Scholar] [CrossRef]
- Wirtz, S.; Neurath, M.F. Mouse models of inflammatory bowel disease. Adv. Drug Deliv. Rev. 2007, 59, 1073–1083. [Google Scholar] [CrossRef]
- Li, M.C.; He, S.H. IL-10 and its related cytokines for treatment of inflammatory bowel disease. World J. Gastroenterol. 2004, 10, 620–625. [Google Scholar] [CrossRef]
- Marek, A.; Brodzicki, J.; Liberek, A.; Korzon, M. TGF-beta (transforming growth factor-beta) in chronic inflammatory conditions—A new diagnostic and prognostic marker? Med. Sci. Monit. 2002, 8, RA145–RA151. [Google Scholar]
- Lee, S.H.; Kwon, J.E.; Cho, M.L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef] [Green Version]
- Li, M.O.; Flavell, R.A. TGF-beta: A master of all T cell trades. Cell 2008, 134, 392–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003, 52, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Shata, M.T. Letter: Pathogenicity of Th17 cells may differ in ulcerative colitis compared with Crohn’s disease. Aliment. Pharmacol. Ther. 2012, 36, 204, author reply 205. [Google Scholar] [CrossRef] [PubMed]
- Geem, D.; Harusato, A.; Flannigan, K.; Denning, T.L. Harnessing regulatory T cells for the treatment of inflammatory bowel disease. Inflamm. Bowel Dis. 2015, 21, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Rubtsov, Y.P.; Rasmussen, J.P.; Chi, E.Y.; Fontenot, J.; Castelli, L.; Ye, X.; Treuting, P.; Siewe, L.; Roers, A.; Henderson WRJr Muller, W.; et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 2008, 28, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, S.; Pizarro, T.T. The Treg/Th17 Axis: A Dynamic Balance Regulated by the Gut Microbiome. Front. Immunol. 2015, 6, 639. [Google Scholar] [CrossRef] [Green Version]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Lepage, P.; Häsler, R.; Spehlmann, M.E.; Rehman, A.; Zvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Doré, J.; Raedler, A.; et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef]
- Singh, B.; Read, S.; Asseman, C.; Malmström, V.; Mottet, C.; Stephens, L.A.; Stepankova, R.; Tlaskalova, H.; Powrie, F. Control of intestinal inflammation by regulatory T cells. Immunol. Rev. 2001, 182, 190–200. [Google Scholar] [CrossRef]
- Fantini, M.C.; Becker, C.; Tubbe, I.; Nikolaev, A.; Lehr, H.A.; Galle, P.; Neurath, M.F. Transforming growth factor beta induced FoxP3+ regulatory T cells suppress Th1 mediated experimental colitis. Gut 2006, 55, 671–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, C.; Dornhoff, H.; Neufert, C.; Fantini, M.C.; Wirtz, S.; Huebner, S.; Nikolaev, A.; Lehr, H.A.; Murphy, A.J.; Valenzuela, D.M.; et al. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J. Immunol. 2006, 177, 2760–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolfi, C.; Rizzo, A.; Franzè, E.; Rotondi, A.; Fantini, M.C.; Sarra, M.; Caruso, R.; Monteleone, I.; Sileri, P.; Franceschilli, L.; et al. Involvement of interleukin-21 in the regulation of colitis-associated colon cancer. J. Exp. Med. 2011, 208, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Flach, M.; Möhle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef] [Green Version]
- Cortez, V.S.; Robinette, M.L.; Colonna, M. Innate lymphoid cells: New insights into function and development. Curr. Opin. Immunol. 2015, 32, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Glover, S.C. Innate Lymphoid Cells in Inflammatory Bowel Disease. Arch. Immunol. Ther. Exp. 2018, 66, 415–421. [Google Scholar] [CrossRef]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Philip, N.H.; Artis, D. New friendships and old feuds: Relationships between innate lymphoid cells and microbial communities. Immunol. Cell Biol. 2013, 91, 225–231. [Google Scholar] [CrossRef]
- Mjösberg, J.; Spits, H. Human innate lymphoid cells. J. Allergy Clin. Immunol. 2016, 138, 1265–1276. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [Green Version]
- Faith, J.J.; McNulty, N.P.; Rey, F.E.; Gordon, J.I. Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 2011, 333, 101–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, W.R.; Gratz, S.W.; Duncan, S.H.; Holtrop, G.; Ince, J.; Scobbie, L.; Duncan, G.; Johnstone, A.M.; Lobley, G.E.; Wallace, R.J.; et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am. J. Clin. Nutr. 2011, 93, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calder, P.C. Polyunsaturated fatty acids and inflammation. Biochem. Soc. Trans. 2005, 33, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.D.; Abreu, M.T. Diet as a Trigger or Therapy for Inflammatory Bowel Diseases. Gastroenterology 2017, 152, 398–414.e6. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.P.; Stanton, C. Health implications of high dietary omega-6 polyunsaturated Fatty acids. J. Nutr. Metab. 2012, 2012, 539426. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 2002, 56, 365–379. [Google Scholar] [CrossRef]
- Gutiérrez, S.; Svahn, S.L.; Johansson, M.E. Effects of Omega-3 Fatty Acids on Immune Cells. Int. J. Mol. Sci. 2019, 20, 5028. [Google Scholar] [CrossRef] [Green Version]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.; Boneh, R.S.; Wine, E. Evolving role of diet in the pathogenesis and treatment of inflammatory bowel diseases. Gut 2018, 67, 1726–1738. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353.e21. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Torbenson, M.; Hamad, A.R.; Soloski, M.J.; Li, Z. High-fat diet modulates non-CD1d-restricted natural killer T cells and regulatory T cells in mouse colon and exacerbates experimental colitis. Clin. Exp. Immunol. 2008, 151, 130–138. [Google Scholar] [CrossRef]
- Agus, A.; Denizot, J.; Thévenot, J.; Martinez-Medina, M.; Massier, S.; Sauvanet, P.; Bernalier-Donadille, A.; Denis, S.; Hofman, P.; Bonnet, R.; et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci. Rep. 2016, 6, 19032. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.A.; Gu, W.; Lee, I.A.; Joh, E.H.; Kim, D.H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 2012, 7, e47713. [Google Scholar] [CrossRef]
- Liu, Z.; Brooks, R.S.; Ciappio, E.D.; Kim, S.J.; Crott, J.W.; Bennett, G.; Greenberg, A.S.; Mason, J.B. Diet-induced obesity elevates colonic TNF-α in mice and is accompanied by an activation of Wnt signaling: A mechanism for obesity-associated colorectal cancer. J. Nutr. Biochem. 2012, 23, 1207–1213. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, P.; Araújo, J.R.; Di Santo, J.P. A Cross-Talk Between Microbiota-Derived Short-Chain Fatty Acids and the Host Mucosal Immune System Regulates Intestinal Homeostasis and Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2018, 24, 558–572. [Google Scholar] [CrossRef] [Green Version]
- Castro, F.; de Souza, H.S.P. Dietary Composition and Effects in Inflammatory Bowel Disease. Nutrients 2019, 11, 1398. [Google Scholar] [CrossRef] [Green Version]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A microbial signature for Crohn’s disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Powell, N.; MacDonald, T.T. Recent advances in gut immunology. Parasite Immunol. 2017, 39, e12430. [Google Scholar] [CrossRef] [Green Version]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Sefik, E.; Geva-Zatorsky, N.; Oh, S.; Konnikova, L.; Zemmour, D.; McGuire, A.M.; Burzyn, D.; Ortiz-Lopez, A.; Lobera, M.; Yang, J.; et al. Individual intestinal symbionts induce a distinct population of RORγ⁺ regulatory T cells. Science 2015, 349, 993–997. [Google Scholar] [CrossRef] [Green Version]
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 2015, 163, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Tan, T.G.; Sefik, E.; Geva-Zatorsky, N.; Kua, L.; Naskar, D.; Teng, F.; Pasman, L.; Ortiz-Lopez, A.; Jupp, R.; Wu, H.J.; et al. Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc. Natl. Acad. Sci. USA 2016, 113, E8141–E8150. [Google Scholar] [CrossRef] [Green Version]
- Russo, G.L. Dietary n-6 and n-3 polyunsaturated fatty acids: From biochemistry to clinical implications in cardiovascular prevention. Biochem. Pharmacol. 2009, 77, 937–946. [Google Scholar] [CrossRef]
- Shoda, R.; Matsueda, K.; Yamato, S.; Umeda, N. Epidemiologic analysis of Crohn disease in Japan: Increased dietary intake of n-6 polyunsaturated fatty acids and animal protein relates to the increased incidence of Crohn disease in Japan. Am. J. Clin. Nutr. 1996, 63, 741–745. [Google Scholar] [CrossRef]
- IBD in EPIC Study Investigators; Tjonneland, A.; Overvad, K.; Bergmann, M.M.; Nagel, G.; Linseisen, J.; Hallmans, G.; Palmqvist, R.; Sjodin, H.; Hagglund, G.; et al. Linoleic acid, a dietary n-6 polyunsaturated fatty acid, and the aetiology of ulcerative colitis: A nested case-control study within a European prospective cohort study. Gut 2009, 58, 1606–1611. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Khalili, H.; Konijeti, G.G.; Higuchi, L.M.; de Silva, P.; Fuchs, C.S.; Willett, W.C.; Richter, J.M.; Chan, A.T. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn’s disease. Gut 2014, 63, 776–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joffre, C.; Dinel, A.L.; Chataigner, M.; Pallet, V.; Layé, S. n-3 Polyunsaturated Fatty Acids and Their Derivates Reduce Neuroinflammation during Aging. Nutrients 2020, 12, 647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, U.N. Arachidonic acid and other unsaturated fatty acids and some of their metabolites function as endogenous antimicrobial molecules: A review. J. Adv. Res. 2018, 11, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Al-Khalaifah, H. Modulatory Effect of Dietary Polyunsaturated Fatty Acids on Immunity, Represented by Phagocytic Activity. Front. Vet. Sci. 2020, 7, 569939. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.G.; Contaifer, D.; Madurantakam, P.; Carbone, S.; Price, E.T.; Van Tassell, B.; Brophy, D.F.; Wijesinghe, D.S. Dietary Bioactive Fatty Acids as Modulators of Immune Function: Implications on Human Health. Nutrients 2019, 11, 2974. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, K.; Nakamura, M.; Odahara, S.; Koido, S.; Katahira, K.; Shiraishi, H.; Ohkusa, T.; Fujise, K.; Tajiri, H. N-3 polyunsaturated fatty acid diet therapy for patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2010, 16, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Lin, X.; Zhao, Q.; Li, J. Fat intake and risk of ulcerative colitis: Systematic review and dose-response meta-analysis of epidemiological studies. J. Gastroenterol. Hepatol. 2017, 32, 19–27. [Google Scholar] [CrossRef]
- Sakamoto, N.; Kono, S.; Wakai, K.; Fukuda, Y.; Satomi, M.; Shimoyama, T.; Inaba, Y.; Miyake, Y.; Sasaki, S.; Okamoto, K.; et al. Dietary risk factors for inflammatory bowel disease: A multicenter case-control study in Japan. Inflamm. Bowel Dis. 2005, 11, 154–163. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Khalili, H.; Song, M.; Higuchi, L.M.; Lochhead, P.; Richter, J.M.; Chan, A.T. Genetic Polymorphisms in Fatty Acid Metabolism Modify the Association Between Dietary n3, n6 Intake and Risk of Ulcerative Colitis: A Prospective Cohort Study. Inflamm. Bowel Dis. 2017, 23, 1898–1904. [Google Scholar] [CrossRef]
- Le Poul, E.; Loison, C.; Struyf, S.; Springael, J.Y.; Lannoy, V.; Decobecq, M.E.; Brezillon, S.; Dupriez, V.; Vassart, G.; Van Damme, J.; et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J. Biol. Chem. 2003, 278, 25481–25489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aljebab, F.; Choonara, I.; Conroy, S. Systematic Review of the Toxicity of Long-Course Oral Corticosteroids in Children. PLoS ONE 2017, 12, e0170259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutgeerts, P.J. Review article: The limitations of corticosteroid therapy in Crohn’s disease. Aliment. Pharmacol. Ther. 2001, 15, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, O.; Cordischi, L.; Cirulli, M.; Paganelli, M.; Labalestra, V.; Uccini, S.; Russo, P.M.; Cucchiara, S. Polymeric diet alone versus corticosteroids in the treatment of active pediatric Crohn’s disease: A randomized controlled open-label trial. Clin. Gastroenterol. Hepatol. 2006, 4, 744–753. [Google Scholar] [CrossRef]
- Hansen, T.; Duerksen, D.R. Enteral Nutrition in the Management of Pediatric and Adult Crohn’s Disease. Nutrients 2018, 10, 537. [Google Scholar] [CrossRef] [Green Version]
- Zachos, M.; Tondeur, M.; Griffiths, A.M. Enteral nutritional therapy for induction of remission in Crohn’s disease. Cochrane Database Syst. Rev. 2007, 4, CD000542. [Google Scholar] [CrossRef]
- Lochs, H.; Dejong, C.; Hammarqvist, F.; Hebuterne, X.; Leon-Sanz, M.; Schütz, T.; van Gemert, W.; van Gossum, A.; Valentini, L.; DGEM (German Society for Nutritional Medicine); et al. ESPEN Guidelines on Enteral Nutrition: Gastroenterology. Clin. Nutr. 2006, 25, 260–274. [Google Scholar] [CrossRef]
- Donnellan, C.F.; Yann, L.H.; Lal, S. Nutritional management of Crohn’s disease. Ther. Adv. Gastroenterol. 2013, 6, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Nakahigashi, M.; Umegae, S.; Kitagawa, T.; Matsumoto, K. Impact of elemental diet on mucosal inflammation in patients with active Crohn’s disease: Cytokine production and endoscopic and histological findings. Inflamm. Bowel Dis. 2005, 11, 580–588. [Google Scholar] [CrossRef]
- Sanderson, I.R.; Croft, N.M. The anti-inflammatory effects of enteral nutrition. JPEN J. Parenter Enter. Nutr. 2005, 29, S134–S140. [Google Scholar] [CrossRef]
- Svolos, V.; Hansen, R.; Nichols, B.; Quince, C.; Ijaz, U.Z.; Papadopoulou, R.T.; Edwards, C.A.; Watson, D.; Alghamdi, A.; Brejnrod, A.; et al. Treatment of Active Crohn’s Disease With an Ordinary Food-based Diet That Replicates Exclusive Enteral Nutrition. Gastroenterology 2019, 156, 1354–1367.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suskind, D.L.; Cohen, S.A.; Brittnacher, M.J.; Wahbeh, G.; Lee, D.; Shaffer, M.L.; Braly, K.; Hayden, H.S.; Klein, J.; Gold, B.; et al. Clinical and Fecal Microbial Changes With Diet Therapy in Active Inflammatory Bowel Disease. J. Clin. Gastroenterol. 2018, 52, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Sigall-Boneh, R.; Pfeffer-Gik, T.; Segal, I.; Zangen, T.; Boaz, M.; Levine, A. Partial enteral nutrition with a Crohn’s disease exclusion diet is effective for induction of remission in children and young adults with Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- SigallBoneh, R.; SarbagiliShabat, C.; Yanai, H.; Chermesh, I.; Ben Avraham, S.; Boaz, M.; Levine, A. Dietary Therapy With the Crohn’s Disease Exclusion Diet is a Successful Strategy for Induction of Remission in Children and Adults Failing Biological Therapy. J. Crohn’s Colitis 2017, 11, 1205–1212. [Google Scholar] [CrossRef] [Green Version]
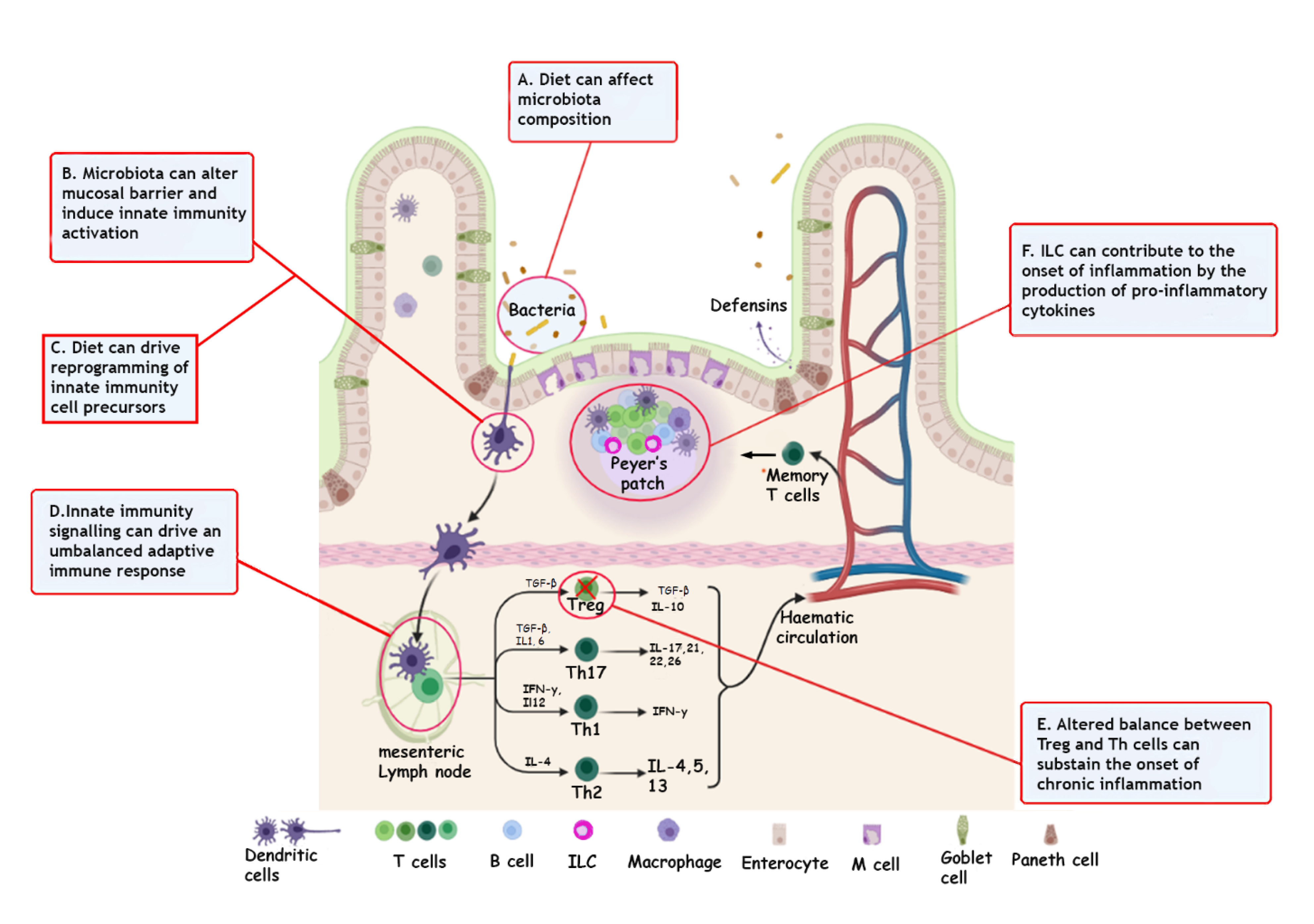

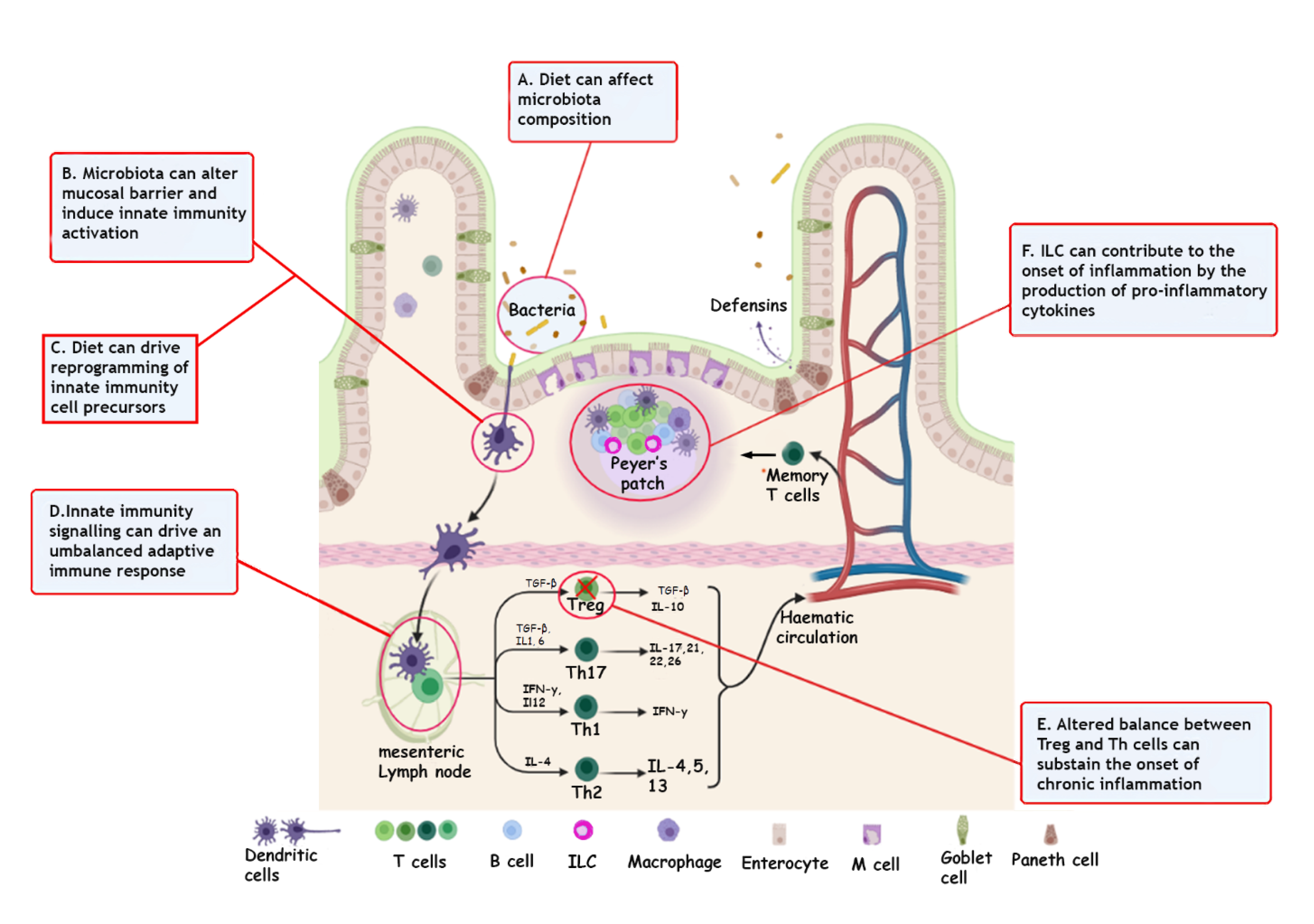{kind=link}
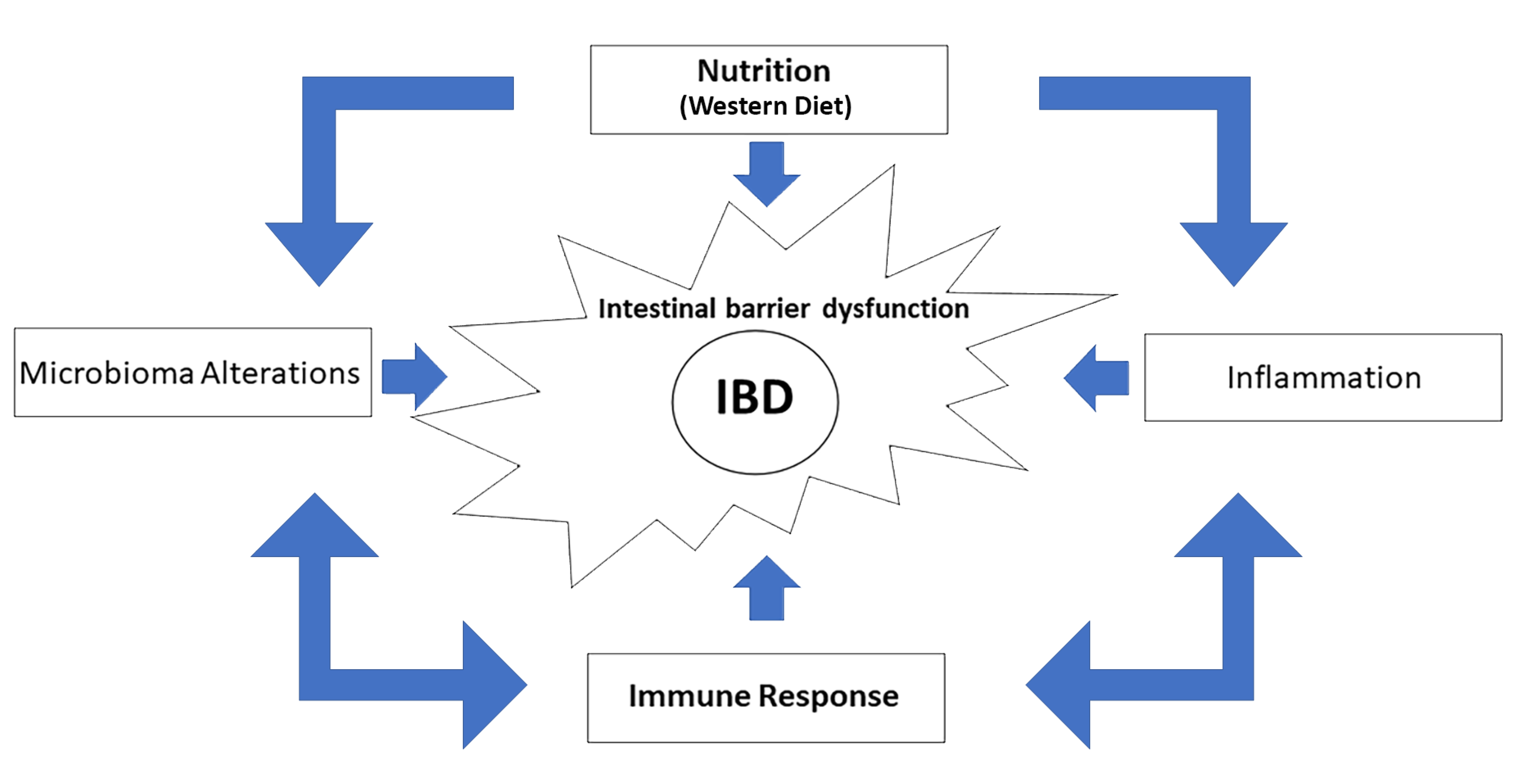
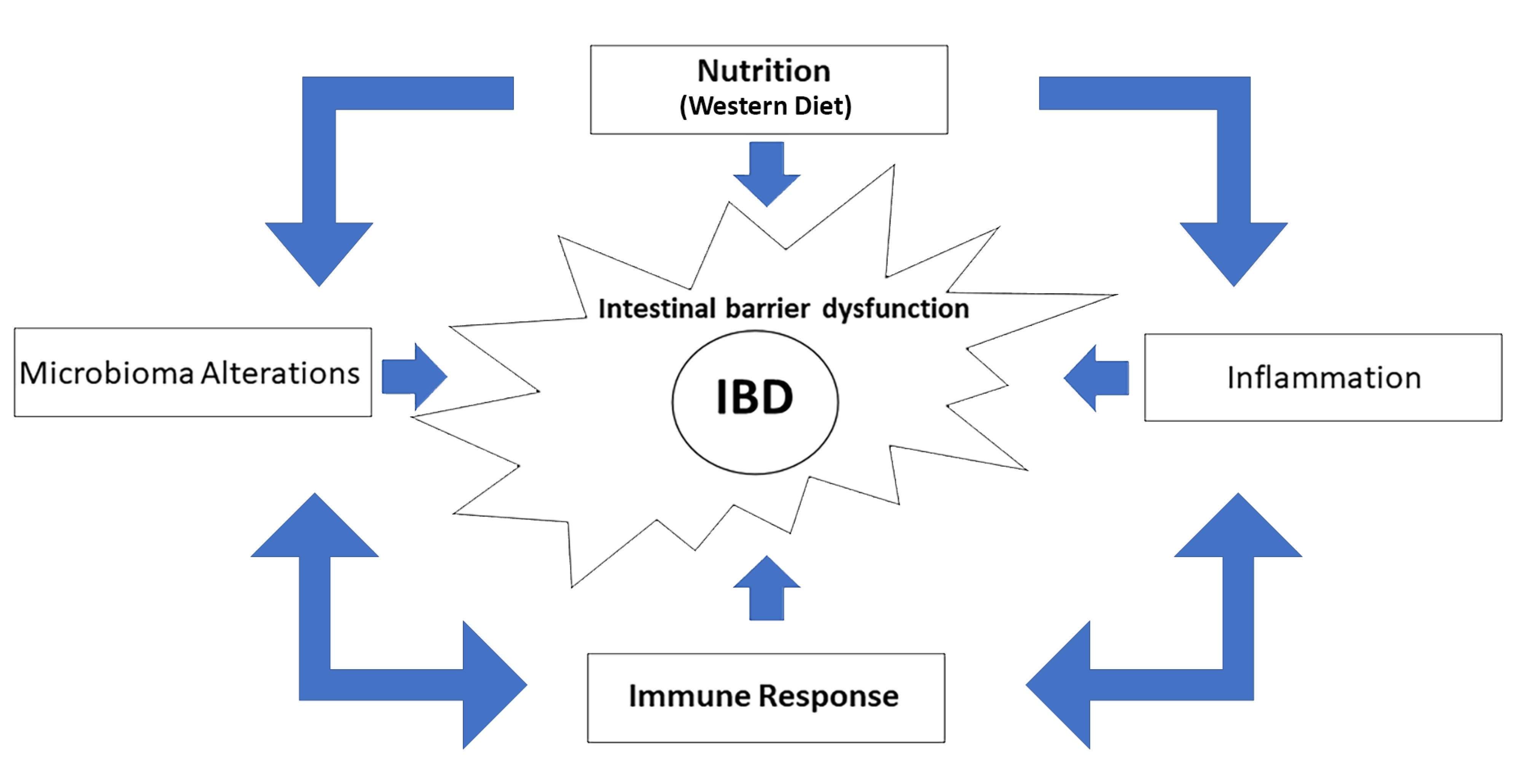{kind=link}
| Cytokines, Immune Cells and Microbioma | |||||||
|---|---|---|---|---|---|---|---|
| TNF | IL-1β | IL-6 | M1 Macrophages | Treg | GRAM Negative Bacteria | References | |
| High consumption of fats and sugars | + | + | + | + | − | + | [14,15,17,25,29,30,36,63,66,67] |
| Regular consumption of omega-3 PUFA, fibers and vegetables | − | − | − | − | + | − | [14,15,50,51,65,66,68,69,70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamburini, B.; La Manna, M.P.; La Barbera, L.; Mohammadnezhad, L.; Badami, G.D.; Shekarkar Azgomi, M.; Dieli, F.; Caccamo, N. Immunity and Nutrition: The Right Balance in Inflammatory Bowel Disease. Cells 2022, 11, 455. https://doi.org/10.3390/cells11030455
Tamburini B, La Manna MP, La Barbera L, Mohammadnezhad L, Badami GD, Shekarkar Azgomi M, Dieli F, Caccamo N. Immunity and Nutrition: The Right Balance in Inflammatory Bowel Disease. Cells. 2022; 11(3):455. https://doi.org/10.3390/cells11030455
Chicago/Turabian StyleTamburini, Bartolo, Marco Pio La Manna, Lidia La Barbera, Leila Mohammadnezhad, Giusto Davide Badami, Mojtaba Shekarkar Azgomi, Francesco Dieli, and Nadia Caccamo. 2022. "Immunity and Nutrition: The Right Balance in Inflammatory Bowel Disease" Cells 11, no. 3: 455. https://doi.org/10.3390/cells11030455
APA StyleTamburini, B., La Manna, M. P., La Barbera, L., Mohammadnezhad, L., Badami, G. D., Shekarkar Azgomi, M., Dieli, F., & Caccamo, N. (2022). Immunity and Nutrition: The Right Balance in Inflammatory Bowel Disease. Cells, 11(3), 455. https://doi.org/10.3390/cells11030455