
Multi-Omics Profiling to Assess Signaling Changes upon VHL Restoration and Identify Putative VHL Substrates in Clear Cell Renal Cell Carcinoma Cell Lines

 , ,
, , 

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Colony and Sphere Formation Assays
2.3. Orthotopic Tumor Model
2.4. Western Blotting
2.5. Co-Immunoprecipitation
2.6. Cell Adhesion Assay
2.7. Targeted Mutagenesis of NFKB2
2.8. RNA Sequencing
2.9. Proteomics
2.10. Ubiquitomics
2.11. Data Extraction from TCGA and Survival Analysis
2.12. Statistical Analysis
3. Results
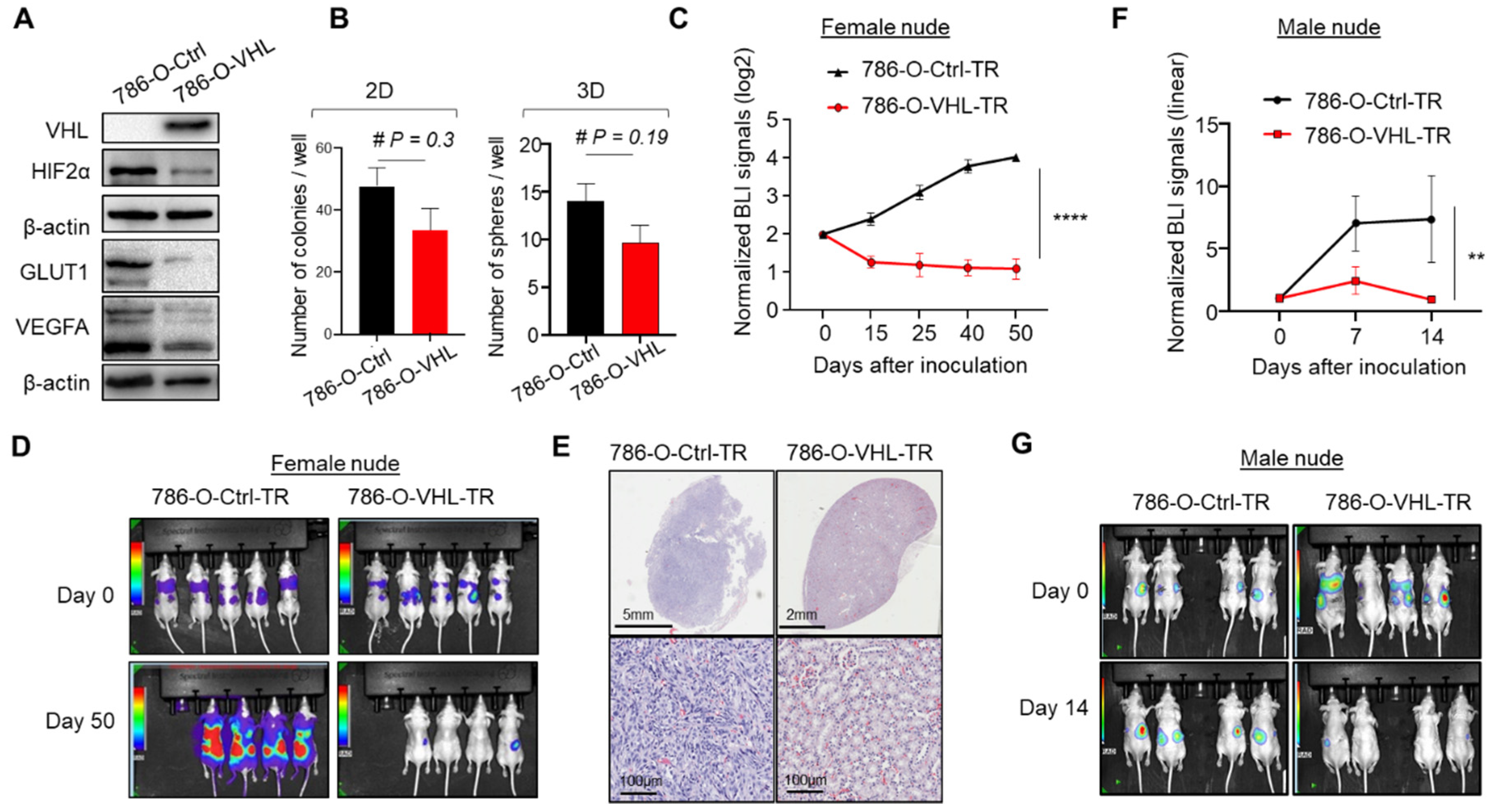
3.1. VHL Restoration in 786-O-Depleted HIF2α and Abrogated Orthotopic Tumor Formation
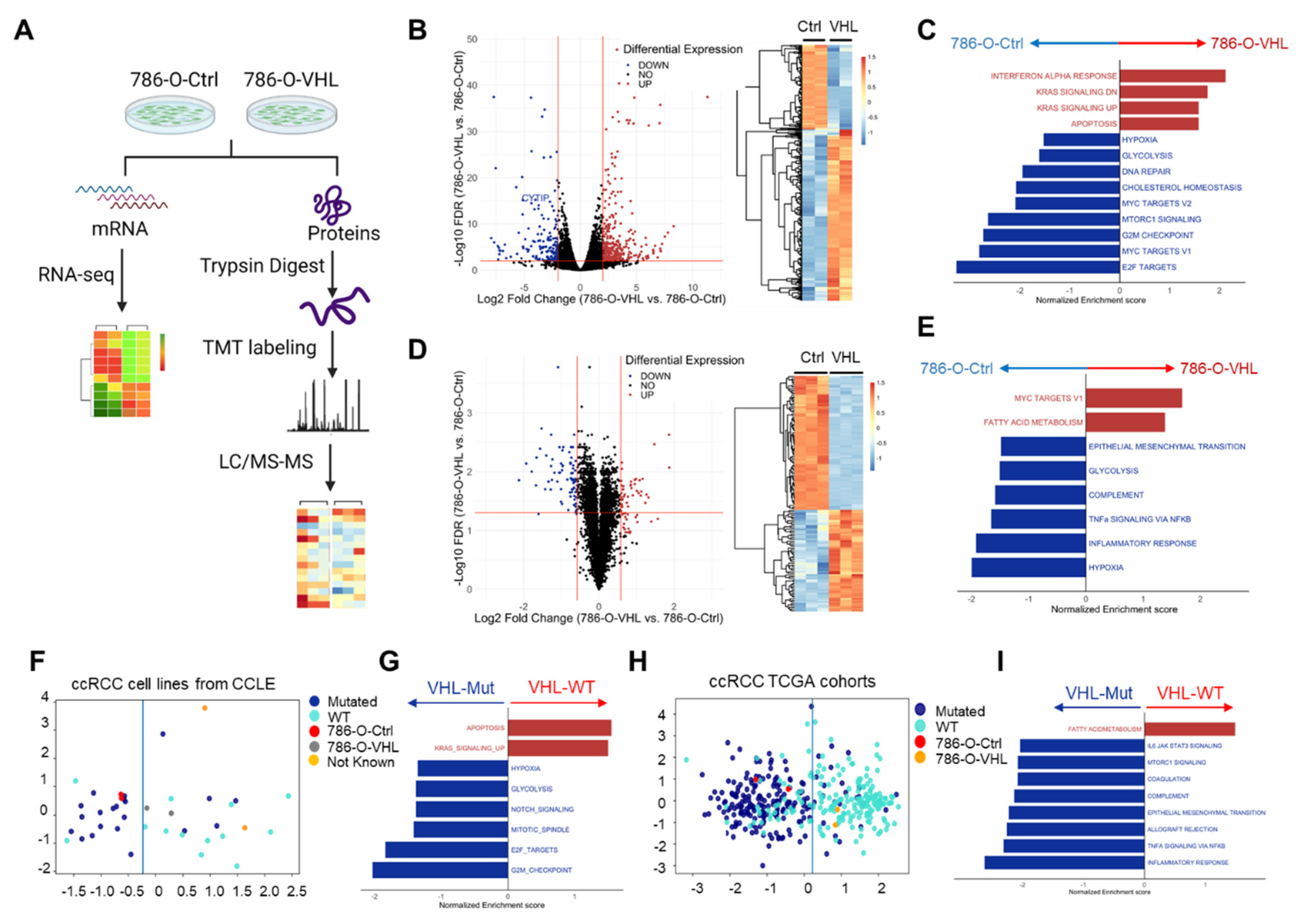
3.2. VHL Restoration Downregulated HIF-Driven Pathways
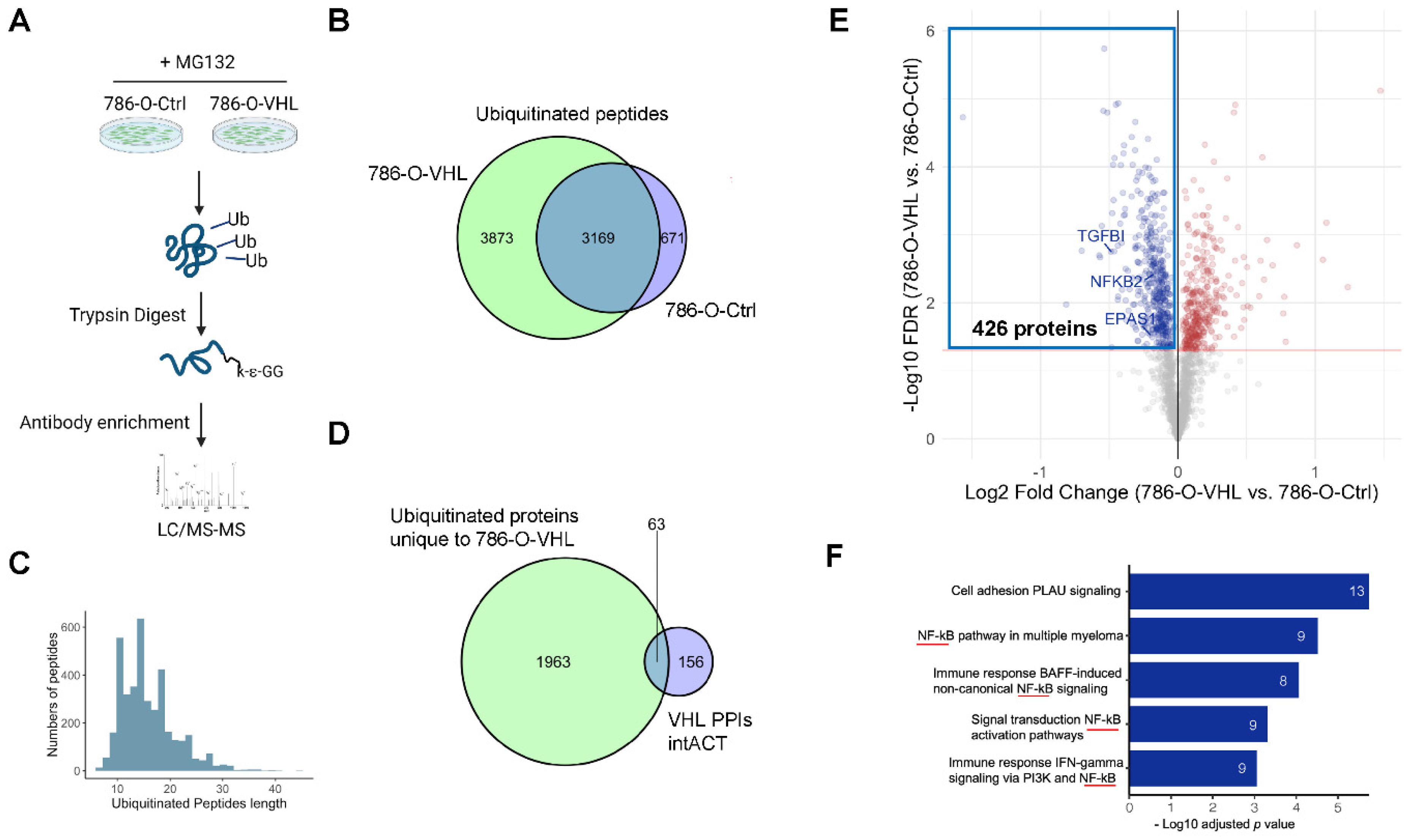
3.3. Ubiquitome Profiling Identified Potential VHL Substrates
3.4. Potential VHL Substrates with Clinical Prognostic Significance
3.5. TGFBI and NFKB2 Are Putative VHL Targeted Proteins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. Von Hippel-Lindau disease. Annu. Rev. Pathol. 2007, 2, 145–173. [Google Scholar] [CrossRef]
- Jonasch, E.; Walker, C.L.; Rathmell, W.K. Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat. Rev. Nephrol. 2021, 17, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, C.E.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of the VHL-ElonginC-ElonginB complex: Implications for VHL tumor suppressor function. Science 1999, 284, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Kang, Y. Hypoxia and hypoxia-inducible factors: Master regulators of metastasis. Clin. Cancer Res. 2010, 16, 5928–5935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Troy, H.; Leek, R.; Chung, Y.L.; Li, J.L.; Raval, R.R.; Turley, H.; Gatter, K.; Pezzella, F.; Griffiths, J.R.; et al. Effects of HIF-1alpha and HIF2alpha on Growth and Metabolism of Clear-Cell Renal Cell Carcinoma 786-0 Xenografts. J. Oncol. 2010, 2010, 757908. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Beroukhim, R.; Schumacher, S.E.; Zhou, J.; Chang, M.; Signoretti, S.; Kaelin, W.G., Jr. Genetic and functional studies implicate HIF1α as a 14q kidney cancer suppressor gene. Cancer Discov. 2011, 1, 222–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raval, R.R.; Lau, K.W.; Tran, M.G.; Sowter, H.M.; Mandriota, S.J.; Li, J.L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell. Biol. 2005, 25, 5675–5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Network, C.G.A.R. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choueiri, T.K.; Bauer, T.M.; Papadopoulos, K.P.; Plimack, E.R.; Merchan, J.R.; McDermott, D.F.; Michaelson, M.D.; Appleman, L.J.; Thamake, S.; Perini, R.F.; et al. Inhibition of hypoxia-inducible factor-2α in renal cell carcinoma with belzutifan: A phase 1 trial and biomarker analysis. Nat. Med. 2021, 27, 802–805. [Google Scholar] [CrossRef]
- Deeks, E.D. Belzutifan: First Approval. Drugs 2021, 81, 1921–1927. [Google Scholar] [CrossRef]
- Guo, J.; Chakraborty, A.A.; Liu, P.; Gan, W.; Zheng, X.; Inuzuka, H.; Wang, B.; Zhang, J.; Zhang, L.; Yuan, M.; et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 2016, 353, 929–932. [Google Scholar] [CrossRef] [Green Version]
- Ohh, M.; Yauch, R.L.; Lonergan, K.M.; Whaley, J.M.; Stemmer-Rachamimov, A.O.; Louis, D.N.; Gavin, B.J.; Kley, N.; Kaelin, W.G., Jr.; Iliopoulos, O. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol. Cell 1998, 1, 959–968. [Google Scholar] [CrossRef]
- Kurban, G.; Duplan, E.; Ramlal, N.; Hudon, V.; Sado, Y.; Ninomiya, Y.; Pause, A. Collagen matrix assembly is driven by the interaction of von Hippel-Lindau tumor suppressor protein with hydroxylated collagen IV alpha 2. Oncogene 2008, 27, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Hasanov, E.; Chen, G.; Chowdhury, P.; Weldon, J.; Ding, Z.; Jonasch, E.; Sen, S.; Walker, C.L.; Dere, R. Ubiquitination and regulation of AURKA identifies a hypoxia-independent E3 ligase activity of VHL. Oncogene 2017, 36, 3450–3463. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsova, A.V.; Meller, J.; Schnell, P.O.; Nash, J.A.; Ignacak, M.L.; Sanchez, Y.; Conaway, J.W.; Conaway, R.C.; Czyzyk-Krzeska, M.F. von Hippel-Lindau protein binds hyperphosphorylated large subunit of RNA polymerase II through a proline hydroxylation motif and targets it for ubiquitination. Proc. Natl. Acad. Sci. USA 2003, 100, 2706–2711. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wu, T.; Simon, J.; Takada, M.; Saito, R.; Fan, C.; Liu, X.D.; Jonasch, E.; Xie, L.; Chen, X.; et al. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science 2018, 361, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Simon, J.M.; Xie, H.; Hu, L.; Wang, J.; Zurlo, G.; Fan, C.; Ptacek, T.S.; Herring, L.; Tan, X.; et al. Genome-wide Screening Identifies SFMBT1 as an Oncogenic Driver in Cancer with VHL Loss. Mol. Cell 2020, 77, 1294–1306.e5. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.J.; Dhanasekaran, S.M.; Petralia, F.; Pan, J.; Song, X.; Hu, Y.; da Veiga Leprevost, F.; Reva, B.; Lih, T.-S.M.; Chang, H.-Y.; et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 2019, 179, 964–983.e31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corona, A.; Blobe, G.C. The role of the extracellular matrix protein TGFBI in cancer. Cell. Signal. 2021, 84, 110028. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Lin, S.C.; Yu, K.J.; Yu, G.; Song, J.H.; Lewis, V.O.; Bird, J.E.; Moon, B.; Lin, P.P.; Tannir, N.M.; et al. BIGH3 Promotes Osteolytic Lesions in Renal Cell Carcinoma Bone Metastasis by Inhibiting Osteoblast Differentiation. Neoplasia 2018, 20, 32–43. [Google Scholar] [CrossRef]
- Lebdai, S.; Verhoest, G.; Parikh, H.; Jacquet, S.F.; Bensalah, K.; Chautard, D.; Leclercq, N.R.; Azzouzi, A.R.; Bigot, P. Identification and Validation of TGFBI as a Promising Prognosis Marker of Clear Cell Renal Cell Carcinoma. Urol. Oncol. Semin. Orig. Investig. 2015, 33, 69. e11-8. [Google Scholar] [CrossRef]
- Shang, D.; Liu, Y.; Yang, P.; Chen, Y.; Tian, Y. TGFBI-promoted adhesion, migration and invasion of human renal cell carcinoma depends on inactivation of von Hippel-Lindau tumor suppressor. Urology 2012, 79, 966.e1–966.e7. [Google Scholar] [CrossRef]
- Ivanov, S.V.; Ivanova, A.V.; Salnikow, K.; Timofeeva, O.; Subramaniam, M.; Lerman, M.I. Two novel VHL targets, TGFBI (BIGH3) and its transactivator KLF10, are up-regulated in renal clear cell carcinoma and other tumors. Biochem. Biophys. Res. Commun. 2008, 370, 536–540. [Google Scholar] [CrossRef] [Green Version]
- Lua, J.; Qayyum, T.; Edwards, J.; Roseweir, A.K. The Prognostic Role of the Non-Canonical Nuclear Factor-Kappa B Pathway in Renal Cell Carcinoma Patients. Urol. Int. 2018, 101, 190–196. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Amir, R.E.; Haecker, H.; Karin, M.; Ciechanover, A. Mechanism of processing of the NF-kappa B2 p100 precursor: Identification of the specific polyubiquitin chain-anchoring lysine residue and analysis of the role of NEDD8-modification on the SCF(beta-TrCP) ubiquitin ligase. Oncogene 2004, 23, 2540–2547. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Rettig, M.B. Mechanism of von Hippel-Lindau protein-mediated suppression of nuclear factor kappa B activity. Mol. Cell. Biol. 2005, 25, 7546–7556. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Minamishima, Y.A.; Yan, Q.; Schlisio, S.; Ebert, B.L.; Zhang, X.; Zhang, L.; Kim, W.Y.; Olumi, A.F.; Kaelin, W.G., Jr. pVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF-kappaB agonist Card9 by CK2. Mol. Cell 2007, 28, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhang, L.; Zhang, X.; Yan, Q.; Minamishima, Y.A.; Olumi, A.F.; Mao, M.; Bartz, S.; Kaelin, W.G., Jr. Hypoxia-inducible factor linked to differential kidney cancer risk seen with type 2A and type 2B VHL mutations. Mol. Cell. Biol. 2007, 27, 5381–5392. [Google Scholar] [CrossRef] [Green Version]
- Ponomarev, V.; Doubrovin, M.; Serganova, I.; Vider, J.; Shavrin, A.; Beresten, T.; Ivanova, A.; Ageyeva, L.; Tourkova, V.; Balatoni, J.; et al. A novel triple-modality reporter gene for whole-body fluorescent, bioluminescent, and nuclear noninvasive imaging. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 740–751. [Google Scholar] [CrossRef]
- Huang, T.; Cheng, X.; Chahoud, J.; Sarhan, A.; Tamboli, P.; Rao, P.; Guo, M.; Manyam, G.; Zhang, L.; Xiang, Y.; et al. Effective combinatorial immunotherapy for penile squamous cell carcinoma. Nat. Commun. 2020, 11, 2124. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.D.; Elliott, A.Y.; Stein, N.; Fraley, E.E. In vitro cultivation of human renal cell cancer. II. Characterization of cell lines. In Vitro 1978, 14, 779–786. [Google Scholar] [CrossRef]
- Iliopoulos, O.; Kibel, A.; Gray, S.; Kaelin, W.G., Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat. Med. 1995, 1, 822–826. [Google Scholar] [CrossRef]
- Vanharanta, S.; Shu, W.; Brenet, F.; Hakimi, A.A.; Heguy, A.; Viale, A.; Reuter, V.E.; Hsieh, J.J.; Scandura, J.M.; Massague, J. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat. Med. 2013, 19, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 307–603. [Google Scholar] [CrossRef]
- Ho Sui, S.J.; Mortimer, J.R.; Arenillas, D.J.; Brumm, J.; Walsh, C.J.; Kennedy, B.P.; Wasserman, W.W. oPOSSUM: Identification of over-represented transcription factor binding sites in co-expressed genes. Nucleic Acids Res. 2005, 33, 3154–3164. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Xie, H.; Liu, X.; Potjewyd, F.; James, L.I.; Wilkerson, E.M.; Herring, L.E.; Xie, L.; Chen, X.; Cabrera, J.C.; et al. TBK1 Is a Synthetic Lethal Target in Cancer with VHL Loss. Cancer Discov. 2020, 10, 460–475. [Google Scholar] [CrossRef]
- Iconomou, M.; Saunders, D.N. Systematic approaches to identify E3 ligase substrates. Biochem. J. 2016, 473, 4083–4101. [Google Scholar] [CrossRef] [Green Version]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Bonicalzi, M.E.; Groulx, I.; de Paulsen, N.; Lee, S. Role of exon 2-encoded beta -domain of the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2001, 276, 1407–1416. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.; Robertson, E.S. Ubiquitin/SUMO modification regulates VHL protein stability and nucleocytoplasmic localization. PLoS ONE 2010, 5, e12636. [Google Scholar] [CrossRef]
- Kurban, G.; Hudon, V.; Duplan, E.; Ohh, M.; Pause, A. Characterization of a von Hippel Lindau pathway involved in extracellular matrix remodeling, cell invasion, and angiogenesis. Cancer Res. 2006, 66, 1313–1319. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, P.; Patel, S.A.; Harewood, L.; Olan, I.; Vojtasova, E.; Syafruddin, S.E.; Zaini, M.N.; Richardson, E.K.; Burge, J.; Warren, A.Y.; et al. NF-κB-Dependent Lymphoid Enhancer Co-option Promotes Renal Carcinoma Metastasis. Cancer Discov. 2018, 8, 850–865. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Ohh, M. The von Hippel-Lindau Tumor Suppressor Protein Sensitizes Renal Cell Carcinoma Cells to Tumor Necrosis Factor-Induced Cytotoxicity By Suppressing the Nuclear Factor-κB-Dependent Antiapoptotic Pathway. Cancer Res. 2003, 63, 7076–7080. [Google Scholar]
- Ho, T.H.; Serie, D.J.; Parasramka, M.; Cheville, J.C.; Bot, B.M.; Tan, W.; Wang, L.; Joseph, R.W.; Hilton, T.; Leibovich, B.C.; et al. Differential gene expression profiling of matched primary renal cell carcinoma and metastases reveals upregulation of extracellular matrix genes. Ann. Oncol. 2017, 28, 604–610. [Google Scholar] [CrossRef]
- Pathan, M.; Keerthikumar, S.; Ang, C.S.; Gangoda, L.; Quek, C.Y.; Williamson, N.A.; Mouradov, D.; Sieber, O.M.; Simpson, R.J.; Salim, A.; et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [Google Scholar] [CrossRef]
- Braun, D.A.; Hou, Y.; Bakouny, Z.; Ficial, M.; Sant’ Angelo, M.; Forman, J.; Ross-Macdonald, P.; Berger, A.C.; Jegede, O.A.; Elagina, L.; et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 2020, 26, 909–918. [Google Scholar] [CrossRef]
- Hascoet, P.; Chesnel, F.; Jouan, F.; Le Goff, C.; Couturier, A.; Darrigrand, E.; Mahe, F.; Rioux-Leclercq, N.; Le Goff, X.; Arlot-Bonnemains, Y. The pVHL(172) isoform is not a tumor suppressor and up-regulates a subset of pro-tumorigenic genes including TGFB1 and MMP13. Oncotarget 2017, 8, 75989–76002. [Google Scholar] [CrossRef]
- Sinha, S.; Mondal, G.; Hwang, E.J.; Han, D.W.; Dutta, S.K.; Iyer, S.; Karumanchi, S.A.; Kim, K.I.; Couch, F.J.; Mukhopadhyay, D. Von Hippel-Lindau gene product directs cytokinesis: A new tumor suppressor function. J. Cell Sci. 2011, 124 Pt 13, 2132–2142. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.H.; Wu, Z.X.; Xie, L.Q.; Li, C.X.; Mao, Y.Q.; Duan, Y.T.; Han, B.; Han, S.F.; Yu, Y.; Lu, H.J.; et al. VHL deficiency augments anthracycline sensitivity of clear cell renal cell carcinomas by down-regulating ALDH2. Nat. Commun. 2017, 8, 15337. [Google Scholar] [CrossRef] [Green Version]
- Lozano, G. Restoring p53 in cancer: The promises and the challenges. J. Mol. Cell. Biol. 2019, 11, 615–619. [Google Scholar] [CrossRef]
- Chen, C.; Tian, A.; Zhao, M.; Ma, X. Adenoviral delivery of VHL suppresses bone sarcoma cell growth through inhibition of Wnt/β-catenin signaling. Cancer Gene Ther. 2019, 26, 83–93. [Google Scholar] [CrossRef]
- Sun, X.; Kanwar, J.R.; Leung, E.; Vale, M.; Krissansen, G.W. Regression of solid tumors by engineered overexpression of von Hippel-Lindau tumor suppressor protein and antisense hypoxia-inducible factor-1alpha. Gene Ther. 2003, 10, 2081–2089. [Google Scholar] [CrossRef]
- Islam, M.A.; Xu, Y.; Tao, W.; Ubellacker, J.M.; Lim, M.; Aum, D.; Lee, G.Y.; Zhou, K.; Zope, H.; Yu, M.; et al. Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat. Biomed. Eng. 2018, 2, 850–864. [Google Scholar] [CrossRef]
- Kong, N.; Tao, W.; Ling, X.; Wang, J.; Xiao, Y.; Shi, S.; Ji, X.; Shajii, A.; Gan, S.T.; Kim, N.Y.; et al. Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci. Transl. Med. 2019, 11, eaaw1565. [Google Scholar] [CrossRef]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and Beyond. Front. Oncol. 2020, 10, 486. [Google Scholar] [CrossRef]
- Ramakrishnan, S.K.; Taylor, M.; Qu, A.; Ahn, S.-H.; Suresh, M.V.; Raghavendran, K.; Gonzalez, F.J.; Shah, Y.M. Loss of von Hippel-Lindau protein (VHL) increases systemic cholesterol levels through targeting hypoxia-inducible factor 2α and regulation of bile acid homeostasis. Mol. Cell. Biol. 2014, 34, 1208–1220. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.I.L.; Watson, I.R.; Der, S.D.; Ohh, M. Loss of VHL Confers Hypoxia-Inducible Factor (HIF)-Dependent Resistance to Vesicular Stomatitis Virus: Role of HIF in Antiviral Response. J. Virol. 2006, 80, 10712–10723. [Google Scholar] [CrossRef] [Green Version]
- Mazumdar, J.; Hickey, M.M.; Pant, D.K.; Durham, A.C.; Sweet-Cordero, A.; Vachani, A.; Jacks, T.; Chodosh, L.A.; Kissil, J.L.; Simon, M.C.; et al. HIF-2α deletion promotes Kras-driven lung tumor development. Proc. Natl. Acad. Sci. USA 2010, 107, 14182–14187. [Google Scholar] [CrossRef] [Green Version]
- Bertout, J.A.; Majmundar, A.J.; Gordan, J.D.; Lam, J.C.; Ditsworth, D.; Keith, B.; Brown, E.J.; Nathanson, K.L.; Simon, M.C. HIF2α inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc. Natl. Acad. Sci. USA 2009, 106, 14391–14396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnaird, A.; Boukouris, A.E.; Saleme, B.; Dromparis, P.; Zervopoulos, S.D.; Gurtu, V.; Sutendra, G.; Michelakis, E.D. Interaction with p53 explains a pro-proliferative function for VHL in cancer. J. Mol. Med. 2020, 98, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Schokrpur, S.; Hu, J.; Moughon, D.L.; Liu, P.; Lin, L.C.; Hermann, K.; Mangul, S.; Guan, W.; Pellegrini, M.; Xu, H.; et al. CRISPR-Mediated VHL Knockout Generates an Improved Model for Metastatic Renal Cell Carcinoma. Sci. Rep. 2016, 6, 29032. [Google Scholar] [CrossRef] [Green Version]
- Bex, C.; Knauth, K.; Dambacher, S.; Buchberger, A. A yeast two-hybrid system reconstituting substrate recognition of the von Hippel-Lindau tumor suppressor protein. Nucleic Acids Res. 2007, 35, e142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.H.; Kim, H.N.; Kim, H.; Yoo, Y.; Shin, H.; Choi, E.Y.; Bae, J.-S.; Lee, W. Acetylated K676 TGFBIp as a severity diagnostic blood biomarker for SARS-CoV-2 pneumonia. Sci. Adv. 2020, 6, eabc1564. [Google Scholar] [CrossRef] [PubMed]
- Grönroos, E.; Hellman, U.; Heldin, C.H.; Ericsson, J. Control of Smad7 stability by competition between acetylation and ubiquitination. Mol. Cell 2002, 10, 483–493. [Google Scholar] [CrossRef]
- Samaras, P.; Schmidt, T.; Frejno, M.; Gessulat, S.; Reinecke, M.; Jarzab, A.; Zecha, J.; Mergner, J.; Giansanti, P.; Ehrlich, H.-C.; et al. ProteomicsDB: A multi-omics and multi-organism resource for life science research. Nucleic Acids Res. 2019, 48, D1153–D1163. [Google Scholar] [CrossRef]
- Perroud, B.; Ishimaru, T.; Borowsky, A.D.; Weiss, R.H. Grade-dependent proteomics characterization of kidney cancer. Mol. Cell. Proteom. MCP 2009, 8, 971–985. [Google Scholar] [CrossRef] [Green Version]
- Oppenheimer, S.R.; Mi, D.; Sanders, M.E.; Caprioli, R.M. Molecular analysis of tumor margins by MALDI mass spectrometry in renal carcinoma. J. Proteome Res. 2010, 9, 2182–2190. [Google Scholar] [CrossRef] [Green Version]
- Senturk, A.; Sahin, A.T.; Armutlu, A.; Kiremit, M.C.; Acar, O.; Erdem, S.; Bagbudar, S.; Esen, T.; Tuncbag, N.; Ozlu, N. Quantitative Proteomics Identifies Secreted Diagnostic Biomarkers as well as Tumor-Dependent Prognostic Targets for Clear Cell Renal Cell Carcinoma. Mol. Cancer Res. 2021, 19, 1322–1337. [Google Scholar] [CrossRef]
- Gerlinger, M.; Santos, C.R.; Spencer-Dene, B.; Martinez, P.; Endesfelder, D.; Burrell, R.A.; Vetter, M.; Jiang, M.; Saunders, R.E.; Kelly, G.; et al. Genome-wide RNA interference analysis of renal carcinoma survival regulators identifies MCT4 as a Warburg effect metabolic target. J. Pathol. 2012, 227, 146–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooney, N.; Mason, S.M.; McDonald, L.; Däbritz, J.H.M.; Campbell, K.J.; Hedley, A.; Howard, S.; Athineos, D.; Nixon, C.; Clark, W.; et al. RUNX1 Is a Driver of Renal Cell Carcinoma Correlating with Clinical Outcome. Cancer Res. 2020, 80, 2325–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | log2 Fold Change (Tumor/Normal) | Adjusted p Values (Tumor vs. Normal) | Protein Name | log2 Fold Change (Tumor/Normal) | Adjusted p Values (Tumor vs. Normal) |
|---|---|---|---|---|---|
| FTL | 2.53 | 4.2 × 10−12 | HMOX1 | 0.74 | 1.2 × 10−29 |
| SLC16A3 | 2.32 | 1.9 × 10−52 | RHBDF2 | 0.74 | 1.6 × 10−38 |
| PLOD2 | 2.29 | 7.4 × 10−33 | PARP9 | 0.74 | 3.3 × 10−38 |
| PYGL | 2.04 | 2.1 × 10−43 | SMC4 | 0.72 | 2.7 × 10−45 |
| SCARB1 | 1.95 | 9.8 × 10−34 | DDX60 | 0.71 | 4 × 10−40 |
| TGM2 | 1.62 | 1.4 × 10−37 | HM13 | 0.7 | 1.4 × 10−23 |
| GYS1 | 1.52 | 1.7 × 10−44 | DENND3 | 0.7 | 2.1 × 10−47 |
| HLA-B | 1.26 | 1.1 × 10−30 | RNF213 | 0.7 | 5.1 × 10−35 |
| NEK6 | 1.24 | 1.3 × 10−51 | NFKB2 | 0.67 | 1.1 × 10−39 |
| DPP9 | 1.12 | 6 × 10−53 | APAF1 | 0.67 | 9.5 × 10−53 |
| APOL2 | 1.11 | 2.8 × 10−24 | RRP1 | 0.64 | 3.1 × 10−53 |
| ERGIC1 | 1.09 | 1.1 × 10−35 | SRM | 0.64 | 6.9 × 10−33 |
| UBE2L6 | 1.07 | 1 × 10−61 | CAD | 0.63 | 5.5 × 10−56 |
| OAS3 | 1.02 | 5.3 × 10−44 | SLC39A14 | 0.61 | 2 × 10−10 |
| ALDOA | 1.01 | 7.6 × 10−57 | HELZ2 | 0.61 | 1.4 × 10−27 |
| PLEKHA2 | 0.98 | 4.1 × 10−49 | TBC1D2 | 0.61 | 7.8 × 10−29 |
| MYO9B | 0.97 | 9.6 × 10−69 | CNDP2 | 0.6 | 3.9 × 10−23 |
| IMPDH1 | 0.95 | 8.3 × 10−49 | CDK17 | 0.59 | 7.3 × 10−45 |
| TGFBI | 0.93 | 2.4 × 10−15 | GFPT1 | 0.59 | 1.9 × 10−29 |
| TRIM22 | 0.92 | 8.9 × 10−49 | ARHGEF1 | 0.59 | 4.5 × 10−55 |
| EHD2 | 0.88 | 8 × 10−28 | NAP1L1 | 0.57 | 3.7 × 10−45 |
| HLA-C | 0.87 | 1.7 × 10−19 | IPO9 | 0.56 | 4.1 × 10−64 |
| RUNX1 | 0.87 | 2.7 × 10−37 | MTHFD2 | 0.54 | 4.8 × 10−16 |
| ANXA2 | 0.86 | 7.7 × 10−49 | ASCC3 | 0.54 | 3.9 × 10−57 |
| PARP14 | 0.85 | 3.8 × 10−48 | COL6A2 | 0.52 | 5.1 × 10−16 |
| FNDC3B | 0.81 | 3.8 × 10−46 | TBC1D2B | 0.51 | 2.5 × 10−36 |
| ASNS | 0.78 | 1.7 × 10−27 | PPIP5K2 | 0.51 | 1.1 × 10−41 |
| AMPD2 | 0.76 | 1.3 × 10−38 | GSDMD | 0.51 | 1.3 × 10−41 |
| LAMB2 | 0.75 | 1.1 × 10−26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Hu, J.; Fang, Y.; Fu, Y.; Liu, B.; Zhang, C.; Feng, S.; Lu, X. Multi-Omics Profiling to Assess Signaling Changes upon VHL Restoration and Identify Putative VHL Substrates in Clear Cell Renal Cell Carcinoma Cell Lines. Cells 2022, 11, 472. https://doi.org/10.3390/cells11030472
Wang X, Hu J, Fang Y, Fu Y, Liu B, Zhang C, Feng S, Lu X. Multi-Omics Profiling to Assess Signaling Changes upon VHL Restoration and Identify Putative VHL Substrates in Clear Cell Renal Cell Carcinoma Cell Lines. Cells. 2022; 11(3):472. https://doi.org/10.3390/cells11030472
Chicago/Turabian StyleWang, Xuechun, Jin Hu, Yihao Fang, Yanbin Fu, Bing Liu, Chao Zhang, Shan Feng, and Xin Lu. 2022. "Multi-Omics Profiling to Assess Signaling Changes upon VHL Restoration and Identify Putative VHL Substrates in Clear Cell Renal Cell Carcinoma Cell Lines" Cells 11, no. 3: 472. https://doi.org/10.3390/cells11030472
APA StyleWang, X., Hu, J., Fang, Y., Fu, Y., Liu, B., Zhang, C., Feng, S., & Lu, X. (2022). Multi-Omics Profiling to Assess Signaling Changes upon VHL Restoration and Identify Putative VHL Substrates in Clear Cell Renal Cell Carcinoma Cell Lines. Cells, 11(3), 472. https://doi.org/10.3390/cells11030472