
A Systemic Review of the Integral Role of TRPM2 in Ischemic Stroke: From Upstream Risk Factors to Ultimate Neuronal Death



Abstract
:1. Introduction
1.1. Stroke
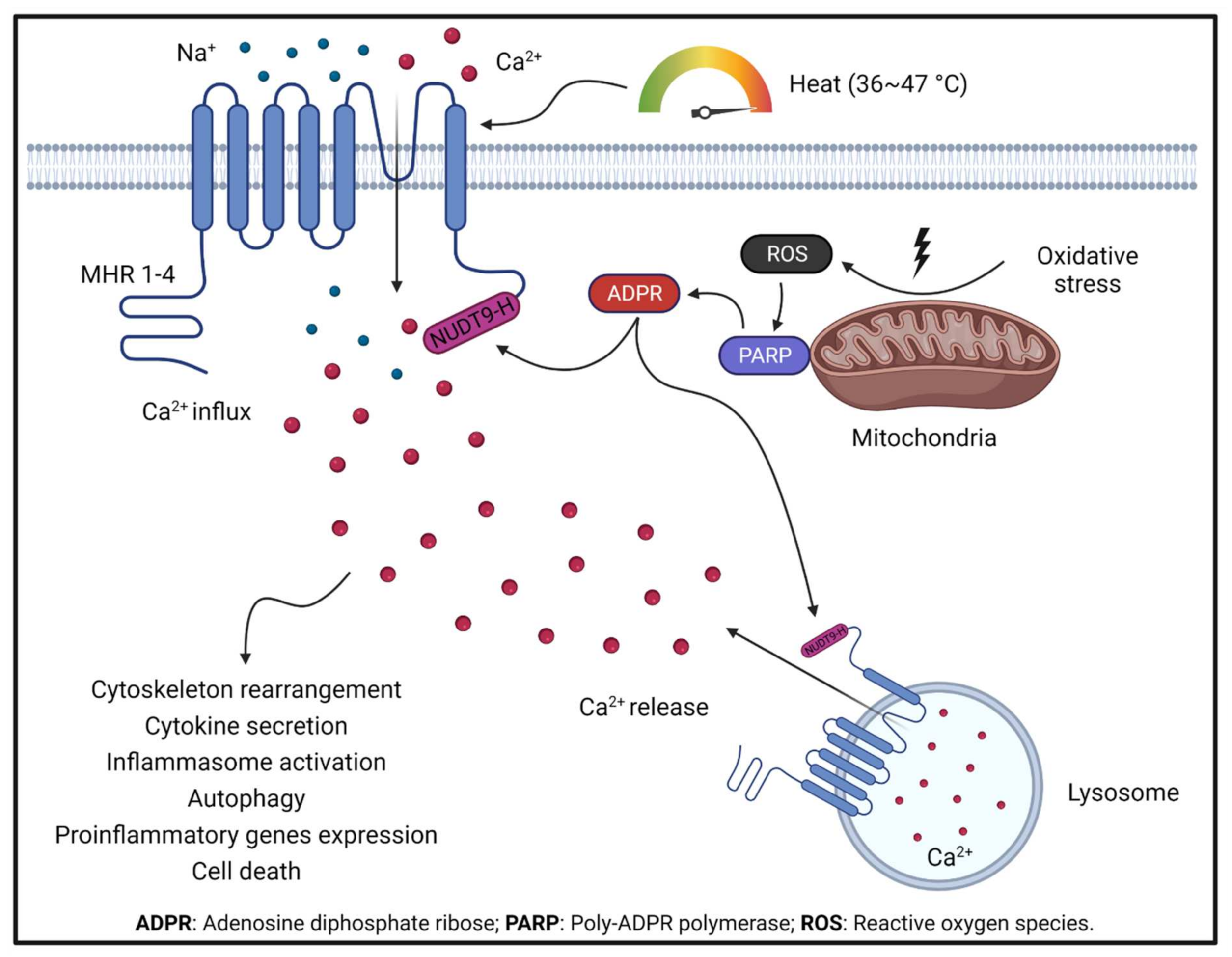
1.2. TRPM2
2. Trpm2 in Diseases Increasing Risk for Ischemic Stroke
2.1. Atrial Fibrillation
2.2. TRPM2 in Hypertension
2.3. TRPM2 in Atherosclerosis
2.4. TRPM2 in Diabetes
2.5. TRPM2 in Thrombosis
3. Mechanisms by Which TRPM2 Increases Brain Injury during Ischemic Stroke
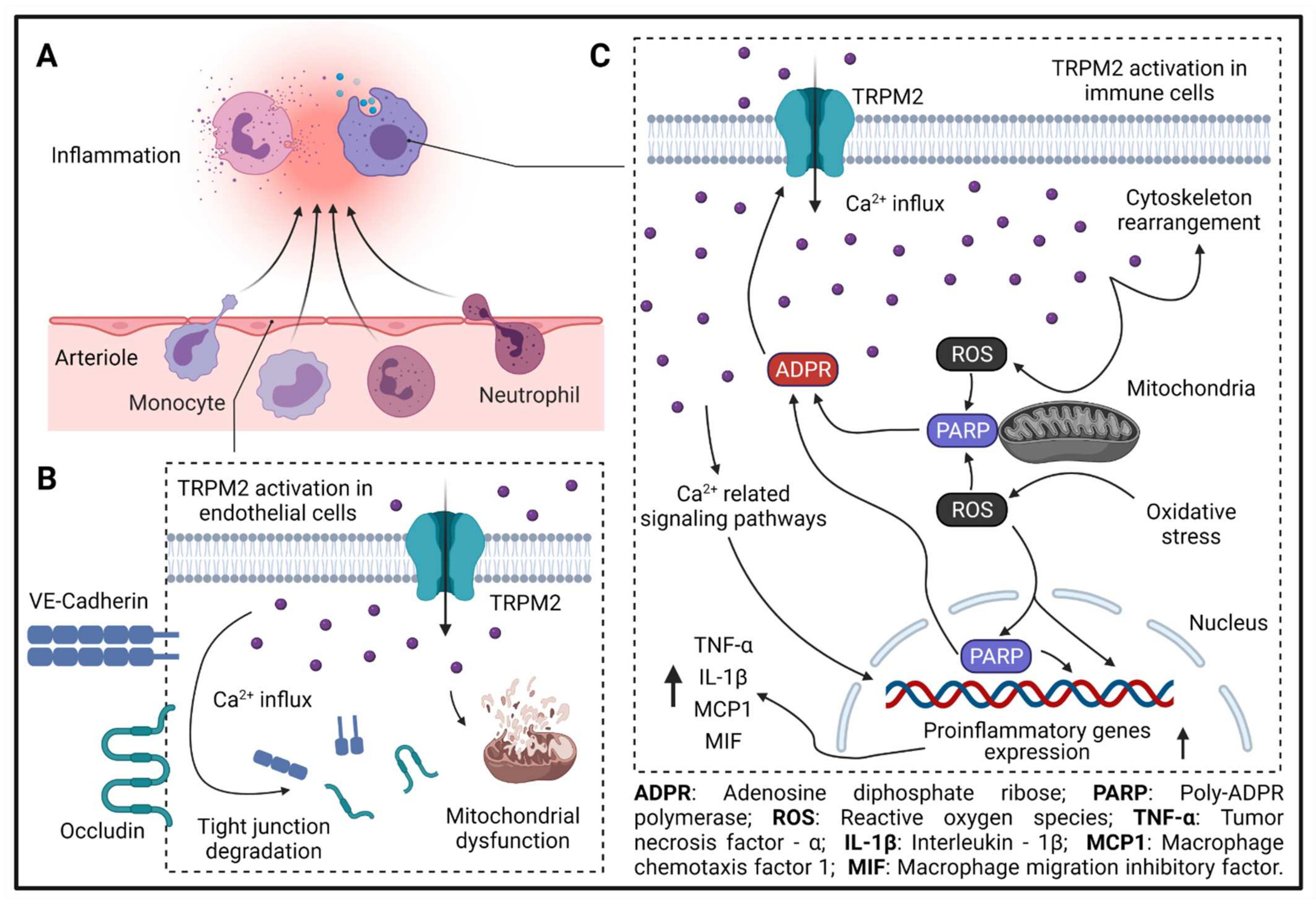
3.1. TRPM2 and Cerebral Endothelial Hyperpermeability
3.2. TRPM2 and Immune-Cell Invasion
3.3. TRPM2 in Glial Cells
3.4. Neuronal TRPM2 and Neuron Death
4. Other TRPM Channels in Ischemic Stroke
5. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, C.O.; Nguyen, M.; Roth, G.A.; Nichols, E.; Alam, T.; Abate, D.; Abd-Allah, F.; Abdelalim, A.; Abraha, H.N.; Abu-Rmeileh, N.M.; et al. Global, regional, and national burden of stroke, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 439–458. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.C.V.; De Silva, D.A.; MacLeod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Prim. 2019, 5, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.V.; Khatri, P. Stroke. Lancet 2020, 396, 129–142. [Google Scholar] [CrossRef]
- Allen, C.L.; Bayraktutan, U. Risk Factors for Ischaemic Stroke. Int. J. Stroke 2008, 3, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Hawkins, K.E.; Doré, S.; Candelario-Jalil, E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am. J. Physiol. Cell Physiol. 2019, 316, C135–C153. [Google Scholar] [CrossRef]
- Planas, A.M. Role of Immune Cells Migrating to the Ischemic Brain. Stroke 2018, 49, 2261–2267. [Google Scholar] [CrossRef]
- Bardutzky, J.; Schwab, S. Antiedema Therapy in Ischemic Stroke. Stroke 2007, 38, 3084–3094. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Chopp, M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog. Neurobiol. 2015, 144, 103–120. [Google Scholar] [CrossRef] [Green Version]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 142. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.H.-C.; Weigel, H.; Cotrina, M.L.; Liu, S.; Bueno, E.; Hansen, A.J.; Hansen, T.W.; Goldman, S.A.; Nedergaard, M. Gap-junction-mediated propagation and amplification of cell injury. Nat. Neurosci. 1998, 1, 494–500. [Google Scholar] [CrossRef]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/Macrophage Polarization Dynamics Reveal Novel Mechanism of Injury Expansion After Focal Cerebral Ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [Green Version]
- Morizawa, Y.M.; Hirayama, Y.; Ohno, N.; Shibata, S.; Shigetomi, E.; Sui, Y.; Nabekura, J.; Sato, K.; Okajima, F.; Takebayashi, H.; et al. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 2017, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Gunzer, M.; Gutzeit, H.O.; Ullrich, O.; Reymann, K.G.; Dinkel, K. Microglia provide neuroprotection after ischemia. FASEB J. 2006, 20, 714–716. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Calixto, A.; Cardona-Gómez, G.P. The Role of Astrocytes in Neuroprotection after Brain Stroke: Potential in Cell Therapy. Front. Mol. Neurosci. 2017, 10, 88. [Google Scholar] [CrossRef] [Green Version]
- Lipton, P. Ischemic Cell Death in Brain Neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar] [CrossRef]
- Cheng, Y.D.; Al-Khoury, L.; Zivin, J.A. Neuroprotection for ischemic stroke: Two decades of success and failure. NeuroRX 2004, 1, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, Y.; Inamura, K.; Miyake, A.; Mochizuki, S.; Yokoi, H.; Matsushime, H.; Furuichi, K. Immunocyte Ca2+ Influx System Mediated by LTRPC2. Science 2001, 293, 1327–1330. [Google Scholar] [CrossRef]
- Perraud, A.-L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599. [Google Scholar] [CrossRef]
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-Permeable Channel Activated by Changes in Redox Status Confers Susceptibility to Cell Death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Montell, C.; Birnbaumer, L.; Flockerzi, V. The TRP Channels, a Remarkably Functional Family. Cell 2002, 108, 595–598. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Voets, T.; Peters, J. TRP Channels in Disease. Sci. STKE 2005, 1–9, [Epub ahead of print]. [Google Scholar] [CrossRef] [PubMed]
- McQuillin, A.; Bass, N.J.; Kalsi, G.; Lawrence, J.; Puri, T.; Choudhury, K.; Detera-Wadleigh, S.D.; Curtis, D.; Gurling, H.M.D. Fine mapping of a susceptibility locus for bipolar and genetically related unipolar affective disorders, to a region containing the C21ORF29 and TRPM2 genes on chromosome 21q22.3. Mol. Psychiatry 2005, 11, 134–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Winkler, P.A.; Sun, W.; Lü, W.; Du, J. Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nature 2018, 562, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Perraud, A.-L.; Shen, B.; Dunn, C.A.; Rippe, K.; Smith, M.K.; Bessman, M.J.; Stoddard, B.L.; Scharenberg, A.M. NUDT9, a Member of the Nudix Hydrolase Family, Is an Evolutionarily Conserved Mitochondrial ADP-ribose Pyrophosphatase. J. Biol. Chem. 2003, 278, 1794–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iordanov, I.; Tóth, B.; Szollosi, A.; Csanády, L. Enzyme activity and selectivity filter stability of ancient TRPM2 channels were simultaneously lost in early vertebrates. eLife 2019, 8, e44556. [Google Scholar] [CrossRef]
- Iordanov, I.; Mihályi, C.; Tóth, B.; Csanády, L. The proposed channel-enzyme transient receptor potential melastatin 2 does not possess ADP ribose hydrolase activity. eLife 2016, 5, e17600. [Google Scholar] [CrossRef]
- Kühn, F.J.P.; Lückhoff, A. Sites of the NUDT9-H Domain Critical for ADP-ribose Activation of the Cation Channel TRPM2. J. Biol. Chem. 2004, 279, 46431–46437. [Google Scholar] [CrossRef] [Green Version]
- Perraud, A.-L.; Schmitz, C.; Scharenberg, A.M. TRPM2 Ca2+ permeable cation channels: From gene to biological function. Cell Calcium 2003, 33, 519–531. [Google Scholar] [CrossRef]
- Lange, I.; Yamamoto, S.; Partida-Sanchez, S.; Mori, Y.; Fleig, A.; Penner, R. TRPM2 Functions as a Lysosomal Ca2+-Release Channel in beta cells. Sci. Signal. 2009, 2, ra23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumoza-Toledo, A.; Lange, I.; Cortado, H.; Bhagat, H.; Mori, Y.; Fleig, A.; Penner, R.; Partida-Sánchez, S. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011, 25, 3529–3542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Xie, J.; Yue, L. Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc. Natl. Acad. Sci. USA 2009, 106, 7239–7244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraft, R.; Grimm, C.; Frenzel, H.; Harteneck, C. Inhibition of TRPM2 cation channels by N -(p -amylcinnamoyl)anthranilic acid. J. Cereb. Blood Flow Metab. 2006, 148, 264–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autzen, H.E.; Myasnikov, A.G.; Campbell, M.G.; Asarnow, D.; Julius, D.; Cheng, Y. Structure of the human TRPM4 ion channel in a lipid nanodisc. Science 2018, 359, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Fliegert, R.; Guse, A.H.; Lü, W.; Du, J. A structural overview of the ion channels of the TRPM family. Cell Calcium 2019, 85, 102111. [Google Scholar] [CrossRef]
- Yin, Y.; Le, S.C.; Hsu, A.L.; Borgnia, M.J.; Yang, H.; Lee, S.-Y. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 2019, 363, eaav9334. [Google Scholar] [CrossRef]
- Zhang, Z.; Tóth, B.; Szollosi, A.; Chen, J.; Csanády, L. Structure of a TRPM2 channel in complex with Ca2+ explains unique gating regulation. eLife 2018, 7, e36409. [Google Scholar] [CrossRef]
- Csanády, L.; Törőcsik, B. Four Ca2+ Ions Activate TRPM2 Channels by Binding in Deep Crevices near the Pore but Intracellularly of the Gate. J. Gen. Physiol. 2009, 133, 189–203. [Google Scholar] [CrossRef]
- Wang, L.; Fu, T.-M.; Zhou, Y.; Xia, S.; Greka, A.; Wu, H. Structures and gating mechanism of human TRPM2. Science 2018, 362, 6421. [Google Scholar] [CrossRef] [Green Version]
- Vilar, B.; Tan, C.-H.; McNaughton, P.A. Heat detection by the TRPM2 ion channel. Nature 2020, 584, E5–E12. [Google Scholar] [CrossRef] [PubMed]
- Kashio, M.; Sokabe, T.; Shintaku, K.; Uematsu, T.; Fukuta, N.; Kobayashi, N.; Mori, Y.; Tominaga, M. Redox signal-mediated sensitization of transient receptor potential melastatin 2 (TRPM2) to temperature affects macrophage functions. Proc. Natl. Acad. Sci. USA 2012, 109, 6745–6750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashio, M.; Tominaga, M. The TRPM2 channel: A thermo-sensitive metabolic sensor. Channels 2017, 11, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Perraud, A.-L.; Takanishi, C.L.; Shen, B.; Kang, S.; Smith, M.K.; Schmitz, C.; Knowles, H.M.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of Free ADP-ribose from Mitochondria Mediates Oxidative Stress-induced Gating of TRPM2 Cation Channels. J. Biol. Chem. 2005, 280, 6138–6148. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Kozai, D.; Kobayashi, R.; Ebert, M.; Mori, Y. Roles of TRPM2 in oxidative stress. Cell Calcium 2011, 50, 279–287. [Google Scholar] [CrossRef]
- Evans, S.S.; Repasky, E.A.; Fisher, D.T. Fever and the thermal regulation of immunity: The immune system feels the heat. Nat. Rev. Immunol. 2015, 15, 335–349. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 2018, 9, 1487. [Google Scholar] [CrossRef] [Green Version]
- Fonfria, E.; Murdock, P.R.; Cusdin, F.S.; Benham, C.D.; Kelsell, R.E.; McNulty, S. Tissue Distribution Profiles of the Human TRPM Cation Channel Family. J. Recept. Signal Transduct. 2006, 26, 159–178. [Google Scholar] [CrossRef]
- Zhong, Z.; Zhai, Y.; Liang, S.; Mori, Y.; Han, R.; Sutterwala, F.S.; Qiao, L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 2013, 4, 1611. [Google Scholar] [CrossRef]
- Mittal, M.; Nepal, S.; Tsukasaki, Y.; Hecquet, C.M.; Soni, D.; Rehman, J.; Tiruppathi, C.; Malik, A.B. Neutrophil Activation of Endothelial Cell-Expressed TRPM2 Mediates Transendothelial Neutrophil Migration and Vascular Injury. Circ. Res. 2017, 121, 1081–1091. [Google Scholar] [CrossRef]
- Di, A.; Gao, X.-P.; Qian, F.; Kawamura, T.; Han, J.; Hecquet, C.M.; Ye, R.D.; Vogel, S.M.; Malik, A.B. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat. Immunol. 2011, 13, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almasi, S.; Kennedy, B.E.; El-Aghil, M.; Sterea, A.M.; Gujar, S.; Partida-Sánchez, S.; El Hiani, Y. TRPM2 channel–mediated regulation of autophagy maintains mitochondrial function and promotes gastric cancer cell survival via the JNK-signaling pathway. J. Biol. Chem. 2018, 293, 3637–3650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Chu, X.; Tong, Q.; Cheung, J.Y.; Conrad, K.; Masker, K.; Miller, B.A. A Novel TRPM2 Isoform Inhibits Calcium Influx and Susceptibility to Cell Death. J. Biol. Chem. 2003, 278, 16222–16229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.A.; Hoffman, N.E.; Merali, S.; Zhang, X.-Q.; Wang, J.; Rajan, S.; Shanmughapriya, S.; Gao, E.; Barrero, C.; Mallilankaraman, K.; et al. TRPM2 Channels Protect against Cardiac Ischemia-Reperfusion Injury: Role of Mitochondria. J. Biol. Chem. 2014, 289, 7615–7629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelderblom, M.; Melzer, N.; Schattling, B.; Göb, E.; Hicking, G.; Arunachalam, P.; Bittner, S.; Ufer, F.; Herrmann, A.M.; Bernreuther, C.; et al. Transient Receptor Potential Melastatin Subfamily Member 2 Cation Channel Regulates Detrimental Immune Cell Invasion in Ischemic Stroke. Stroke A J. Cereb. Circ. 2014, 45, 3395–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, M.; Yang, W.; Ainscough, J.; Hu, X.-P.; Li, X.; Sedo, A.; Zhang, X.; Chen, Z.; Beech, D.; Sivaprasadarao, A.; et al. TRPM2 channel deficiency prevents delayed cytosolic Zn2+ accumulation and CA1 pyramidal neuronal death after transient global ischemia. Cell Death Dis. 2014, 5, e1541. [Google Scholar] [CrossRef]
- Zong, P.; Feng, J.; Yue, Z.; Wu, G.; Sun, B.; He, Y.; Miller, B.; Albert, S.Y.; Su, Z.; Mori, Y.; et al. Functional Coupling of TRPM2 and NMDARs exacerbates excitotoxicity in ischemic brain injury. bioRxiv 2021. [Google Scholar]
- Park, L.; Wang, G.; Moore, J.; Girouard, H.; Zhou, P.; Anrather, J.; Iadecola, C. The key role of transient receptor potential melastatin-2 channels in amyloid-β-induced neurovascular dysfunction. Nat. Commun. 2014, 5, 5318. [Google Scholar] [CrossRef] [Green Version]
- Ostapchenko, V.G.; Chen, M.; Guzman, M.S.; Xie, Y.-F.; LaVine, N.; Fan, J.; Beraldo, F.H.; Martyn, A.C.; Belrose, J.C.; Mori, Y.; et al. The Transient Receptor Potential Melastatin 2 (TRPM2) Channel Contributes to β-Amyloid Oligomer-Related Neurotoxicity and Memory Impairment. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 15157–15169. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, N.E.; Miller, B.A.; Wang, J.; Elrod, J.W.; Rajan, S.; Gao, E.; Song, J.; Zhang, X.-Q.; Hirschler-Laszkiewicz, I.; Shanmughapriya, S.; et al. Ca2+ entry via Trpm2 is essential for cardiac myocyte bioenergetics maintenance. Am. J. Physiol. Circ. Physiol. 2015, 308, H637–H650. [Google Scholar] [CrossRef] [Green Version]
- Düzen, I.V.; Yavuz, F.; Vuruskan, E.; Saracoglu, E.; Poyraz, F.; Göksülük, H.; Candemir, B.; Demiryürek, S. Leukocyte TRP channel gene expressions in patients with non-valvular atrial fibrillation. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Alves-Lopes, R.; Neves, K.B.; Anagnostopoulou, A.; Rios, F.J.; Lacchini, S.; Montezano, A.C.; Touyz, R.M. Crosstalk Between Vascular Redox and Calcium Signaling in Hypertension Involves TRPM2 (Transient Receptor Potential Melastatin 2) Cation Channel. Hypertension 2020, 75, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Zong, P.; Feng, J.; Yue, Z.; Albert, S.Y.; Mori, Y.; Yue, L. TRPM2 deficiency protects against atherosclerosis by inhibiting TRPM2-CD36 inflammatory axis in macrophages. bioRxiv 2021. [Google Scholar]
- Hecquet, C.M.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Role of TRPM2 Channel in Mediating H2O2 -Induced Ca2+ Entry and Endothelial Hyperpermeability. Circ. Res. 2008, 102, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Uchida, K.; Dezaki, K.; Damdindorj, B.; Inada, H.; Shiuchi, T.; Mori, Y.; Yada, T.; Minokoshi, Y.; Tominaga, M. Lack of TRPM2 Impaired Insulin Secretion and Glucose Metabolisms in Mice. Diabetes 2010, 60, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Wang, W.; Tadagavadi, R.K.; Briley, N.E.; Love, M.I.; Miller, B.A.; Reeves, W.B. TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J. Clin. Investig. 2014, 124, 4989–5001. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.A. TRPM2 in Cancer. Cell Calcium 2019, 80, 8–17. [Google Scholar] [CrossRef]
- Dardiotis, E.; Aloizou, A.-M.; Markoula, S.; Siokas, V.; Tsarouhas, K.; Tzanakakis, G.; Libra, M.; Kyritsis, A.P.; Brotis, A.; Aschner, M.; et al. Cancer-associated stroke: Pathophysiology, detection and management (Review). Int. J. Oncol. 2019, 54, 779–796. [Google Scholar] [CrossRef] [Green Version]
- Adams, H.P., Jr.; Bendixen, B.H.; Kappelle, L.J.; Biller, J.; Love, B.B.; Gordon, D.L.; Marsh, E.E., 3rd. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993, 24, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Marini, C.; De Santis, F.; Sacco, S.; Russo, T.; Olivieri, L.; Totaro, R.; Carolei, A. Contribution of Atrial Fibrillation to Incidence and Outcome of Ischemic Stroke: Results from a Population-Based Study. Stroke 2005, 36, 1115–1119. [Google Scholar] [CrossRef] [Green Version]
- Nattel, S.; Harada, M. Atrial Remodeling and Atrial Fibrillation: Recent advances and translational perspectives. J. Am. Coll. Cardiol. 2014, 63, 2335–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nattel, S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin. Electrophysiol. 2017, 3, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Staerk, L.; Sherer, J.A.; Ko, D.; Benjamin, E.; Helm, R.H. Atrial Fibrillation: Epidemiology, Pathophysiology, and Clinical Outcomes. Circ. Res. 2017, 120, 1501–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Hu, Y.-F.; Chen, Y.-J.; Lin, Y.-J.; Chen, S.-A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243. [Google Scholar] [CrossRef]
- Kakae, M.; Miyanohara, J.; Morishima, M.; Nagayasu, K.; Mori, Y.; Shirakawa, H.; Kaneko, S.; Morsishima, M. Pathophysiological role of TRPM2 in age-related cognitive impairment in mice. Neuroscience 2019, 408, 204–213. [Google Scholar] [CrossRef]
- Belrose, J.C.; Xie, Y.-F.; Gierszewski, L.J.; MacDonald, J.F.; Jackson, M.F. Loss of glutathione homeostasis associated with neuronal senescence facilitates TRPM2 channel activation in cultured hippocampal pyramidal neurons. Mol. Brain 2012, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Yue, Z.; Du, J.; Xia, J.; Qin, X.; Zhang, Y.; He, Y.; Fusco, D.; Liang, B.; Yue, L. A Potential Role of TRPM2 Mediated Inflammation in Heart Disease. Circulation 2018, 128 (Suppl. 22). [Google Scholar]
- Korantzopoulos, P.; Letsas, K.; Fragakis, N.; Tse, G.; Liu, T. Oxidative stress and atrial fibrillation: An update. Free Radic. Res. 2018, 52, 1199–1209. [Google Scholar] [CrossRef]
- Mihm, M.J.; Yu, F.; Carnes, C.; Reiser, P.J.; McCarthy, P.M.; Van Wagoner, D.R.; Bauer, J.A. Impaired Myofibrillar Energetics and Oxidative Injury During Human Atrial Fibrillation. Circulation 2001, 104, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Adam, O.; Lavall, D.; Theobald, K.; Hohl, M.; Grube, M.; Ameling, S.; Sussman, M.A.; Rosenkranz, S.; Kroemer, H.K.; Schäfers, H.-J.; et al. Rac1-Induced Connective Tissue Growth Factor Regulates Connexin 43 and N-Cadherin Expression in Atrial Fibrillation. J. Am. Coll. Cardiol. 2010, 55, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Armillei, M.K.; Yu, A.S.; Liang, B.T.; Runnels, L.W.; Yue, L. Ca2+ Signaling in Cardiac Fibroblasts and Fibrosis-Associated Heart Diseases. J. Cardiovasc. Dev. Dis. 2019, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, M.; Luo, X.; Qi, X.Y.; Tadevosyan, A.; Maguy, A.; Ördög, B.; Ledoux, J.; Kato, T.; Naud, P.; Voigt, N.; et al. Transient Receptor Potential Canonical-3 Channel–Dependent Fibroblast Regulation in Atrial Fibrillation. Circulation 2012, 126, 2051–2064. [Google Scholar] [CrossRef]
- Denham, N.; Pearman, C.; Caldwell, J.L.; Madders, G.W.P.; Eisner, D.; Trafford, A.W.; Dibb, K.M. Calcium in the Pathophysiology of Atrial Fibrillation and Heart Failure. Front. Physiol. 2018, 9, 1380. [Google Scholar] [CrossRef] [Green Version]
- Burashnikov, A.; Antzelevitch, C. Reinduction of Atrial Fibrillation Immediately After Termination of the Arrhythmia Is Mediated by Late Phase 3 Early Afterdepolarization–Induced Triggered Activity. Circulation 2003, 107, 2355–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Yeh, Y.-H.; Chartier, D.; Xiao, L.; Tsuji, Y.; Brundel, B.J.; Kodama, I.; Nattel, S. The Calcium/Calmodulin/Kinase System and Arrhythmogenic Afterdepolarizations in Bradycardia-Related Acquired Long-QT Syndrome. Circ. Arrhythmia Electrophysiol. 2009, 2, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Hankey, G.J. Potential New Risk Factors for Ischemic Stroke: What is their potential? Stroke 2006, 37, 2181–2188. [Google Scholar] [CrossRef] [Green Version]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Howard, V.J.; Madsen, T.E.; Kleindorfer, D.O.; Judd, S.E.; Rhodes, J.D.; Soliman, E.Z.; Kissela, B.M.; Safford, M.M.; Moy, C.S.; McClure, L.A.; et al. Sex and Race Differences in the Association of Incident Ischemic Stroke With Risk Factors. JAMA Neurol. 2019, 76, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Gorgui, J.; Gorshkov, M.; Khan, N.; Daskalopoulou, S.S. Hypertension as a Risk Factor for Ischemic Stroke in Women. Can. J. Cardiol. 2014, 30, 774–782. [Google Scholar] [CrossRef]
- Harrison, D.G.; Coffman, T.M.; Wilcox, C.S. Pathophysiology of Hypertension: The Mosaic Theory and Beyond. Circ. Res. 2021, 128, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Van Varik, B.J.; Rennenberg, R.J.M.W.; Reutelingsperger, C.P.; Kroon, A.A.; De Leeuw, P.W.; Schurgers, L.J. Mechanisms of arterial remodeling: Lessons from genetic diseases. Front. Genet. 2012, 3, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. In Hypertension Basic Research to Clinical Practice; Springer: Berlin/Heidelberg, Germany, 2016; pp. 511–540. [Google Scholar] [CrossRef]
- Satoh, K.; Nigro, P.; Berk, B.C. Oxidative Stress and Vascular Smooth Muscle Cell Growth: A Mechanistic Linkage by Cyclophilin A. Antioxidants Redox Signal. 2010, 12, 675–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortuño, A.; José, G.S.; Moreno, M.U.; Díez, J.; Zalba, G. Oxidative stress and vascular remodelling. Exp. Physiol. 2005, 90, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N.; Dabla, P.K. Oxidative Stress and Antioxidants in Hypertension–A Current Review. Curr. Hypertens. Rev. 2015, 11, 132–142. [Google Scholar] [CrossRef]
- Griendling, K.K.; Camargo, L.L.; Rios, F.J.; Alves-Lopes, R.; Montezano, A.C.; Touyz, R.M. Oxidative Stress and Hypertension. Circ. Res. 2021, 128, 993–1020. [Google Scholar] [CrossRef]
- Tran, Q.-K.; Ohashi, K.; Watanabe, H. Calcium signalling in endothelial cells. Cardiovasc. Res. 2000, 48, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Amberg, G.C.; Navedo, M.F. Calcium dynamics in vascular smooth muscle. Microcirculation 2013, 20, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Kuchan, M.J.; Frangos, J.A. Role of calcium and calmodulin in flow-induced nitric oxide production in endothelial cells. Am. J. Physiol. Physiol. 1994, 266, C628–C636. [Google Scholar] [CrossRef]
- Ding, Y.; Vaziri, N.D. Calcium channel blockade enhances nitric oxide synthase expression by cultured endothelial cells. Hypertension 1998, 32, 718–723. [Google Scholar] [CrossRef] [Green Version]
- Batova, S.; Dewever, J.; Balligand, J.-L.; Godfraind, T.; Dessy, C.; Feron, O. The calcium channel blocker amlodipine promotes the unclamping of eNOS from caveolin in endothelial cells. Cardiovasc. Res. 2006, 71, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Resnick, L. The cellular ionic basis of hypertension and allied clinical conditions. Prog. Cardiovasc. Dis. 1999, 42, 1–22. [Google Scholar] [CrossRef]
- Sonkusare, S.; Palade, P.T.; Marsh, J.D.; Telemaque, S.; Pesic, A.; Rusch, N.J. Vascular calcium channels and high blood pressure: Pathophysiology and therapeutic implications. Vasc. Pharmacol. 2006, 44, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Guo, W.; Hao, B.; Shi, X.; Lu, Y.; Wong, C.W.; Ma, V.W.; Yip, T.T.; Au, J.S.; Hao, Q.; et al. Mechanistic study of TRPM2-Ca2+-CAMK2-BECN1 signaling in oxidative stress-induced autophagy inhibition. Autophagy 2016, 12, 1340–1354. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Li, J.; Ko, W.-H.; Kwan, Y.-W.; Jiang, L.; Sun, L.; Yao, X. TRPM2 promotes autophagic degradation in vascular smooth muscle cells. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- McCarthy, C.G.; Wenceslau, C.F.; Calmasini, F.B.; Klee, N.S.; Brands, M.W.; Joe, B.; Webb, R.C. Reconstitution of autophagy ameliorates vascular function and arterial stiffening in spontaneously hypertensive rats. Am. J. Physiol. Circ. Physiol. 2019, 317, H1013–H1027. [Google Scholar] [CrossRef]
- Amarenco, P.; Cohen, A.; Tzourio, C.; Bertrand, B.; Hommel, M.; Besson, G.; Chauvel, C.; Touboul, P.-J.T.; Bousser, M.-G. Atherosclerotic Disease of the Aortic Arch and the Risk of Ischemic Stroke. New Engl. J. Med. 1994, 331, 1474–1479. [Google Scholar] [CrossRef]
- Joh, J.H.; Cho, S. Cardiovascular risk of carotid atherosclerosis: Global consensus beyond societal guidelines. Lancet Glob. Health 2020, 8, e625–e626. [Google Scholar] [CrossRef]
- Banerjee, C.; Chimowitz, M.I. Stroke Caused by Atherosclerosis of the Major Intracranial Arteries. Circ. Res. 2017, 120, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiner, I.; Eisfeld, J.; Warnstedt, M.U.; Radukina, N.; Jüngling, E.; Lückhoff, A. Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem. J. 2006, 398, 225–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morad, H.; Luqman, S.; Tan, C.-H.; Swann, V.; McNaughton, P.A. TRPM2 ion channels steer neutrophils towards a source of hydrogen peroxide. Sci. Rep. 2021, 11, 1–16. [Google Scholar] [CrossRef]
- Wittmann, C.; Chockley, P.; Singh, S.K.; Pase, L.; Lieschke, G.; Grabher, C. Hydrogen Peroxide in Inflammation: Messenger, Guide, and Assassin. Adv. Hematol. 2012, 2012, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, W.; Jung, D.Y.; Ko, H.J.; Lee, Y.; Friedline, R.H.; Lee, E.; Jun, J.; Ma, Z.; Kim, F.; et al. TRPM2 Ca2+ channel regulates energy balance and glucose metabolism. Am. J. Physiol. Metab. 2012, 302, E807–E816. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008, 14, 738–747. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Li, W.; Park, Y.M.; Rahaman, S.O. Mechanisms of cell signaling by the scavenger receptor CD36: Implications in atherosclerosis and thrombosis. Trans. Am. Clin. Clim. Assoc. 2010, 121, 206–220. [Google Scholar]
- Moore, K.J.; Freeman, M.W. Scavenger Receptors in Atherosclerosis: Beyond lipid uptake. Arter. Thromb. Vasc. Biol. 2006, 26, 1702–1711. [Google Scholar] [CrossRef] [Green Version]
- Baldrighi, M.; Mallat, Z.; Li, X. NLRP3 inflammasome pathways in atherosclerosis. Atherosclerosis 2017, 267, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Liu, Y.; Wang, X.; New, L.; Han, J.; Brunk, U.T. Activation of the p38 MAP kinase pathway is required for foam cell formation from macrophages exposed to oxidized LDL. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2002, 110, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Ricci, R.; Sumara, G.; Sumara, I.; Rozenberg, I.; Kurrer, M.; Akhmedov, A.; Hersberger, M.; Eriksson, U.; Eberli, F.R.; Becher, B.; et al. Requirement of JNK2 for Scavenger Receptor A-Mediated Foam Cell Formation in Atherogenesis. Science 2004, 306, 1558–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahaman, S.O.; Lennon, D.J.; Febbraio, M.; Podrez, E.A.; Hazen, S.L.; Silverstein, R.L. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006, 4, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beceiro, S.; Radin, J.N.; Chatuvedi, R.; Piazuelo, M.B.; Horvarth, D.J.; Cortado, H.; Gu, Y.; Dixon, B.; Gu, C.; Lange, I.; et al. TRPM2 ion channels regulate macrophage polarization and gastric inflammation during Helicobacter pylori infection. Mucosal Immunol. 2016, 10, 493–507. [Google Scholar] [CrossRef]
- Knowles, H.; Heizer, J.W.; Li, Y.; Chapman, K.; Ogden, C.A.; Andreasen, K.; Shapland, E.; Kucera, G.; Mogan, J.; Humann, J.; et al. Transient Receptor Potential Melastatin 2 (TRPM2) ion channel is required for innate immunity against Listeria monocytogenes. Proc. Natl. Acad. Sci. USA 2011, 108, 11578–11583. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Ainscough, J.F.; Yang, W.; Sedo, A.; Yu, S.-P.; Mei, Z.-Z.; Sivaprasadarao, A.; Beech, D.J.; Jiang, L.-H. A differential role of macrophage TRPM2 channels in Ca2+ signaling and cell death in early responses to H2O2. Am. J. Physiol. Physiol. 2013, 305, C61–C69. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361, Erratum in Nature 2010, 466, 652. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Ovbiagele, B.; Feng, W. Diabetes and Stroke: Epidemiology, Pathophysiology, Pharmaceuticals and Outcomes. Am. J. Med. Sci. 2016, 351, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Kissela, B.M.; Khoury, J.; Kleindorfer, D.; Woo, D.; Schneider, A.; Alwell, K.; Miller, R.; Ewing, I.; Moomaw, C.J.; Szaflarski, J.P.; et al. Epidemiology of Ischemic Stroke in Patients With Diabetes: The greater Cincinnati/Northern Kentucky Stroke Study. Diabetes Care 2005, 28, 355–359. [Google Scholar] [CrossRef] [Green Version]
- Gæde, P.; Lund-Andersen, H.; Parving, H.-H.; Pedersen, O. Effect of a Multifactorial Intervention on Mortality in Type 2 Diabetes. New Engl. J. Med. 2008, 358, 580–591. [Google Scholar] [CrossRef] [Green Version]
- A DiMeglio, L.; Evans-Molina, C.; Oram, R. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Grodsky, G.M.; Bennett, L.L. Cation Requirements for Insulin Secretion in the Isolated Perfused Pancreas. Diabetes 1966, 15, 910. [Google Scholar] [CrossRef] [PubMed]
- Gromada, J.; Høy, M.; Renström, E.; Bokvist, K.; Eliasson, L.; Göpel, S.; Rorsman, P. CaM kinase II-dependent mobilization of secretory granules underlies acetylcholine-induced stimulation of exocytosis in mouse pancreatic B-cells. J. Physiol. 1999, 518, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Togashi, K.; Hara, Y.; Tominaga, T.; Higashi, T.; Konishi, Y.; Mori, Y.; Tominaga, M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006, 25, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Bari, M.R.; Akbar, S.; Eweida, M.; Kühn, F.J.; Gustafsson, A.J.; Lückhoff, A.; Islam, S. H2O2-induced Ca2+ influx and its inhibition by N-(p-amylcinnamoyl) anthranilic acid in the β-cells: Involvement of TRPM2 channels. J. Cell. Mol. Med. 2009, 13, 3260–3267. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Espinoza-Jiménez, A.; Navarrete-Peón, A.; Terrazas, L.I. Alternatively Activated Macrophages in Types 1 and 2 Diabetes. Mediat. Inflamm. 2012, 2012, 815953. [Google Scholar] [CrossRef]
- Drews, G.; Krippeit-Drews, P.; Düfer, M. Oxidative stress and beta-cell dysfunction. Pflug. Arch. 2010, 460, 703–718. [Google Scholar] [CrossRef]
- Domínguez, C.; Ruiz, E.; Gussinye, M.; Carrascosa, A. Oxidative Stress at Onset and in Early Stages of Type 1 Diabetes in Children and Adolescents. Diabetes Care 1998, 21, 1736–1742. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Prim. 2015, 1, 15019. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef] [PubMed]
- Appari, M.; Channon, K.M.; McNeill, E. Metabolic Regulation of Adipose Tissue Macrophage Function in Obesity and Diabetes. Antioxidants Redox Signal. 2018, 29, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Kraakman, M.; Murphy, A.; Jandeleit-Dahm, K.; Kammoun, H.L. Macrophage Polarization in Obesity and Type 2 Diabetes: Weighing Down Our Understanding of Macrophage Function. Front. Immunol. 2014, 5, 470. [Google Scholar] [CrossRef] [PubMed]
- Orliaguet, L.; Dalmas, E.; Drareni, K.; Venteclef, N.; Alzaid, F. Mechanisms of Macrophage Polarization in Insulin Signaling and Sensitivity. Front. Endocrinol. 2020, 11, 62. [Google Scholar] [CrossRef] [Green Version]
- Di Gregorio, G.B.; Yao-Borengasser, A.; Rasouli, N.; Varma, V.; Lu, T.; Miles, L.M.; Ranganathan, G.; Peterson, C.A.; McGehee, R.E.; Kern, P.A. Expression of CD68 and Macrophage Chemoattractant Protein-1 Genes in Human Adipose and Muscle Tissues: Association with cytokine expression, insulin resistance, and reduction by pioglitazone. Diabetes 2005, 54, 2305–2313. [Google Scholar] [CrossRef] [Green Version]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [Green Version]
- Koppaka, S.; Kehlenbrink, S.; Carey, M.; Li, W.; Sanchez, E.; Lee, D.-E.; Lee, H.; Chen, J.; Carrasco, E.; Kishore, P.; et al. Reduced Adipose Tissue Macrophage Content Is Associated With Improved Insulin Sensitivity in Thiazolidinedione-Treated Diabetic Humans. Diabetes 2013, 62, 1843–1854. [Google Scholar] [CrossRef] [Green Version]
- Wagner, D.D.; Burger, P.C. Platelets in Inflammation and Thrombosis. Arter. Thromb. Vasc. Biol. 2003, 23, 2131–2137. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Nesbitt, W.; Giuliano, S.; Kulkarni, S.; Dopheide, S.M.; Harper, I.; Jackson, S.P. Intercellular calcium communication regulates platelet aggregation and thrombus growth. J. Cell Biol. 2003, 160, 1151–1161. [Google Scholar] [CrossRef] [Green Version]
- Varga-Szabo, D.; Braun, A.; Nieswandt, B. Calcium signaling in platelets. J. Thromb. Haemost. 2009, 7, 1057–1066. [Google Scholar] [CrossRef]
- Freedman, J.E. Oxidative Stress and Platelets. Arter. Thromb. Vasc. Biol. 2008, 28, s11–s16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, R.N.; Tolhurst, G.; Walmsley, G.; Vizuete-Forster, M.; Miller, N.; Mahaut-Smith, M.P. Molecular and electrophysiological characterization of transient receptor potential ion channels in the primary murine megakaryocyte. J. Physiol. 2006, 576, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Authi, K.S. TRP Channels in Platelet Function. Handb. Exp. Pharm. 2007, 179, 425–443. [Google Scholar] [CrossRef]
- Isenberg, J.S.; Romeo, M.J.; Yu, C.; Nghiem, K.; Monsale, J.; Rick, M.E.; Wink, D.A.; Frazier, W.A.; Roberts, D.D. Thrombospondin-1 stimulates platelet aggregation by blocking the antithrombotic activity of nitric oxide/cGMP signaling. Blood 2008, 111, 613–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.-N.; Kim, G.-M.; Chen, J.-J.; Cheung, W.-M.; He, Y.Y.; Hsu, C.Y. Differential Regulation of Thrombospondin-1 and Thrombospondin-2 After Focal Cerebral Ischemia/Reperfusion. Stroke 2003, 34, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Saver, J.L.; Starkman, S.; Eckstein, M.; Stratton, S.; Pratt, F.D.; Hamilton, S.; Conwit, R.; Liebeskind, D.S.; Sung, G.; Kramer, I.; et al. Prehospital Use of Magnesium Sulfate as Neuroprotection in Acute Stroke. New Engl. J. Med. 2015, 372, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matei, N.; Camara, J.; Zhang, J.H. The Next Step in the Treatment of Stroke. Front. Neurol. 2021, 11, 582605. [Google Scholar] [CrossRef]
- Hawkins, B.; Davis, T. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Bell, A.H.; Miller, S.L.; Castillo-Melendez, M.; Malhotra, A. The Neurovascular Unit: Effects of Brain Insults During the Perinatal Period. Front. Neurosci. 2020, 13, 1452. [Google Scholar] [CrossRef]
- Kniesel, U.; Wolburg, H. Tight Junctions of the Blood–Brain Barrier. Cell. Mol. Neurobiol. 2000, 20, 57–76. [Google Scholar] [CrossRef] [PubMed]
- Kassner, A.; Merali, Z. Assessment of Blood–Brain Barrier Disruption in Stroke. Stroke 2015, 46, 3310–3315. [Google Scholar] [CrossRef] [PubMed]
- Andjelkovic, A.V.; Xiang, J.; Stamatovic, S.M.; Hua, Y.; Xi, G.; Wang, M.M.; Keep, R.F. Endothelial Targets in Stroke: Translating Animal Models to Human. Arter. Thromb. Vasc. Biol. 2019, 39, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Fields, R.D. Physiological function of microglia. Neuron Glia Biol. 2011, 7, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Guruswamy, R.; ElAli, A. Complex Roles of Microglial Cells in Ischemic Stroke Pathobiology: New Insights and Future Directions. Int. J. Mol. Sci. 2017, 18, 496. [Google Scholar] [CrossRef] [Green Version]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Fonfria, E.; Mattei, C.; Hill, K.; Brown, J.T.; Randall, A.; Benham, C.D.; Skaper, S.D.; Campbell, C.A.; Crook, B.; Murdock, P.R.; et al. TRPM2 Is Elevated in the tMCAO Stroke Model, Transcriptionally Regulated, and Functionally Expressed in C13 Microglia. J. Recept. Signal Transduct. 2006, 26, 179–198. [Google Scholar] [CrossRef]
- Kraft, R.; Grimm, C.; Grosse, K.; Hoffmann, A.; Sauerbruch, S.; Kettenmann, H.; Schultz, G.; Harteneck, C. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am. J. Physiol. Physiol. 2004, 286, C129–C137. [Google Scholar] [CrossRef]
- Mortadza, S.A.S.; Sim, J.A.; Neubrand, V.E.; Jiang, L.-H. A critical role of TRPM2 channel in Aβ42 -induced microglial activation and generation of tumor necrosis factor-α. Glia 2018, 66, 562–575. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Cho, T.; Jantaratnotai, N.; Wang, Y.T.; McGeer, E.; McGeer, P.L. Depletion of GSH in glial cells induces neurotoxicity: Relevance to aging and degenerative neurological diseases. FASEB J. 2010, 24, 2533–2545. [Google Scholar] [CrossRef]
- Miyanohara, J.; Kakae, M.; Nagayasu, K.; Nakagawa, T.; Mori, Y.; Arai, K.; Shirakawa, H.; Kaneko, S. TRPM2 Channel Aggravates CNS Inflammation and Cognitive Impairment via Activation of Microglia in Chronic Cerebral Hypoperfusion. J. Neurosci. 2018, 38, 3520–3533. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Park, E.-M.; Febbraio, M.; Anrather, J.; Park, L.; Racchumi, G.; Silverstein, R.L.; Iadecola, C. The Class B Scavenger Receptor CD36 Mediates Free Radical Production and Tissue Injury in Cerebral Ischemia. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 2504–2512. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Oberheim, N.; Cotrina, M.L.; Nedergaard, M. Astrocytes and Ischemic Injury. Stroke 2009, 40, S8–S12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreto, G.E.; White, R.E.; Ouyang, Y.; Xu, L.; Giffard, R.G. Astrocytes: Targets for Neuroprotection in Stroke. Central Nerv. Syst. Agents Med. Chem. 2011, 11, 164–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy-O’Reilly, M.; McCullough, L.D. Astrocytes fuel the fire of lymphocyte toxicity after stroke. Proc. Natl. Acad. Sci. USA 2017, 114, 425–427. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Lu, J.; Shao, A.; Zhang, J.H.; Zhang, J. Glial Cells: Role of the Immune Response in Ischemic Stroke. Front. Immunol. 2020, 11, 294. [Google Scholar] [CrossRef]
- Yamada, T.; Kawahara, K.; Kosugi, T.; Tanaka, M. Nitric Oxide Produced During Sublethal Ischemia Is Crucial for the Preconditioning-Induced Down-Regulation of Glutamate Transporter GLT-1 in Neuron/Astrocyte Co-Cultures. Neurochem. Res. 2006, 31, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.-X.; Zhu, Y.-F.; Chang, H.-F.; Liang, Y. Nanoceria restrains PM2.5-induced metabolic disorder and hypothalamus inflammation by inhibition of astrocytes activation related NF-kappaB pathway in Nrf2 deficient mice. Free Radic. Biol. Med. 2016, 99, 259–272. [Google Scholar] [CrossRef]
- Weilinger, N.L.; Lohman, A.W.; Rakai, B.D.; Ma, E.M.M.; Bialecki, J.; Maslieieva, V.; Rilea, T.; Bandet, M.V.; Ikuta, N.T.; Scott, L.; et al. Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat. Neurosci. 2016, 19, 432–442. [Google Scholar] [CrossRef]
- Kaneko, S.; Kawakami, S.; Hara, Y.; Wakamori, M.; Itoh, E.; Minami, T.; Takada, Y.; Kume, T.; Katsuki, H.; Mori, Y.; et al. A Critical Role of TRPM2 in Neuronal Cell Death by Hydrogen Peroxide. J. Pharmacol. Sci. 2006, 101, 66–76. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Quillinan, N.; Yang, Y.-F.; Nakayama, S.; Cheng, J.; Kelley, M.; Herson, P. TRPM2 channel activation following in vitro ischemia contributes to male hippocampal cell death. Neurosci. Lett. 2012, 530, 41–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olah, M.E.; Jackson, M.F.; Li, H.; Perez, Y.; Sun, H.-S.; Kiyonaka, S.; Mori, Y.; Tymianski, M.; Macdonald, J.F. Ca2+-dependent induction of TRPM2 currents in hippocampal neurons. J. Physiol. 2009, 587, 965–979. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Turlova, E.; Li, F.; Bao, M.-H.; Szeto, V.; Wong, R.; Abussaud, A.; Wang, H.; Zhu, S.; Gao, X.; et al. Transient receptor potential melastatin 2 channels (TRPM2) mediate neonatal hypoxic-ischemic brain injury in mice. Exp. Neurol. 2017, 296, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Verma, S.; Nakayama, S.; Quillinan, N.; Grafe, M.R.; Hurn, P.D.; Herson, P.S. Sex Differences in Neuroprotection Provided by Inhibition of TRPM2 Channels following Experimental Stroke. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2011, 31, 2160–2168. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; A Macey, T.; Quillinan, N.; Klawitter, J.; Perraud, A.-L.L.; Traystman, R.J.; Herson, P.S. Androgen and PARP-1 Regulation of TRPM2 Channels after Ischemic Injury. J. Cereb. Blood Flow Metab. 2013, 33, 1549–1555. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.J.; Tymianski, M. Targeting NMDA receptors in stroke: New hope in neuroprotection. Mol. Brain 2018, 11, 1–14. [Google Scholar] [CrossRef]
- Sanz-Clemente, A.; Nicoll, R.A.; Roche, K.W. Diversity in NMDA Receptor Composition: Many regulators, many consequences. Neuroscientist 2012, 19, 62–75. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef]
- Martel, M.-A.; Ryan, T.; Bell, K.F.; Fowler, J.; McMahon, A.; Al-Mubarak, B.; Komiyama, N.H.; Horsburgh, K.; Kind, P.; Grant, S.G.; et al. The Subtype of GluN2 C-terminal Domain Determines the Response to Excitotoxic Insults. Neuron 2012, 74, 543–556. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [Green Version]
- Alim, I.; Teves, L.; Li, R.; Mori, Y.; Tymianski, M. Modulation of NMDAR Subunit Expression by TRPM2 Channels Regulates Neuronal Vulnerability to Ischemic Cell Death. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 17264–17277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, S.; Vest, R.; Traystman, R.J.; Herson, P.S. Sexually Dimorphic Response of TRPM2 Inhibition Following Cardiac Arrest-Induced Global Cerebral Ischemia in Mice. J. Mol. Neurosci. 2013, 51, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Dietz, R.M.; Cruz-Torres, I.; Strnad, F.; Garske, A.K.; Moreno, M.; Venna, V.R.; Quillinan, N.; Herson, P.S. Extended therapeutic window of a novel peptide inhibitor of TRPM2 channels following focal cerebral ischemia. Exp. Neurol. 2016, 283, 151–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vennekens, R.; Nilius, B. Insights into TRPM4 Function, Regulation and Physiological Role. In Transient Receptor Potential (TRP) Channels; Springer: Berlin/Heidelberg, Germany, 2007; pp. 269–285. [Google Scholar] [CrossRef]
- Gerzanich, V.; Woo, S.K.; Vennekens, R.; Tsymbalyuk, O.; Ivanova, S.; Ivanov, A.; Geng, Z.; Chen, Z.; Nilius, B.; Flockerzi, V.; et al. De novo expression of Trpm4 initiates secondary hemorrhage in spinal cord injury. Nat. Med. 2009, 15, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.; Ng, G.; Yu, C.Y.; Fhu, C.K.; Yu, D.; Vennekens, R.; Nilius, B.; Soong, T.W.; Liao, P. TRPM4 inhibition promotes angiogenesis after ischemic stroke. Pflügers Arch. -Eur. J. Physiol. 2013, 466, 563–576. [Google Scholar] [CrossRef]
- Yan, J.; Bengtson, C.P.; Buchthal, B.; Hagenston, A.M.; Bading, H. Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science 2020, 370. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Xiong, Z.-G. TRPM7 is a unique target for therapeutic intervention of stroke. Int. J. Physiol. Pathophysiol. Pharmacol. 2017, 9, 211–216. [Google Scholar]
- Inoue, K.; Branigan, D.; Xiong, Z.-G. Zinc-induced Neurotoxicity Mediated by Transient Receptor Potential Melastatin 7 Channels. J. Biol. Chem. 2010, 285, 7430–7439. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.-S.; Jackson, M.F.; Martin, L.J.; Jansen, K.; Teves, L.; Cui, H.; Kiyonaka, S.; Mori, Y.; Jones, M.; Forder, J.P.; et al. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat. Neurosci. 2009, 12, 1300–1307. [Google Scholar] [CrossRef]
- Singh, A.P.; Biswas, A.; Shukla, A.W.; Maiti, P. Targeted therapy in chronic diseases using nanomaterial-based drug delivery vehicles. Signal Transduct. Target. Ther. 2019, 4, 1–21. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Model (s) | Target (s) | Mechanism (s) |
|---|---|---|---|
| Rat | In vitro H2O2 treatment | Neuron | Knockdown of TRPM2 using siRNA inhibited H2O2−induced neuronal death [181]. |
| Mouse | In vitro OGD In vivo tMCAO | Neuron | Knockdown of TRPM2 using shRNA inhibited OGD-induced neuronal death, and reduced infarction size after MCAO [185]. |
| Mouse | In vitro OGD In vivo BCCAO | Neuron | Global knockout of TRPM2 inhibited increase of intracellular Zn2+ and ROS production, and attenuated neuronal death after global ischemia [56]. |
| Mouse | In vitro H2O2 treatment In vivo tMCAO | Neuron | Global knockout of TRPM2 inhibited increase of neuro-excitability in response to H2O2, and attenuated neuronal death and brain injury after tMCAO by promoting pro-survival signaling while inhibiting pro-apoptotic signaling [192]. |
| Mouse | In vivo neonatal hypoxic ischemic brain injury model | Neuron | Global knockout of TRPM2 attenuated neuronal death and reduced infarct size after hypoxic–ischemic brain injury partially by regulating GSK-3β signaling [184]. |
| Mouse | In vivo CA/CPR | Neuron | Inhibition of TRPM2 using clotrimazole reduced neuronal death in male mice, but not in female mice [193]. |
| Mouse | In vivo tMCAO | Neuron | Inhibition of TRPM2 using a peptide inhibitor reduced infarction size after MCAO [194]. |
| Mouse | In vivo tMCAO | Immune cells | Transplantation of bone marrow from global TRPM2 knockout mice into wild-type mice, or inhibition of TRPM2 using ACA reduced infarction size in wild-type mice after MCAO [55]. |
| Mouse | In vivo BCAS | Microglia | Global knockout of TRPM2 inhibited brain damage and cognitive dysfunction [172]. |
| Human | In vitro BSO treatment | Microglia and astrocytes | Knockdown of TRPM2 using siRNA attenuated the inflammatory responses in human microglia and astrocytes [171]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zong, P.; Lin, Q.; Feng, J.; Yue, L. A Systemic Review of the Integral Role of TRPM2 in Ischemic Stroke: From Upstream Risk Factors to Ultimate Neuronal Death. Cells 2022, 11, 491. https://doi.org/10.3390/cells11030491
Zong P, Lin Q, Feng J, Yue L. A Systemic Review of the Integral Role of TRPM2 in Ischemic Stroke: From Upstream Risk Factors to Ultimate Neuronal Death. Cells. 2022; 11(3):491. https://doi.org/10.3390/cells11030491
Chicago/Turabian StyleZong, Pengyu, Qiaoshan Lin, Jianlin Feng, and Lixia Yue. 2022. "A Systemic Review of the Integral Role of TRPM2 in Ischemic Stroke: From Upstream Risk Factors to Ultimate Neuronal Death" Cells 11, no. 3: 491. https://doi.org/10.3390/cells11030491
APA StyleZong, P., Lin, Q., Feng, J., & Yue, L. (2022). A Systemic Review of the Integral Role of TRPM2 in Ischemic Stroke: From Upstream Risk Factors to Ultimate Neuronal Death. Cells, 11(3), 491. https://doi.org/10.3390/cells11030491