
Impact of Organelle Transport Deficits on Mitophagy and Autophagy in Niemann–Pick Disease Type C



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Neural Differentiation
2.2. Live Cell Staining and Imaging
2.3. Analysis of Organelle Transport
- -
- Track duration: time between the first and the last spot of a track;
- -
- Mean speed: mean speed over the time course of track;
- -
- Maximal speed: maximal speed between two spots of a track;
- -
- Minimal speed: minimal speed between two spots of a track;
- -
- Median speed: median of all velocities between two spots;
- -
- Track displacement/track length: distance between the first and last spot of a track;
- -
- Total distance travelled: sum of all distances between two consecutive spots;
- -
- Maximal distance travelled: furthest distance from the starting point of a track;
- -
- Confinement ratio:
- Z(PXMut) = Z-Score of parameter x (e.g., mean speed) of the NPC1-deficient cell line (Mut) compared to the control cell line (ctrl). PXMut = mean of parameter X of the NPC1-deficient cell line;
- PXCtrl = mean of parameter X of the control cell line;
- SDXMut = standard deviation of parameter X of the NPC1-deficient cell line;
- SDXCtrl = standard deviation of parameter X of the control cell line;
- NXMut = sample size of parameter X of the NPC1-deficient cell line;
- NXCtrl = sample size of parameter X of the control cell line.
2.4. Immunofluorescence and Fluorescence Microscopy
2.5. Colocalization Analysis
2.6. Determination of Mitochondrial Morphology
2.7. Western Blot Analysis
2.8. Analysis of Mitochondrial Membrane Potential
2.9. Statistical Analysis
3. Results
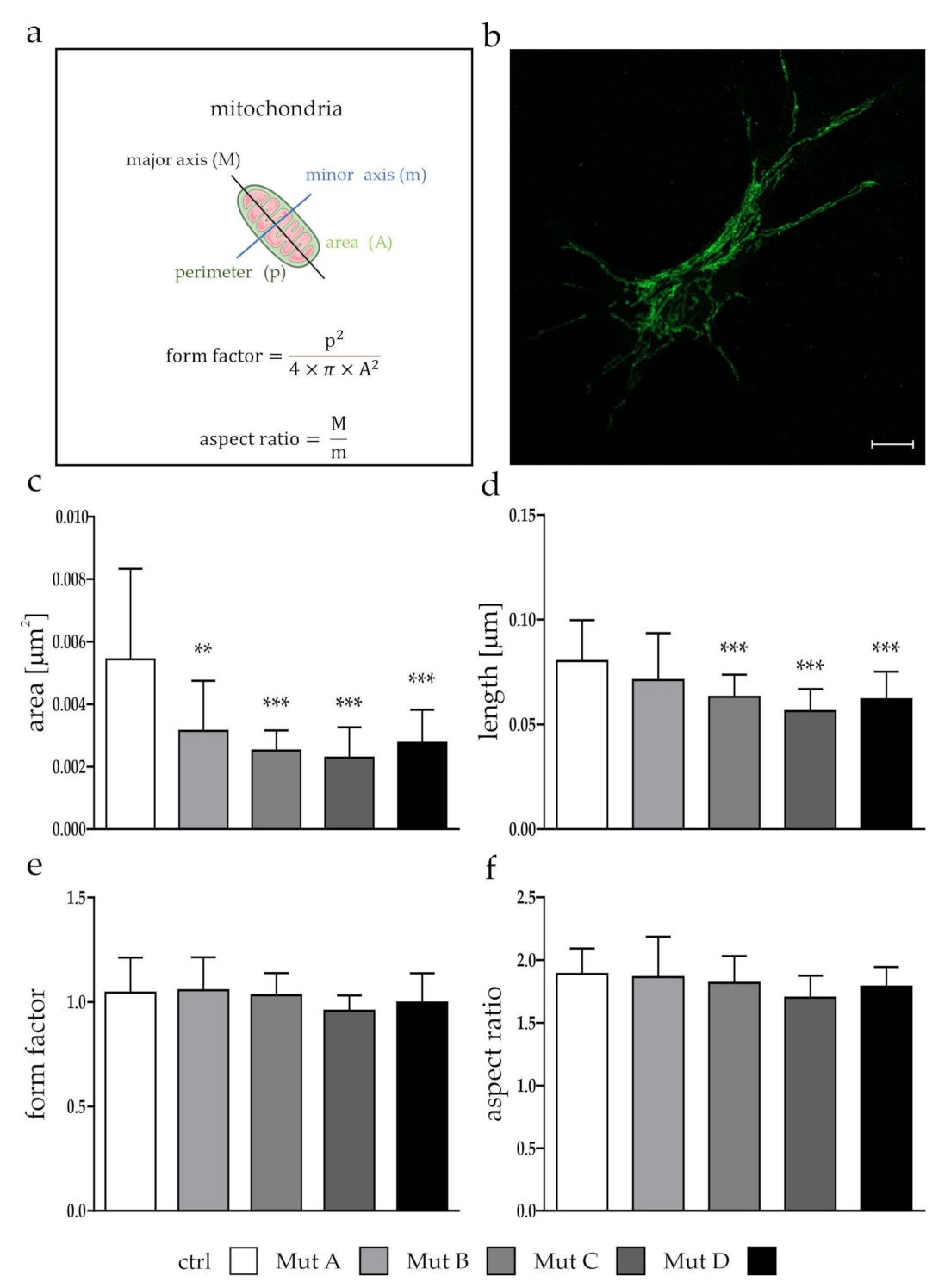
3.1. Mitochondria Were Smaller, but Showed No Differences in Function
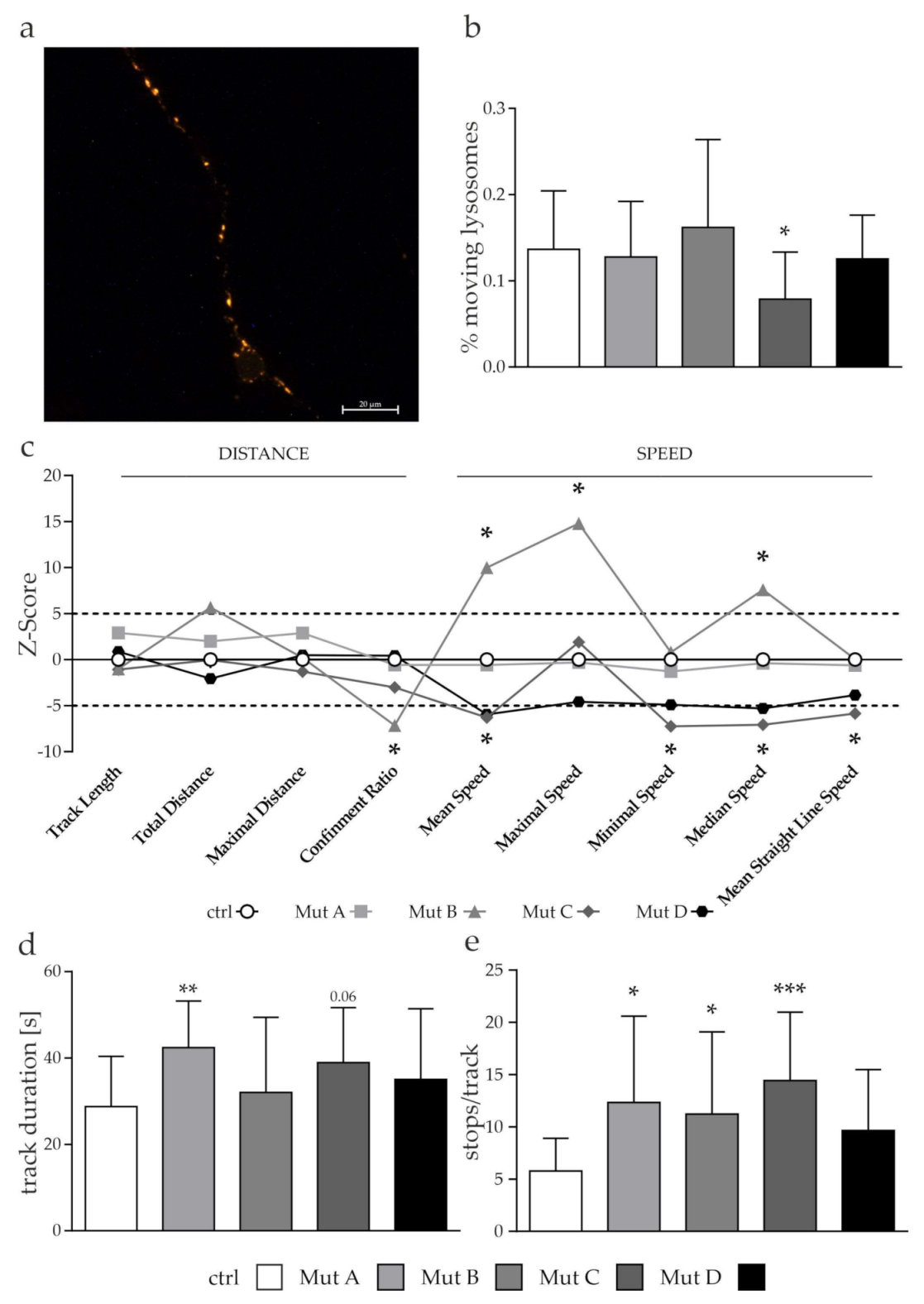
3.2. The Transport Speed of Moving Mitochondria was Reduced Due to an Increased Amount of Stops
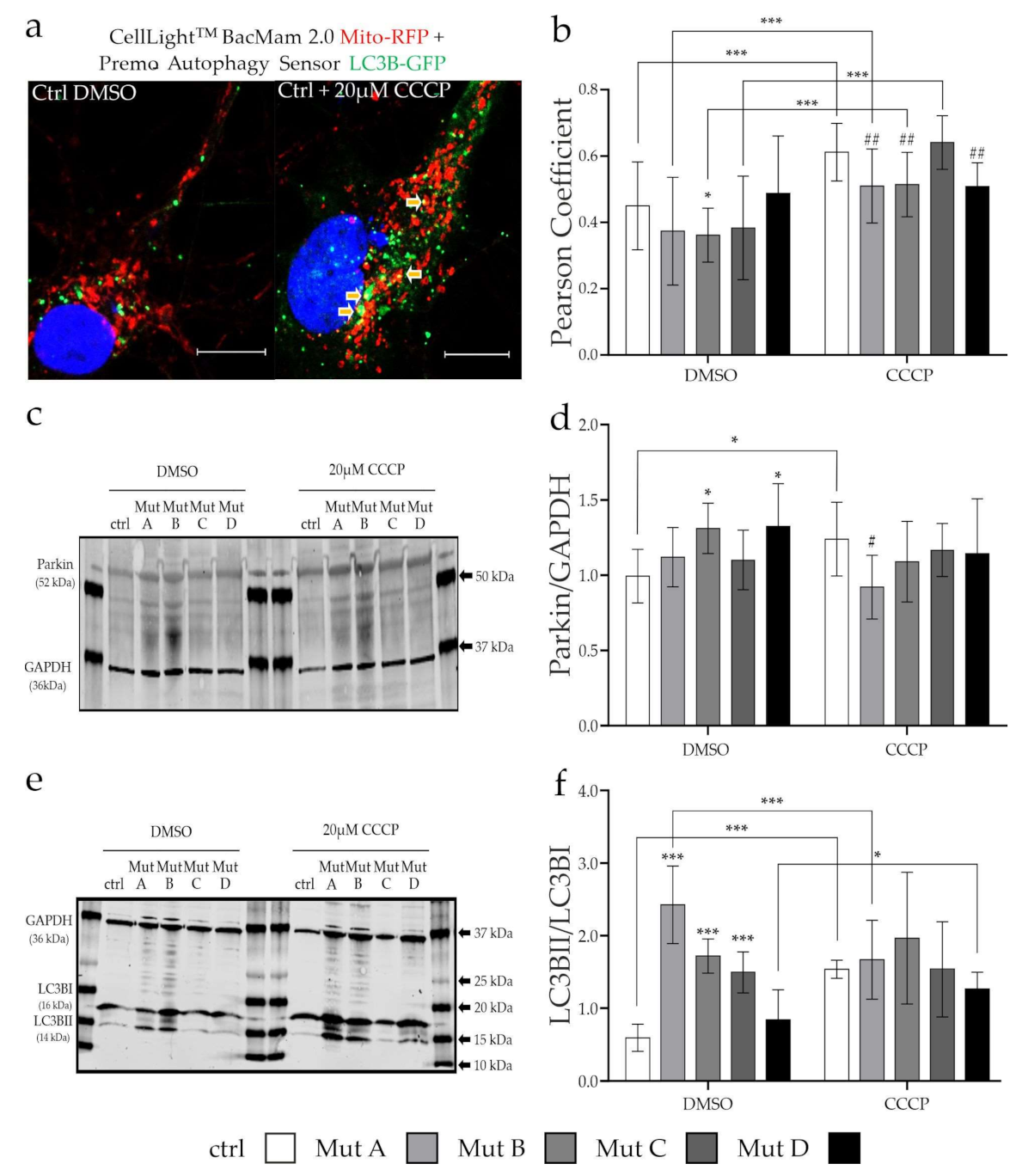
3.3. Degradation of Mitochondria via Mitophagy Is Reduced
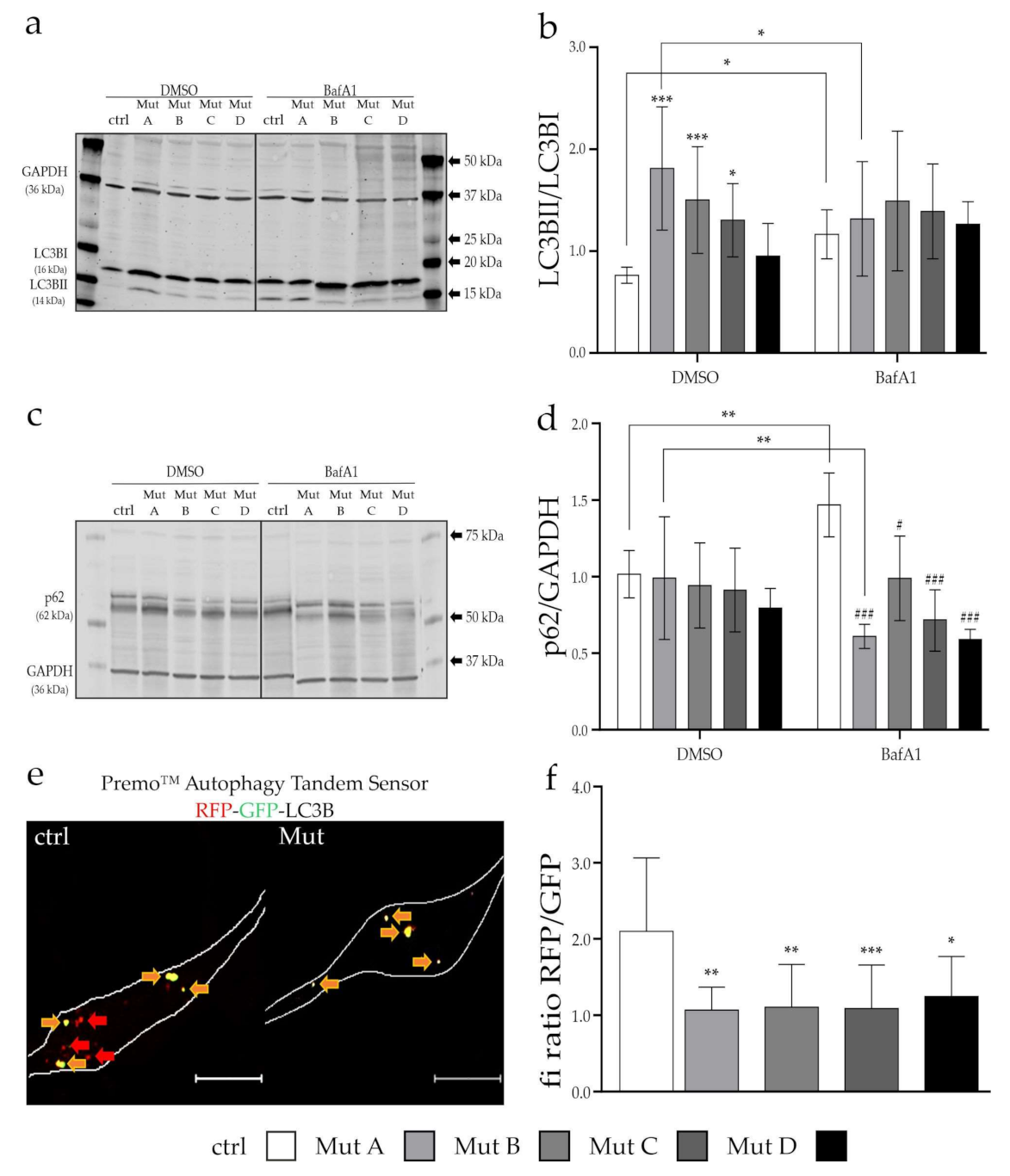
3.4. Initiation and Progression of Autophagy Was Affected
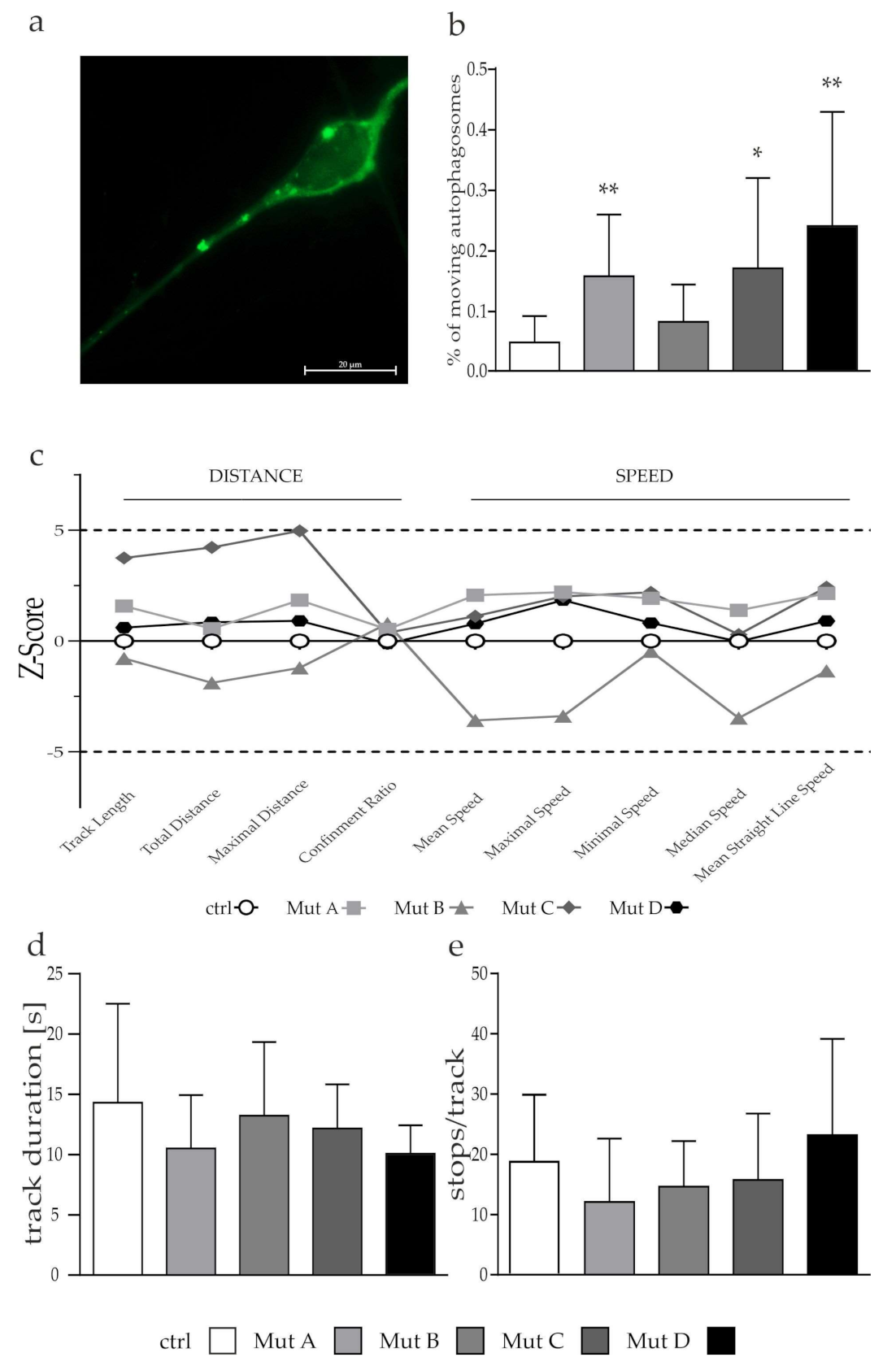
3.5. Lysosomes but Not Autophagosomes Showed a Tendency of a Reduced Transport
4. Discussion
4.1. Analysis of Mitochondrial Morphology and Function
4.2. Impact of Hampered Mitochondrial Transport on Mitophagy in NP-C1- and NP-C2-Deficient Cells
4.3. Impairement of Autophagy in NPC1- and NPC2-Deficient Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| ATG4 | Autophagy-related protein 4 |
| ATP | Adenosine triphosphate |
| AVs | Autophagic vesicles |
| BafA1 | Bafilomycin A1 |
| BSA | Bovine serum albumin |
| CCCP | Carbonyl cyanide 3-chlorophenylhydrazone |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| DMSO | Dimethyl sulfoxide |
| EGF | Epidermal growth factor |
| FGF2 | Basic fibroblast growth factor |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GFP | Green-fluorescent protein |
| HD | Huntington’s disease |
| Hsp60 | Heat-shock protein 60 |
| iPSC | Induced pluripotent stem cell |
| Lamp2 | Lysosome-associated membrane protein 2 |
| LBPA | Lysobisphosphatidic acid |
| LC3BI/II | Microtubule-associated proteins 1A/1B light chain 3B |
| LRRK2 | Leucine-rich repeat kinase 2 |
| LSM | Laser scanning microscope |
| mGluR5 | Group I metabotropic glutamate receptor 5 |
| Miro1/2 | Mitochondrial rho GTPase 1 or 2 |
| MLN64 | Metastatic lymph node 64 protein |
| MMP | Mitochondrial membrane potential |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NDCs | Neuronal differentiated cells |
| NF-L | Neurofilament light chain |
| NP-C 1/2 | Niemann–Pick disease type C 1/2 |
| NPCs | Neural progenitor cells |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| OPTN | Optineurin |
| PBS | Phosphate-buffered saline |
| PD | Parkinson’s disease |
| PFA | Paraformaldehyde |
| PGC-1α | Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha |
| PINK1 | Serine/threonine kinase PTEN-induced kinase 1 |
| PSA-NCAM | Polysialylated-neural cell adhesion molecule |
| RFP | Red-fluorescent protein |
| ROS | Reactive oxygen species |
| SCAR20 | Spinocerebellar ataxia 20 |
| SNARE | Soluble N-ethylmaleimide-sensitive-factor attachment receptors |
| SQSTM1/p62 | Sequestosome-1 |
| SNX14 | Sorting nexin 14 |
| TBK1 | TANK-binding kinase 1 |
| Tom20 | Mitochondrial import receptor subunit TOM20 homolog |
| ULK-1 | mTOR/Unc-51-like kinase |
References
- Pfeffer, S.R. NPC intracellular cholesterol transporter 1 (NPC1)-mediated cholesterol export from lysosomes. J. Biol. Chem. 2019, 294, 1706–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanier, M.T. Niemann-Pick diseases. Handb. Clin. Neurol. 2013, 113, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Cologna, S.M.; Rosenhouse-Dantsker, A. Insights into the Molecular Mechanisms of Cholesterol Binding to the NPC1 and NPC2 Proteins. In Direct Mechanisms in Cholesterol Modulation of Protein Function; Rosenhouse-Dantsker, A., Bukiya, A.N., Eds.; Springer: Cham, Switzerland, 2019; pp. 139–160. [Google Scholar]
- Yu, W.; Gong, J.-S.; Ko, M.; Garver, W.S.; Yanagisawa, K.; Michikawa, M. Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J. Biol. Chem. 2005, 280, 11731–11739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woś, M.; Szczepanowska, J.; Pikuła, S.; Tylki-Szymańska, A.; Zabłocki, K.; Bandorowicz-Pikuła, J. Mitochondrial dysfunction in fibroblasts derived from patients with Niemann-Pick type C disease. Arch. Biochem. Biophys. 2016, 593, 50–59. [Google Scholar] [CrossRef] [PubMed]
- López-Doménech, G.; Higgs, N.F.; Vaccaro, V.; Roš, H.; Arancibia-Cárcamo, I.L.; MacAskill, A.F.; Kittler, J.T. Loss of Dendritic Complexity Precedes Neurodegeneration in a Mouse Model with Disrupted Mitochondrial Distribution in Mature Dendrites. Cell Rep. 2016, 17, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popov, L.-D. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Günther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef]
- Zhang, H.; Hong, Y.; Yang, W.; Wang, R.; Yao, T.; Wang, J.; Liu, K.; Yuan, H.; Xu, C.; Zhou, Y.; et al. SNX14 deficiency-induced defective axonal mitochondrial transport in Purkinje cells underlies cerebellar ataxia and can be reversed by valproate. Natl. Sci. Rev. 2021, 8, nwab024. [Google Scholar] [CrossRef]
- Bastien, J.; Menon, S.; Messa, M.; Nyfeler, B. Molecular targets and approaches to restore autophagy and lysosomal capacity in neurodegenerative disorders. Mol. Asp. Med. 2021, 82, 101018. [Google Scholar] [CrossRef] [PubMed]
- Safiulina, D.; Kuum, M.; Choubey, V.; Gogichaishvili, N.; Liiv, J.; Hickey, M.A.; Cagalinec, M.; Mandel, M.; Zeb, A.; Liiv, M.; et al. Miro proteins prime mitochondria for Parkin translocation and mitophagy. EMBO J. 2019, 38, e99384. [Google Scholar] [CrossRef]
- Sarkar, S.; Carroll, B.; Buganim, Y.; Maetzel, D.; Ng, A.H.M.; Cassady, J.P.; Cohen, M.A.; Chakraborty, S.; Wang, H.; Spooner, E.; et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 2013, 5, 1302–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Roney, J.C.; Li, S.; Farfel-Becker, T.; Huang, N.; Sun, T.; Xie, Y.; Cheng, X.-T.; Lin, M.-Y.; Platt, F.M.; Sheng, Z.-H. Lipid-mediated motor-adaptor sequestration impairs axonal lysosome delivery leading to autophagic stress and dystrophy in Niemann-Pick type C. Dev. Cell 2021, 56, 1452–1468. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, A.V.; Kang, I.; de Pace, R.; Wassif, C.A.; Fujiwara, H.; Kell, P.; Jiang, X.; Ory, D.S.; Bonifacino, J.S.; Ward, M.E.; et al. A human iPSC-derived inducible neuronal model of Niemann-Pick disease, type C1. BMC Biol. 2021, 19, 218. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Völkner, C.; Liedtke, M.; Petters, J.; Lukas, J.; Escobar, H.M.; Knuebel, G.; Bullerdiek, J.; Holzmann, C.; Hermann, A.; Frech, M.J. Generation of an iPSC line (AKOSi004-A) from fibroblasts of a female adult NPC1 patient, carrying the compound heterozygous mutation p.Val1023Serfs*15/p.Gly992Arg and of an iPSC line (AKOSi005-A) from a female adult control individual. Stem Cell Res. 2020, 50, 102127. [Google Scholar] [CrossRef]
- Völkner, C.; Liedtke, M.; Petters, J.; Huth, K.; Knuebel, G.; Murua Escobar, H.; Bullerdiek, J.; Lukas, J.; Hermann, A.; Frech, M.J. Generation of an iPSC line (AKOSi006-A) from fibroblasts of an NPC1 patient, carrying the homozygous mutation p.I1061T (c.3182 T C) and a control iPSC line (AKOSi007-A) using a non-integrating Sendai virus system. Stem Cell Res. 2020, 49, 102056. [Google Scholar] [CrossRef]
- Völkner, C.; Liedtke, M.; Hermann, A.; Frech, M.J. Pluripotent Stem Cells for Disease Modeling and Drug Discovery in Niemann-Pick Type C1. Int. J. Mol. Sci. 2021, 22, 710. [Google Scholar] [CrossRef]
- Liedtke, M.; Völkner, C.; Jürs, A.V.; Peter, F.; Rabenstein, M.; Hermann, A.; Frech, M.J. Pathophysiological In Vitro Profile of Neuronal Differentiated Cells Derived from Niemann-Pick Disease Type C2 Patient-Specific iPSCs Carrying the NPC2 Mutations c.58GT/c.140GT. Int. J. Mol. Sci. 2021, 22, 4009. [Google Scholar] [CrossRef]
- Pal, A.; Glaß, H.; Naumann, M.; Kreiter, N.; Japtok, J.; Sczech, R.; Hermann, A. High content organelle trafficking enables disease state profiling as powerful tool for disease modelling. Sci. Data 2018, 5, 180241. [Google Scholar] [CrossRef] [Green Version]
- Jürs, A.V.; Völkner, C.; Liedtke, M.; Huth, K.; Lukas, J.; Hermann, A.; Frech, M.J. Oxidative Stress and Alterations in the Antioxidative Defense System in Neuronal Cells Derived from NPC1 Patient-Specific Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2020, 21, 7667. [Google Scholar] [CrossRef] [PubMed]
- Servier. SMART Servier Medical Art. Available online: https://smart.servier.com (accessed on 7 January 2022).
- Caruso Bavisotto, C.; Alberti, G.; Vitale, A.M.; Paladino, L.; Campanella, C.; Rappa, F.; Gorska, M.; Conway de Macario, E.; Cappello, F.; Macario, A.J.L.; et al. Hsp60 Post-translational Modifications: Functional and Pathological Consequences. Front. Mol. Biosci. 2020, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Zhang, B.; Lee, V.M.-Y.; Trojanowski, J.Q. Axonal transport defects: A common theme in neurodegenerative diseases. Acta Neuropathol. 2005, 109, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Ma, K.; Han, J.; Chen, Q.; Zhu, Y. Monitoring Mitophagy in Mammalian Cells. Methods Enzymol. 2017, 588, 187–208. [Google Scholar] [CrossRef]
- Wang, J.-Q.; Zhu, S.; Wang, Y.; Wang, F.; An, C.; Jiang, D.; Gao, L.; Tu, Y.; Zhu, X.; Wang, Y.; et al. Miro2 supplies a platform for Parkin translocation to damaged mitochondria. Sci. Bull. 2019, 64, 730–747. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.; Martinez-Lopez, N.; Otten, E.G.; Carroll, B.; Maetzel, D.; Singh, R.; Sarkar, S.; Korolchuk, V.I. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Biophys. Acta 2016, 1861, 269–284. [Google Scholar] [CrossRef] [Green Version]
- Theocharopoulou, G. The ubiquitous role of mitochondria in Parkinson and other neurodegenerative diseases. AIMS Neurosci. 2020, 7, 43–65. [Google Scholar] [CrossRef]
- Kennedy, B.E.; Madreiter, C.T.; Vishnu, N.; Malli, R.; Graier, W.F.; Karten, B. Adaptations of energy metabolism associated with increased levels of mitochondrial cholesterol in Niemann-Pick type C1-deficient cells. J. Biol. Chem. 2014, 289, 16278–16289. [Google Scholar] [CrossRef] [Green Version]
- Charman, M.; Kennedy, B.E.; Osborne, N.; Karten, B. MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick Type C1 protein. J. Lipid Res. 2010, 51, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Balboa, E.; Castro, J.; Pinochet, M.-J.; Cancino, G.I.; Matías, N.; Sáez, P.J.; Martínez, A.; Álvarez, A.R.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; et al. MLN64 induces mitochondrial dysfunction associated with increased mitochondrial cholesterol content. Redox Biol. 2017, 12, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.E.; Charman, M.; Karten, B. Niemann-Pick Type C2 protein contributes to the transport of endosomal cholesterol to mitochondria without interacting with NPC1. J. Lipid Res. 2012, 53, 2632–2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, K.; Sinha, M.; Le Pham, C.L.; Jana, S.; Chanda, D.; Cappai, R.; Chakrabarti, S. Alpha-synuclein induced membrane depolarization and loss of phosphorylation capacity of isolated rat brain mitochondria: Implications in Parkinson’s disease. FEBS Lett. 2010, 584, 1571–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossy-Wetzel, E.; Petrilli, A.; Knott, A.B. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008, 31, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassereau, J.; Casasnovas, C.; Gueguen, N.; Malinge, M.-C.; Guillet, V.; Reynier, P.; Bonneau, D.; Amati-Bonneau, P.; Banchs, I.; Volpini, V.; et al. Simultaneous MFN2 and GDAP1 mutations cause major mitochondrial defects in a patient with CMT. Neurology 2011, 76, 1524–1526. [Google Scholar] [CrossRef] [Green Version]
- Ordonez, M.P. Defective mitophagy in human Niemann-Pick Type C1 neurons is due to abnormal autophagy activation. Autophagy 2012, 8, 1157–1158. [Google Scholar] [CrossRef] [Green Version]
- Nekrasova, O.E.; Mendez, M.G.; Chernoivanenko, I.S.; Tyurin-Kuzmin, P.A.; Kuczmarski, E.R.; Gelfand, V.I.; Goldman, R.D.; Minin, A.A. Vimentin intermediate filaments modulate the motility of mitochondria. Mol. Biol. Cell 2011, 22, 2282–2289. [Google Scholar] [CrossRef]
- Perrot, R.; Julien, J.-P. Real-time imaging reveals defects of fast axonal transport induced by disorganization of intermediate filaments. FASEB J. 2009, 23, 3213–3225. [Google Scholar] [CrossRef]
- Peter, F.; Rost, S.; Rolfs, A.; Frech, M.J. Activation of PKC triggers rescue of NPC1 patient specific iPSC derived glial cells from gliosis. Orphanet J. Rare Dis. 2017, 12, 145. [Google Scholar] [CrossRef]
- Ikenaka, K.; Katsuno, M.; Kawai, K.; Ishigaki, S.; Tanaka, F.; Sobue, G. Disruption of axonal transport in motor neuron diseases. Int. J. Mol. Sci. 2012, 13, 1225–1238. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.-H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossmann, D.; Berenguer-Escuder, C.; Bellet, M.E.; Scheibner, D.; Bohler, J.; Massart, F.; Rapaport, D.; Skupin, A.; Fouquier d’Hérouël, A.; Sharma, M.; et al. Mutations in RHOT1 Disrupt Endoplasmic Reticulum-Mitochondria Contact Sites Interfering with Calcium Homeostasis and Mitochondrial Dynamics in Parkinson’s Disease. Antioxid. Redox Signal. 2019, 31, 1213–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roca-Agujetas, V.; Barbero-Camps, E.; de Dios, C.; Podlesniy, P.; Abadin, X.; Morales, A.; Marí, M.; Trullàs, R.; Colell, A. Cholesterol alters mitophagy by impairing optineurin recruitment and lysosomal clearance in Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, 4439–4448. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef] [Green Version]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers 2019, 11, 1422. [Google Scholar] [CrossRef] [Green Version]
- Davis, O.B.; Shin, H.R.; Lim, C.-Y.; Wu, E.Y.; Kukurugya, M.; Maher, C.F.; Perera, R.M.; Ordonez, M.P.; Zoncu, R. NPC1-mTORC1 Signaling Couples Cholesterol Sensing to Organelle Homeostasis and Is a Targetable Pathway in Niemann-Pick Type C. Dev. Cell 2021, 56, 260–276. [Google Scholar] [CrossRef]
- Ibrahim, K.S.; McLaren, C.J.; Abd-Elrahman, K.S.; Ferguson, S.S.G. Optineurin deletion disrupts metabotropic glutamate receptor 5-mediated regulation of ERK1/2, GSK3β/ZBTB16, mTOR/ULK1 signaling in autophagy. Biochem. Pharmacol. 2021, 185, 114427. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Yang, F.; Rabenstein, M.; Wang, Z.; Frech, M.J.; Wree, A.; Bräuer, A.U.; Witt, M.; Gläser, A.; Hermann, A.; et al. Stimulation of mGluR1/5 Improves Defective Internalization of AMPA Receptors in NPC1 Mutant Mouse. Cereb. Cortex 2020, 30, 1465–1480. [Google Scholar] [CrossRef] [PubMed]
- Maetzel, D.; Sarkar, S.; Wang, H.; Abi-Mosleh, L.; Xu, P.; Cheng, A.W.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Rep. 2014, 2, 866–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meske, V.; Erz, J.; Priesnitz, T.; Ohm, T.-G. The autophagic defect in Niemann-Pick disease type C neurons differs from somatic cells and reduces neuronal viability. Neurobiol. Dis. 2014, 64, 88–97. [Google Scholar] [CrossRef]
- Fernández, Á.F.; López-Otín, C. The functional and pathologic relevance of autophagy proteases. J. Clin. Invest. 2015, 125, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, T.; Noda, N.N. Autophagy-regulating protease Atg4: Structure, function, regulation, and inhibition. J. Antibiot. 2017, 71, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.-Q.; Ni, T.; Hong, B.; Wang, H.-Y.; Jiang, F.-J.; Zou, S.; Chen, Y.; Zheng, X.-L.; Klionsky, D.J.; Liang, Y.; et al. Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy 2012, 8, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Kriegenburg, F.; Ungermann, C.; Reggiori, F. Coordination of Autophagosome-Lysosome Fusion by Atg8 Family Members. Curr. Biol. 2018, 28, R512–R518. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Ilnytska, O.; Lai, K.; Gorshkov, K.; Schultz, M.L.; Tran, B.N.; Jeziorek, M.; Kunkel, T.J.; Azaria, R.D.; McLoughlin, H.S.; Waghalter, M.; et al. Enrichment of NPC1-deficient cells with the lipid LBPA stimulates autophagy, improves lysosomal function, and reduces cholesterol storage. J. Biol. Chem. 2021, 297, 100813. [Google Scholar] [CrossRef]
- Liao, W.; Wang, Z.; Fu, Z.; Ma, H.; Jiang, M.; Xu, A.; Zhang, W. p62/SQSTM1 protects against cisplatin-induced oxidative stress in kidneys by mediating the cross talk between autophagy and the Keap1-Nrf2 signalling pathway. Free Radic. Res. 2019, 53, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Sadovnikova, I.S.; Starkov, N.N.; Starkov, A.A.; Popov, V.N. p62-Nrf2-p62 Mitophagy Regulatory Loop as a Target for Preventive Therapy of Neurodegenerative Diseases. Brain Sci. 2020, 10, 847. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.-T.; Zhou, B.; Lin, M.-Y.; Cai, Q.; Sheng, Z.-H. Axonal autophagosomes use the ride-on service for retrograde transport toward the soma. Autophagy 2015, 11, 1434–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liedtke, M.; Völkner, C.; Hermann, A.; Frech, M.J. Impact of Organelle Transport Deficits on Mitophagy and Autophagy in Niemann–Pick Disease Type C. Cells 2022, 11, 507. https://doi.org/10.3390/cells11030507
Liedtke M, Völkner C, Hermann A, Frech MJ. Impact of Organelle Transport Deficits on Mitophagy and Autophagy in Niemann–Pick Disease Type C. Cells. 2022; 11(3):507. https://doi.org/10.3390/cells11030507
Chicago/Turabian StyleLiedtke, Maik, Christin Völkner, Andreas Hermann, and Moritz J. Frech. 2022. "Impact of Organelle Transport Deficits on Mitophagy and Autophagy in Niemann–Pick Disease Type C" Cells 11, no. 3: 507. https://doi.org/10.3390/cells11030507
APA StyleLiedtke, M., Völkner, C., Hermann, A., & Frech, M. J. (2022). Impact of Organelle Transport Deficits on Mitophagy and Autophagy in Niemann–Pick Disease Type C. Cells, 11(3), 507. https://doi.org/10.3390/cells11030507