
Mitochondria-Endoplasmic Reticulum Interplay Regulates Exo-Cytosis in Human Neuroblastoma Cells


Abstract
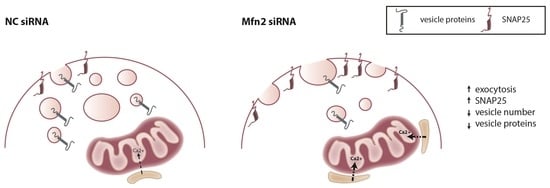
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Ca2+ Imaging
2.3. Cell Culture, Transfections, and Treatments
2.4. Electron Microscopy
2.5. Gene Expression Analysis
2.6. Lactate Dehydrogenase Assay (LDH) Assay
2.7. Oxygen Consumption and Total ATP Levels
2.8. SDS-PAGE and Immunoblotting
2.9. SypHy Imaging
2.10. Statistical Analysis
3. Results
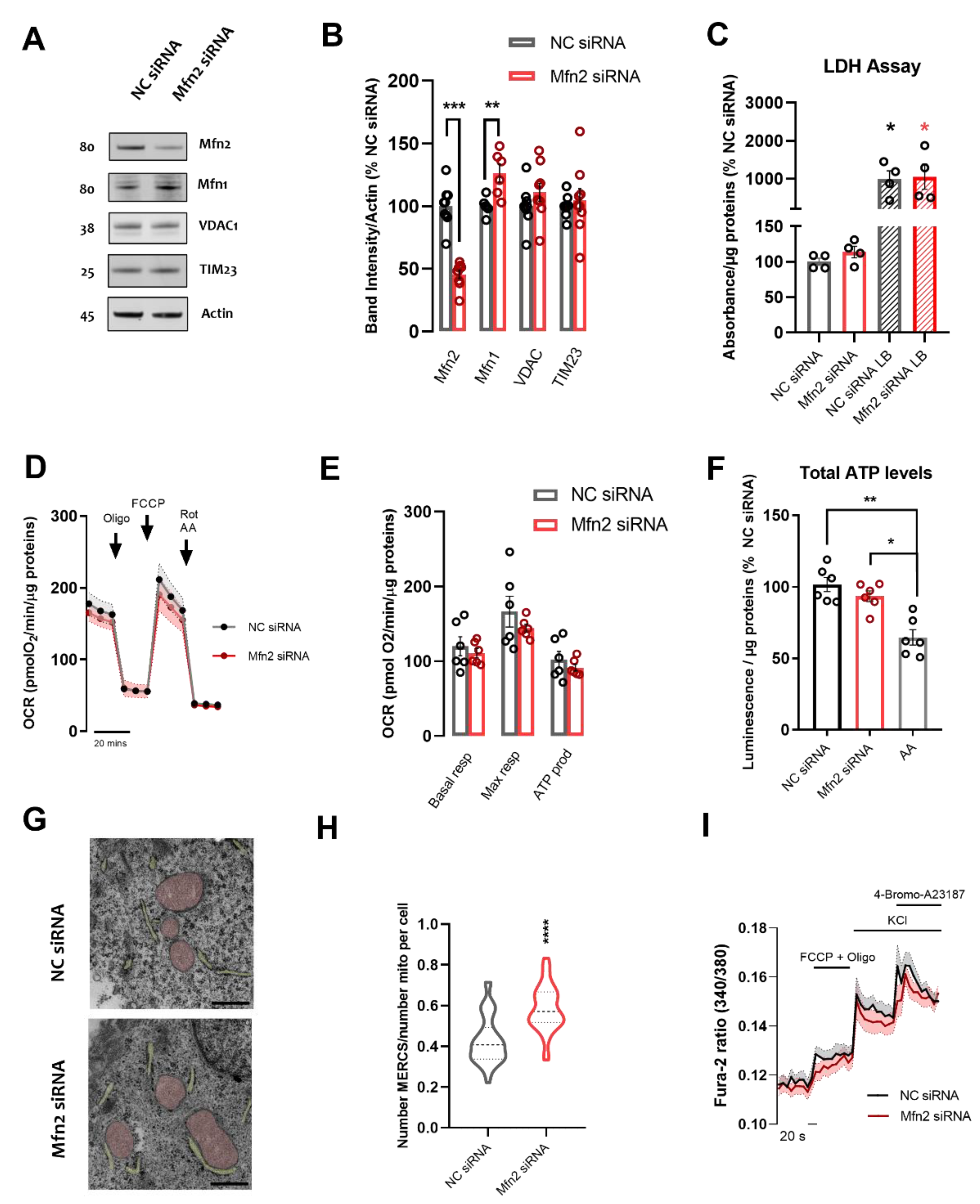
3.1. Mfn2 Acts as a Negative Regulator of MERCS in SH-SY5Y Cells
3.2. Upregulation of ER to Mitochondria Proximity Depletes Number of Vesicles, Increases SNAP25 Protein Levels and Boosts Exocytosis
3.3. SNAP25 Downregulation Abrogated Mfn2 KD-Mediated Increased Exocytosis
3.4. Ca2+-Shuttling between ER and Mitochondria Is Important in Modulating Vesicle Release Mechanisms
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raturi, A.; Gutiérrez, T.; Ortiz-Sandoval, C.; Ruangkittisakul, A.; Herrera-Cruz, M.S.; Rockley, J.P.; Gesson, K.; Ourdev, D.; Lou, P.-H.; Lucchinetti, E.; et al. TMX1 determines cancer cell metabolism as a thiol-based modulator of ER–mitochondria Ca2+ flux. J. Cell Biol. 2016, 214, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with High Ca2+ Close to IP3-Sensitive Channels that Are Sensed by Neighboring Mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Lee, S.; Wang, W.; Hwang, J.; Namgung, U.; Min, K.-T. Increased ER–mitochondria tethering promotes axon regeneration. Proc. Natl. Acad. Sci. USA 2019, 116, 16074–16079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Várnai, P.; Hajnóczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol. Cell 2016, 63, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, J.E. Newly made phosphatidylserine and phosphatidylethanolamine are preferentially translocated between rat liver mitochondria and endoplasmic reticulum. J. Biol. Chem. 1991, 266, 89–97. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER–mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Iwasawa, R.; Mahul-Mellier, A.-L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2010, 30, 556–568. [Google Scholar] [CrossRef] [Green Version]
- Hirabayashi, Y.; Kwon, S.-K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 2017, 358, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Stoica, R.; De Vos, K.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J.; et al. ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; Nagashima, S.; Tokuyama, T.; Amo, T.; Matsuki, Y.; Ishido, S.; Kudo, Y.; McBride, H.M.; Fukuda, T.; Matsushita, N.; et al. MITOL Regulates Endoplasmic Reticulum-Mitochondria Contacts via Mitofusin2. Mol. Cell 2013, 51, 20–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alford, S.C.; Ding, Y.; Simmen, T.; Campbell, R.E. Dimerization-Dependent Green and Yellow Fluorescent Proteins. ACS Synth. Biol. 2012, 1, 569–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göbel, J.; Engelhardt, E.; Pelzer, P.; Sakthivelu, V.; Jahn, H.M.; Jevtic, M.; Folz-Donahue, K.; Kukat, C.; Schauss, A.; Frese, C.K.; et al. Mitochondria-Endoplasmic Reticulum Contacts in Reactive Astrocytes Promote Vascular Remodeling. Cell Metab. 2020, 31, 791–808.e8. [Google Scholar] [CrossRef]
- Schneeberger, M.; Dietrich, M.O.; Sebastián, D.; Imbernón, M.; Castaño, C.; Garcia, A.; Esteban, Y.; Gonzalez-Franquesa, A.; Rodríguez, I.C.; Bortolozzi, A.; et al. Mitofusin 2 in POMC Neurons Connects ER Stress with Leptin Resistance and Energy Imbalance. Cell 2013, 155, 172–187. [Google Scholar] [CrossRef] [Green Version]
- Cosson, P.; Marchetti, A.; Ravazzola-Schreyer, M.; Orci, L. Mitofusin-2 Independent Juxtaposition of Endoplasmic Reticulum and Mitochondria: An Ultrastructural Study. PLoS ONE 2012, 7, e46293. [Google Scholar] [CrossRef] [Green Version]
- Leal, N.S.; Schreiner, B.; Pinho, C.M.; Filadi, R.; Wiehager, B.; Karlström, H.; Pizzo, P.; Ankarcrona, M. Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid beta-peptide production. J. Cell Mol. Med. 2016, 20, 1686–1695. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum–mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.T.; Garcin, P.O.; Fu, M.; Masoudi, M.; St-Pierre, P.; Panté, N.; Nabi, I.R. Distinct mechanisms controlling rough and smooth endoplasmic reticulum contacts with mitochondria. J. Cell Sci. 2015, 128, 2759–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmon, M.; Larkman, P.; Hardingham, G.; Jackson, M.; Skehel, P. A Bi-fluorescence complementation system to detect associations between the Endoplasmic reticulum and mitochondria. Sci. Rep. 2017, 7, 17467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieri, D.; Vicario, M.; Giacomello, M.; Vallese, F.; Filadi, R.; Wagner, T.; Pozzan, T.; Pizzo, P.; Scorrano, L.; Brini, M.; et al. SPLICS: A split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ. 2017, 25, 1131–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filadi, R.; Greotti, E.; Pizzo, P. Highlighting the endoplasmic reticulum-mitochondria connection: Focus on Mitofusin 2. Pharmacol. Res. 2018, 128, 42–51. [Google Scholar] [CrossRef]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef] [Green Version]
- Südhof, T.C. The synaptic vesicle cycle. Annu. Rev. Neurosci. 2004, 27, 509–547. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Pozzan, T. Microdomains of Intracellular Ca2+: Molecular Determinants and Functional Consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Wong, C.-O.; Chen, K.; Lin, Y.Q.; Chao, Y.; Duraine, L.; Lu, Z.; Yoon, W.H.; Sullivan, J.M.; Broadhead, G.T.; Sumner, C.J.; et al. A TRPV Channel in Drosophila Motor Neurons Regulates Presynaptic Resting Ca2+ Levels, Synapse Growth, and Synaptic Transmission. Neuron 2014, 84, 764–777. [Google Scholar] [CrossRef] [Green Version]
- Chakroborty, S.; Kim, J.; Schneider, C.; Jacobson, C.; Molgó, J.; Stutzmann, G.E. Early presynaptic and postsynaptic calcium signaling abnormalities mask underlying synaptic depression in presymptomatic Alzheimer’s disease mice. J. Neurosci. 2012, 32, 8341–8353. [Google Scholar] [CrossRef] [Green Version]
- Futatsugi, A.; Nakamura, T.; Yamada, M.K.; Ebisui, E.; Nakamura, K.; Uchida, K.; Kitaguchi, T.; Takahashi-Iwanaga, H.; Noda, T.; Aruga, J.; et al. IP 3 Receptor Types 2 and 3 Mediate Exocrine Secretion Underlying Energy Metabolism. Science 2005, 309, 2232–2234. [Google Scholar] [CrossRef]
- Chen, M.; Van Hook, M.J.; Thoreson, W.B. Ca2+ Diffusion through Endoplasmic Reticulum Supports Elevated Intraterminal Ca2+ Levels Needed to Sustain Synaptic Release from Rods in Darkness. J. Neurosci. 2015, 35, 11364–11373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.-G.; Zucker, R.S. Mitochondrial Involvement in Post-Tetanic Potentiation of Synaptic Transmission. Neuron 1997, 18, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Billups, B.; Forsythe, I. Presynaptic Mitochondrial Calcium Sequestration Influences Transmission at Mammalian Central Synapses. J. Neurosci. 2002, 22, 5840–5847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, G.; Barrett, E.F. Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J. Physiol. 2003, 548, 425–438. [Google Scholar] [CrossRef]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-Driven Local ATP Synthesis Is Required for Synaptic Function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Qiao, H.; Pan, P.-Y.; Chen, Y.; Sheng, Z.-H. Motile Axonal Mitochondria Contribute to the Variability of Presynaptic Strength. Cell Rep. 2013, 4, 413–419. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Hedskog, L.; Petersen, C.A.H.; Winblad, B.; Ankarcrona, M. Dimebon (Latrepirdine) Enhances Mitochondrial Function and Protects Neuronal Cells from Death. J. Alzheimers Dis. 2010, 21, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Pivovarova, N.B.; Pozzo-Miller, L.D.; Hongpaisan, J.; Andrews, S.B. Correlated Calcium Uptake and Release by Mitochondria and Endoplasmic Reticulum of CA3 Hippocampal Dendrites after Afferent Synaptic Stimulation. J. Neurosci. 2002, 22, 10653–10661. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Perez-Nievas, B.G.; Glennon, E.; Lau, D.; Paillusson, S.; Mórotz, G.M.; Calì, T.; Pizzo, P.; Noble, W.; Miller, C.C.J. The VAPB-PTPIP51 endoplasmic reticulum-mitochondria tethering proteins are present in neuronal synapses and regulate synaptic activity. Acta Neuropathol. Commun. 2019, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granseth, B.; Odermatt, B.; Royle, S.; Lagnado, L. Clathrin-Mediated Endocytosis Is the Dominant Mechanism of Vesicle Retrieval at Hippocampal Synapses. Neuron 2006, 51, 773–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallilankaraman, K.; Cárdenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenár, T.; Csordás, G.; Madireddi, P.; Yang, J.; Müller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodall, A.R.; Danks, K.; Walker, J.H.; Ball, S.G.; Vaughan, P.F.T. Occurrence of two types of secretory vesicles in the human neuroblastoma SH-SY5Y. J. Neurochem. 2002, 68, 1542–1552. [Google Scholar] [CrossRef]
- Påhlman, S.; Ruusala, A.-I.; Abrahamsson, L.; Mattsson, M.E.; Esscher, T. Retinoic acid-induced differentiation of cultured human neuroblastoma cells: A comparison with phorbolester-induced differentiation. Cell Differ. 1984, 14, 135–144. [Google Scholar] [CrossRef]
- Ou, X.M.; Partoens, P.M.; Wang, J.M.; Walker, J.H.; Danks, K.; Vaughan, P.F.; De Potter, W.P. The storage of noradrenaline, neuropeptide Y and chromogranins in and stoichiometric release from large dense cored vesicles of the undifferentiated human neuroblastoma cell line SH-SY5Y. Int. J. Mol. Med. 1998, 1, 105–117. [Google Scholar] [CrossRef]
- Taglialatela, M.; Secondo, A.; Fresi, A.; Rosati, B.; Pannaccione, A.; Castaldo, P.; Giorgio, G.; Wanke, E.; Annunziato, L. Inhibition of depolarization-induced [3H]noradrenaline release from SH-SY5Y human neuroblastoma cells by some second-generation H1 receptor antagonists through blockade of store-operated Ca2+ channels (SOCs)11Abbreviations: hERG, human Ether-a-go-go Related Gene; SOC, Ca2+ currents activated by [Ca2+]i store depletion; NE, norepinephrine; [K+]e, extracellular K+ concentration; [Ca2+]i, intracellular Ca2+ concentration; HBS, HEPES-buffered saline; SERCA, sarcoplasmic-endoplasmic reticulum calcium ATPase; and TTX, tetrodotoxin. Biochem. Pharmacol. 2001, 62, 1229–1238. [Google Scholar]
- Leal, N.S.; Dentoni, G.; Schreiner, B.; Naia, L.; Piras, A.; Graff, C.; Cattaneo, A.; Meli, G.; Hamasaki, M.; Nilsson, P.; et al. Amyloid Β-Peptide Increases Mitochondria-Endoplasmic Reticulum Contact Altering Mitochondrial Function and Autophagosome Formation in Alzheimer’s Disease-Related Models. Cells 2020, 9, 2552. [Google Scholar] [CrossRef]
- Ding, X.; Liu, S.; Tian, M.; Zhang, W.; Zhu, T.; Li, D.; Wu, J.; Deng, H.; Jia, Y.; Xie, W.; et al. Activity-induced histone modifications govern Neurexin-1 mRNA splicing and memory preservation. Nat. Neurosci. 2017, 20, 690–699. [Google Scholar] [CrossRef]
- Hoerder-Suabedissen, A.; Korrell, K.V.; Hayashi, S.; Jeans, A.; Ramirez, D.M.O.; Grant, E.; Christian, H.C.; Kavalali, E.T.; Wilson, M.C.; Molnár, Z. Cell-Specific Loss of SNAP25 from Cortical Projection Neurons Allows Normal Development but Causes Subsequent Neurodegeneration. Cereb. Cortex 2018, 29, 2148–2159. [Google Scholar] [CrossRef]
- Cahill, A.L.; Herring, B.E.; Fox, A.P. Stable silencing of SNAP-25 in PC12 cells by RNA interference. BMC Neurosci. 2006, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefani, D.; Patron, M.; Rizzuto, R. Structure and function of the mitochondrial calcium uniporter complex. Biochim. Biophys. Acta 2015, 1853, 2006–2011. [Google Scholar] [CrossRef] [PubMed]
- Gafni, J.; Munsch, J.A.; Lam, T.H.; Catlin, M.C.; Costa, L.G.; Molinski, T.F.; Pessah, I.N. Xestospongins: Potent Membrane Permeable Blockers of the Inositol 1,4,5-Trisphosphate Receptor. Neuron 1997, 19, 723–733. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Naia, L.; Pinho, C.M.; Dentoni, G.; Liu, J.; Leal, N.S.; Ferreira, D.M.S.; Schreiner, B.; Filadi, R.; Fão, L.; Connolly, N.M.C.; et al. Neuronal cell-based high-throughput screen for enhancers of mitochondrial function reveals luteolin as a modulator of mitochondria-endoplasmic reticulum coupling. BMC Biol. 2021, 19, 1–21. [Google Scholar] [CrossRef]
- Di Marco, G.; Vallese, F.; Jourde, B.; Bergsdorf, C.; Sturlese, M.; De Mario, A.; Techer-Etienne, V.; Haasen, D.; Oberhauser, B.; Schleeger, S.; et al. A High-Throughput Screening Identifies MICU1 Targeting Compounds. Cell Rep. 2020, 30, 2321–2331.e6. [Google Scholar] [CrossRef] [Green Version]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum–mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef]
- Shirokova, O.M.; Pchelin, P.V.; Mukhina, I.V. MERCs. The Novel Assistant to Neurotransmission? Front. Neurosci. 2020, 14, 1169. [Google Scholar] [CrossRef]
- Guillaud, L.; Dimitrov, D.; Takahashi, T. Presynaptic morphology and vesicular composition determine vesicle dynamics in mouse central synapses. eLife 2017, 6, e24845. [Google Scholar] [CrossRef] [PubMed]
- Babu, L.P.A.; Wang, H.-Y.; Eguchi, K.; Guillaud, L.; Takahashi, T. Microtubule and Actin Differentially Regulate Synaptic Vesicle Cycling to Maintain High-Frequency Neurotransmission. J. Neurosci. 2019, 40, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Sobue, K.; Kanda, K.; Harada, A.; Yorifuji, H. The cytoskeletal architecture of the presynaptic terminal and molecular structure of synapsin 1. J. Cell Biol. 1989, 108, 111–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantano, S.; Montecucco, C. The blockade of the neurotransmitter release apparatus by botulinum neurotoxins. Cell. Mol. Life Sci. 2013, 71, 793–811. [Google Scholar] [CrossRef]
- Sørensen, J.B.; Nagy, G.; Varoqueaux, F.; Nehring, R.B.; Brose, N.; Wilson, M.C.; Neher, E. Differential Control of the Releasable Vesicle Pools by SNAP-25 Splice Variants and SNAP-23. Cell 2003, 114, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Mohrmann, R.; de Wit, H.; Verhage, M.; Neher, E.; Sørensen, J.B. Fast Vesicle Fusion in Living Cells Requires at Least Three SNARE Complexes. Science 2010, 330, 502–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toft-Bertelsen, T.L.; Ziomkiewicz, I.; Houy, S.; Pinheiro, P.S.; Sørensen, J.B. Regulation of Ca2+ channels by SNAP-25 via recruitment of syntaxin-1 from plasma membrane clusters. Mol. Biol. Cell 2016, 27, 3329–3341. [Google Scholar] [CrossRef]
- Lin, Y.; Hou, X.; Shen, W.-J.; Hanssen, R.; Khor, V.K.; Cortez, Y.; Roseman, A.N.; Azhar, S.; Kraemer, F.B. SNARE-Mediated Cholesterol Movement to Mitochondria Supports Steroidogenesis in Rodent Cells. Mol. Endocrinol. 2016, 30, 234–247. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.; Yan, S.; Yu, Q.; Chen, D.; Yan, S.S. Mfn2 is Required for Mitochondrial Development and Synapse Formation in Human Induced Pluripotent Stem Cells/hiPSC Derived Cortical Neurons. Sci. Rep. 2016, 6, 31462. [Google Scholar] [CrossRef] [Green Version]
- Nelson, S.B.; Turrigiano, G.G. Synaptic depression: A key player in the cortical balancing act. Nat. Neurosci. 1998, 1, 539–541. [Google Scholar] [CrossRef]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, X.; Nguyen, T.T.; Choi, S.-K.; Xu, S.; Das, R.; Cha, S.-K.; Kim, N.; Han, J.; Wiederkehr, A.; Wollheim, C.B.; et al. Essential Role of Mitochondrial Ca2+ Uniporter in the Generation of Mitochondrial pH Gradient and Metabolism-Secretion Coupling in Insulin-releasing Cells. J. Biol. Chem. 2015, 290, 4086–4096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER–mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2511. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; He, X.-P.; Russell, J.; Lu, B. Ca2+ influx–independent synaptic potentiation mediated by mitochondrial Na+-Ca2+ exchanger and protein kinase C. J. Cell Biol. 2003, 163, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.-S.; Tian, J.-H.; Pan, P.-Y.; Zald, P.; Li, C.; Deng, C.; Sheng, Z.-H. Docking of Axonal Mitochondria by Syntaphilin Controls Their Mobility and Affects Short-Term Facilitation. Cell 2008, 132, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.K.; Sando, R., III; Lewis, T.L.; Hirabayashi, Y.; Maximov, A.; Polleux, F. LKB1 Regulates Mitochondria-Dependent Presynaptic Calcium Clearance and Neurotransmitter Release Properties at Excitatory Synapses along Cortical Axons. PLoS Biol. 2016, 14, e1002516. [Google Scholar] [CrossRef] [Green Version]
- Low, J.T.; Shukla, A.; Behrendorff, N.; Thorn, P. Exocytosis, dependent on Ca2+ release from Ca2+ stores, is regulated by Ca2+ microdomains. J. Cell Sci. 2010, 123, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, J.; Viniegra, S.; Gimenez-Molina, Y.; García-Martinez, V.; Expósito-Romero, G.; del Mar Frances, M.; García-Sancho, J.; Gutiérrez, L.M. The position of mitochondria and ER in relation to that of the secretory sites in chromaffin cells. J. Cell Sci. 2014, 127 Pt 23, 5105–5114. [Google Scholar]
- Montero, M.; Alonso, M.T.; Carnicero, E.; Cuchillo-Ibáñez, I.; Albillos, A.; Garcia, A.G.; Garcia-Sancho, J.; Alvarez, J. Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat. Cell Biol. 1999, 2, 57–61. [Google Scholar] [CrossRef]
- García, A.G.; García-De-Diego, A.M.; Gandia, L.; Borges, R.; Garcia-Sancho, J. Calcium Signaling and Exocytosis in Adrenal Chromaffin Cells. Physiol. Rev. 2006, 86, 1093–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, D.J. Mitochondrial dysfunction, oxidative stress, regulation of exocytosis and their relevance to neurodegenerative diseases. J. Neurochem. 2007, 104, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xue, R.; Ong, W.-Y.; Chen, P. Roles of Cholesterol in Vesicle Fusion and Motion. Biophys. J. 2009, 97, 1371–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrbough, J.; Broadie, K. Lipid regulation of the synaptic vesicle cycle. Nat. Rev. Neurosci. 2005, 6, 139–150. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dentoni, G.; Naia, L.; Ankarcrona, M. Mitochondria-Endoplasmic Reticulum Interplay Regulates Exo-Cytosis in Human Neuroblastoma Cells. Cells 2022, 11, 514. https://doi.org/10.3390/cells11030514
Dentoni G, Naia L, Ankarcrona M. Mitochondria-Endoplasmic Reticulum Interplay Regulates Exo-Cytosis in Human Neuroblastoma Cells. Cells. 2022; 11(3):514. https://doi.org/10.3390/cells11030514
Chicago/Turabian StyleDentoni, Giacomo, Luana Naia, and Maria Ankarcrona. 2022. "Mitochondria-Endoplasmic Reticulum Interplay Regulates Exo-Cytosis in Human Neuroblastoma Cells" Cells 11, no. 3: 514. https://doi.org/10.3390/cells11030514