
Put in a “Ca2+ll” to Acute Myeloid Leukemia

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. The Role of Calcium Homeostasis in AML Cell Proliferation and Differentiation
2.1. Calcium and Cell Cycle Regulation in AML
2.2. Calcium Channels and Proliferation in AML
2.3. Implication of Notch and Ca2+ Signaling in AML Proliferation
2.4. Calcium Involvement in AML Differentiation
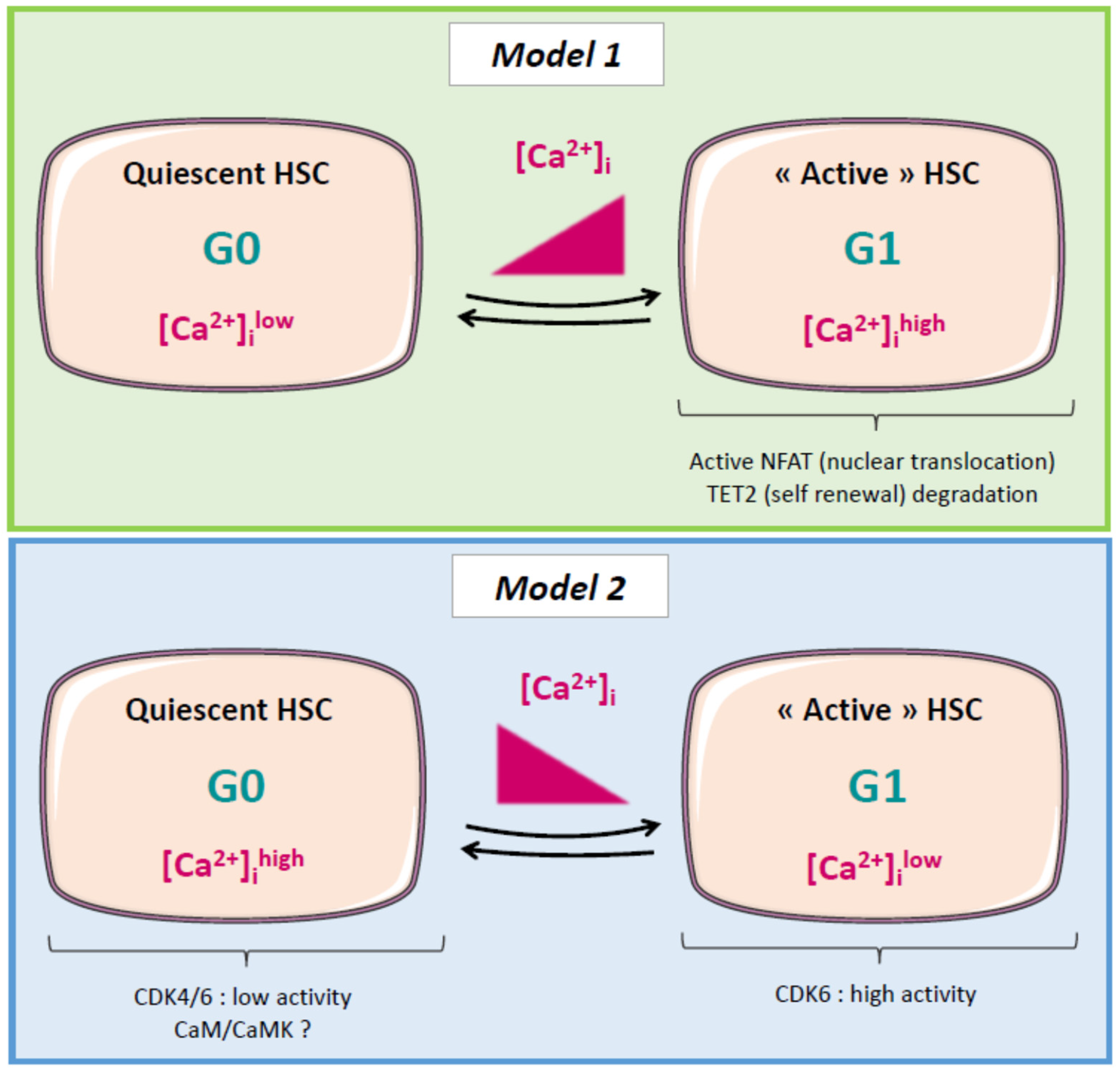
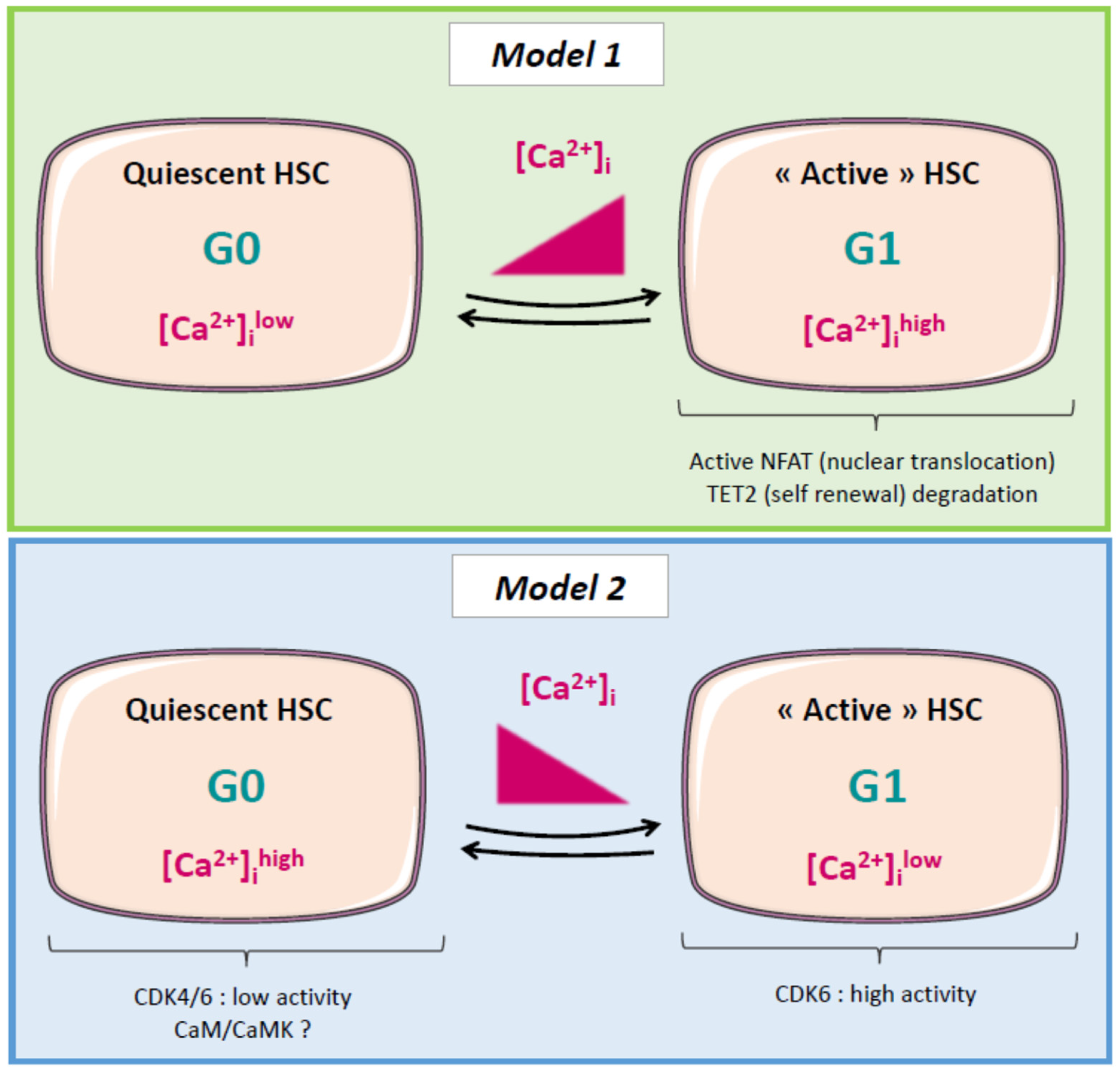
2.5. Calcium and Cell Cycle Regulation in Normal and Cancer Stem Cells (CSCs)
2.6. Leukemic Stem Cells (LSCs), Relapse, and Calcium: A Possible Link?
2.7. Future Directions
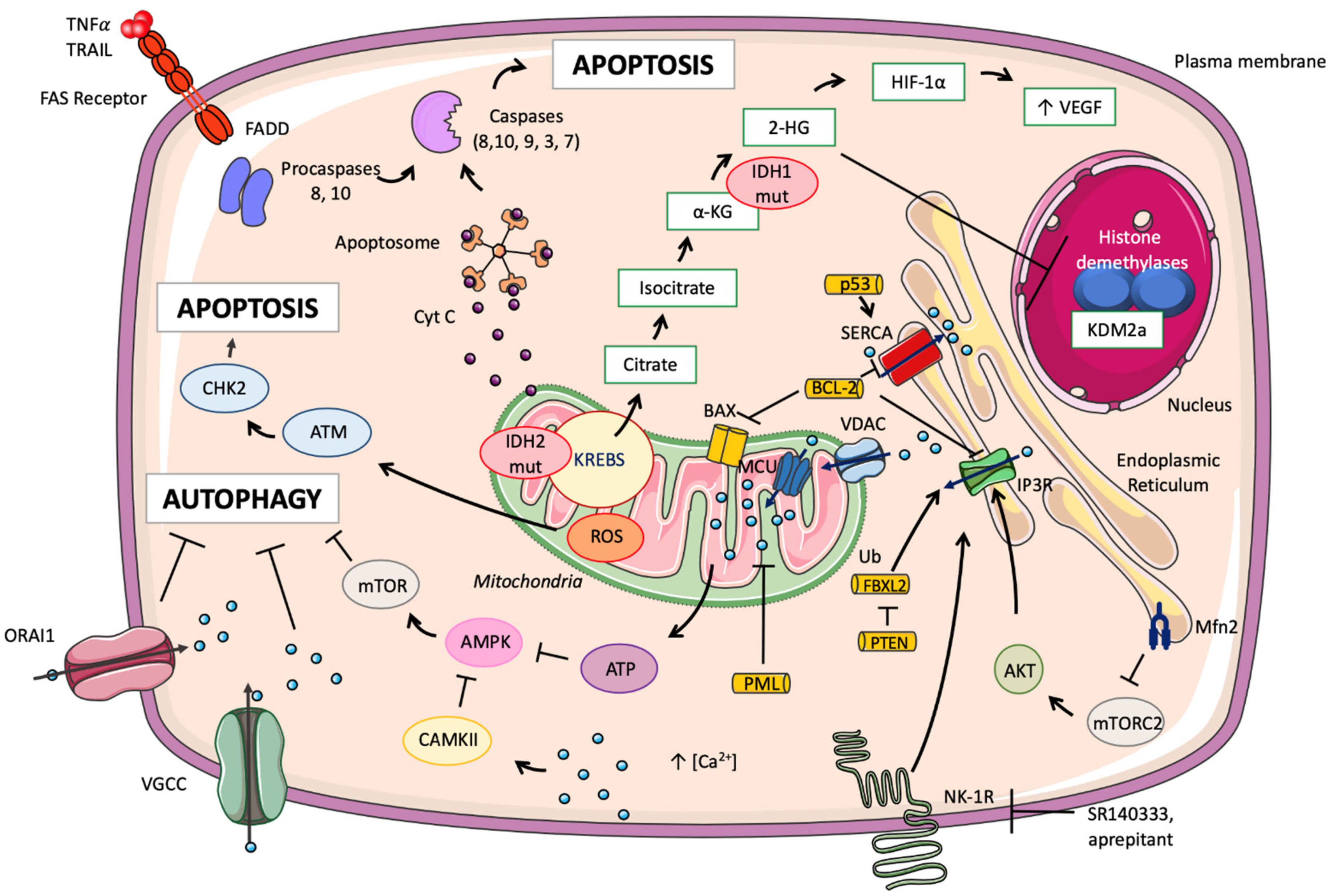
3. Mitochondria, Calcium, and AML
3.1. Normal Hematopoiesis
3.2. LSCs and AML
3.3. Mitochondrial Calcium and Its Implication in Cancer Mechanisms
3.4. Isocitrate Dehydrogenase in AML
4. Calcium, Microenvironment, and AML Cells
4.1. Role of the Endosteal Niche
4.2. Modulation by Retinoic Acid (RA)
4.3. The Vascular Niche
4.4. Immune Escape
5. Calcium Signaling in AML Treatment: A New Hope?
5.1. Chemotherapies, Calcium, and Mitochondria
5.2. Modulation of ER Calcium Stores
5.3. Chemotherapies Impacting Calcium Influx
5.4. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Medinger, M.; Passweg, J.R. Acute Myeloid Leukaemia Genomics. Br. J. Haematol. 2017, 179, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Bixby, D.; Perl, A.; Bhatt, V.R.; Altman, J.K.; Appelbaum, F.R.; de Lima, M.; Fathi, A.T.; Foran, J.M.; Gojo, I.; et al. NCCN Guidelines Insights: Acute Myeloid Leukemia, Version 2.2021. J. Natl. Compr. Canc. Netw. 2021, 19, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium Signalling: Dynamics, Homeostasis and Remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, A.G.; Guimarães, E.S.; Andrade, L.M.; Menezes, G.B.; Fatima Leite, M. Decoding Calcium Signaling across the Nucleus. Physiology 2014, 29, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323. [Google Scholar] [CrossRef] [PubMed]
- Cabanas, H.; Harnois, T.; Magaud, C.; Cousin, L.; Constantin, B.; Bourmeyster, N.; Déliot, N. Deregulation of Calcium Homeostasis in Bcr-Abl-Dependent Chronic Myeloid Leukemia. Oncotarget 2018, 9, 26309–26327. [Google Scholar] [CrossRef] [Green Version]
- Luchsinger, L.L.; Strikoudis, A.; Danzl, N.M.; Bush, E.C.; Finlayson, M.O.; Satwani, P.; Sykes, M.; Yazawa, M.; Snoeck, H.-W. Harnessing Hematopoietic Stem Cell Low Intracellular Calcium Improves Their Maintenance In Vitro. Cell Stem Cell 2019, 25, 225–240. [Google Scholar] [CrossRef]
- Fukushima, T.; Tanaka, Y.; Hamey, F.K.; Chang, C.-H.; Oki, T.; Asada, S.; Hayashi, Y.; Fujino, T.; Yonezawa, T.; Takeda, R.; et al. Discrimination of Dormant and Active Hematopoietic Stem Cells by G(0) Marker Reveals Dormancy Regulation by Cytoplasmic Calcium. Cell Rep. 2019, 29, 4144–4158.e7. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Li, L. Osteoblast Ablation Burns out Functional Stem Cells. Blood 2015, 125, 2590–2591. [Google Scholar] [CrossRef] [Green Version]
- Bowers, M.; Zhang, B.; Ho, Y.; Agarwal, P.; Chen, C.-C.; Bhatia, R. Osteoblast Ablation Reduces Normal Long-Term Hematopoietic Stem Cell Self-Renewal but Accelerates Leukemia Development. Blood 2015, 125, 2678–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becchetti, A. Ion Channels and Transporters in Cancer. 1. Ion Channels and Cell Proliferation in Cancer. Am. J. Physiol. Cell Physiol. 2011, 301, C255–C265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Déliot, N.; Constantin, B. Plasma Membrane Calcium Channels in Cancer: Alterations and Consequences for Cell Proliferation and Migration. Biochim. Biophys. Acta 2015, 1848, 2512–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chafouleas, J.G.; Lagacé, L.; Bolton, W.E.; Boyd, A.E., 3rd; Means, A.R. Changes in Calmodulin and Its MRNA Accompany Reentry of Quiescent (G0) Cells into the Cell Cycle. Cell 1984, 36, 73–81. [Google Scholar] [CrossRef]
- Rasmussen, C.D.; Means, A.R. Calmodulin, Cell Growth and Gene Expression. Trends Neurosci. 1989, 12, 433–438. [Google Scholar] [CrossRef]
- Takuwa, N.; Zhou, W.; Kumada, M.; Takuwa, Y. Ca(2+)-Dependent Stimulation of Retinoblastoma Gene Product Phosphorylation and P34cdc2 Kinase Activation in Serum-Stimulated Human Fibroblasts. J. Biol. Chem. 1993, 268, 138–145. [Google Scholar] [CrossRef]
- Yen, A.; Freeman, L.; Powers, V.; Van Sant, R.; Fishbaugh, J. Cell Cycle Dependence of Calmodulin Levels during HL-60 Proliferation and Myeloid Differentiation. No Changes during Pre-Commitment. Exp. Cell Res. 1986, 165, 139–151. [Google Scholar] [CrossRef]
- Kahl, C.R.; Means, A.R. Regulation of Cell Cycle Progression by Calcium/Calmodulin-Dependent Pathways. Endocr. Rev. 2003, 24, 719–736. [Google Scholar] [CrossRef] [Green Version]
- Monaco, S.; Rusciano, M.R.; Maione, A.S.; Soprano, M.; Gomathinayagam, R.; Todd, L.R.; Campiglia, P.; Salzano, S.; Pastore, L.; Leggiero, E.; et al. A Novel Crosstalk between Calcium/Calmodulin Kinases II and IV Regulates Cell Proliferation in Myeloid Leukemia Cells. Cell Signal. 2015, 27, 204–214. [Google Scholar] [CrossRef] [Green Version]
- Kang, X.; Cui, C.; Wang, C.; Wu, G.; Chen, H.; Lu, Z.; Chen, X.; Wang, L.; Huang, J.; Geng, H.; et al. CAMKs Support Development of Acute Myeloid Leukemia. J. Hematol. Oncol. 2018, 11, 30. [Google Scholar] [CrossRef] [Green Version]
- Tomono, M.; Toyoshima, K.; Ito, M.; Amano, H.; Kiss, Z. Inhibitors of Calcineurin Block Expression of Cyclins A and E Induced by Fibroblast Growth Factor in Swiss 3T3 Fibroblasts. Arch. Biochem. Biophys. 1998, 353, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, M.; Schatz, A.; Wagner, M.; Michl, P.; Linhart, T.; Adler, G.; Gress, T.M.; Ellenrieder, V. Overexpression of C-Myc in Pancreatic Cancer Caused by Ectopic Activation of NFATc1 and the Ca2+/Calcineurin Signaling Pathway. EMBO J. 2006, 25, 3714–3724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels and the Hallmarks of Cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Padma, S.; Subramanyam, C. Clinical Significance of Serum Calcineurin in Acute Leukemia. Clin. Chim. Acta 2002, 321, 17–21. [Google Scholar] [CrossRef]
- Chen, S.-J.; Bao, L.; Keefer, K.; Shanmughapriya, S.; Chen, L.; Lee, J.; Wang, J.; Zhang, X.-Q.; Hirschler-Laszkiewicz, I.; Merali, S.; et al. Transient Receptor Potential Ion Channel TRPM2 Promotes AML Proliferation and Survival through Modulation of Mitochondrial Function, ROS, and Autophagy. Cell Death Dis. 2020, 11, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Babicheva, A.; Zhao, T.; Ayon, R.J.; Rodriguez, M.; Rahimi, S.; Balistrieri, F.; Harrington, A.; Shyy, J.Y.-J.; Thistlethwaite, P.A.; et al. Notch Enhances Ca(2+) Entry by Activating Calcium-Sensing Receptors and Inhibiting Voltage-Gated K(+) Channels. Am. J. Physiol. Cell Physiol. 2020, 318, C954–C968. [Google Scholar] [CrossRef]
- Shi, J.; Fu, L.; Wang, W. High Expression of Inositol 1,4,5-Trisphosphate Receptor, Type 2 (ITPR2) as a Novel Biomarker for Worse Prognosis in Cytogenetically Normal Acute Myeloid Leukemia. Oncotarget 2015, 6, 5299–5309. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, H.; Yamamura, A.; Ko, E.A.; Pohl, N.M.; Smith, K.A.; Zeifman, A.; Powell, F.L.; Thistlethwaite, P.A.; Yuan, J.X.-J. Activation of Notch Signaling by Short-Term Treatment with Jagged-1 Enhances Store-Operated Ca(2+) Entry in Human Pulmonary Arterial Smooth Muscle Cells. Am. J. Physiol. Cell Physiol. 2014, 306, C871–C878. [Google Scholar] [CrossRef] [Green Version]
- Tohda, S.; Sakano, S.; Ohsawa, M.; Murakami, N.; Nara, N. A Novel Cell Line Derived from de Novo Acute Myeloblastic Leukaemia with Trilineage Myelodysplasia Which Proliferates in Response to a Notch Ligand, Delta-1 Protein. Br. J. Haematol. 2002, 117, 373–378. [Google Scholar] [CrossRef]
- Li, G.-H.; Fan, Y.-Z.; Liu, X.-W.; Zhang, B.-F.; Yin, D.-D.; He, F.; Huang, S.-Y.; Kang, Z.-J.; Xu, H.; Liu, Q.; et al. Notch Signaling Maintains Proliferation and Survival of the HL60 Human Promyelocytic Leukemia Cell Line and Promotes the Phosphorylation of the Rb Protein. Mol. Cell. Biochem. 2010, 340, 7–14. [Google Scholar] [CrossRef]
- Kannan, S.; Sutphin, R.M.; Hall, M.G.; Golfman, L.S.; Fang, W.; Nolo, R.M.; Akers, L.J.; Hammitt, R.A.; McMurray, J.S.; Kornblau, S.M.; et al. Notch Activation Inhibits AML Growth and Survival: A Potential Therapeutic Approach. J. Exp. Med. 2013, 210, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Lobry, C.; Ntziachristos, P.; Ndiaye-Lobry, D.; Oh, P.; Cimmino, L.; Zhu, N.; Araldi, E.; Hu, W.; Freund, J.; Abdel-Wahab, O.; et al. Notch Pathway Activation Targets AML-Initiating Cell Homeostasis and Differentiation. J. Exp. Med. 2013, 210, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Tohda, S.; Kogoshi, H.; Murakami, N.; Sakano, S.; Nara, N. Diverse Effects of the Notch Ligands Jagged1 and Delta1 on the Growth and Differentiation of Primary Acute Myeloblastic Leukemia Cells. Exp. Hematol. 2005, 33, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Launay, S.; Giannì, M.; Kovàcs, T.; Bredoux, R.; Bruel, A.; Gélébart, P.; Zassadowski, F.; Chomienne, C.; Enouf, J.; Papp, B. Lineage-Specific Modulation of Calcium Pump Expression during Myeloid Differentiation. Blood 1999, 93, 4395–4405. [Google Scholar] [CrossRef] [PubMed]
- Laouedj, M.; Tardif, M.R.; Gil, L.; Raquil, M.-A.; Lachhab, A.; Pelletier, M.; Tessier, P.A.; Barabé, F. S100A9 Induces Differentiation of Acute Myeloid Leukemia Cells through TLR4. Blood 2017, 129, 1980–1990. [Google Scholar] [CrossRef] [Green Version]
- Chapekar, M.S.; Hartman, K.D.; Knode, M.C.; Glazer, R.I. Synergistic Effect of Retinoic Acid and Calcium Ionophore A23187 on Differentiation, c-Myc Expression, and Membrane Tyrosine Kinase Activity in Human Promyelocytic Leukemia Cell Line HL-60. Mol. Pharmacol. 1987, 31, 140–145. [Google Scholar] [PubMed]
- Li, H.; Xu, J.; Zhou, Y.; Liu, X.; Shen, L.E.; Zhu, Y.U.; Li, Z.; Wang, X.; Guo, Q.; Hui, H. PLSCR1/IP3R1/Ca(2+) Axis Contributes to Differentiation of Primary AML Cells Induced by Wogonoside. Cell Death Dis. 2017, 8, e2768. [Google Scholar] [CrossRef]
- O’Reilly, D.; Buchanan, P. Calcium Channels and Cancer Stem Cells. Cell Calcium 2019, 81, 21–28. [Google Scholar] [CrossRef]
- Snoeck, H.-W. Calcium Regulation of Stem Cells. EMBO Rep. 2020, 21, e50028. [Google Scholar] [CrossRef]
- Horsley, V.; Aliprantis, A.O.; Polak, L.; Glimcher, L.H.; Fuchs, E. NFATc1 Balances Quiescence and Proliferation of Skin Stem Cells. Cell 2008, 132. [Google Scholar] [CrossRef] [Green Version]
- Aulestia, F.J.; Néant, I.; Dong, J.; Haiech, J.; Kilhoffer, M.-C.; Moreau, M.; Leclerc, C. Quiescence Status of Glioblastoma Stem-like Cells Involves Remodelling of Ca(2+) Signalling and Mitochondrial Shape. Sci. Rep. 2018, 8, 9731. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Kahsay, A.; Pinton, P. Mitochondrial Calcium Homeostasis in Hematopoietic Stem Cell: Molecular Regulation of Quiescence, Function, and Differentiation. Int. Rev. Cell Mol. Biol. 2021, 362, 111–140. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, T.; Hashimoto, M.; Matsumura, T.; Nakamura-Ishizu, A.; Suda, T. Ca(2+)-Mitochondria Axis Drives Cell Division in Hematopoietic Stem Cells. J. Exp. Med. 2018, 215, 2097–2113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Su, J.; Jeong, M.; Ko, M.; Huang, Y.; Park, H.J.; Guzman, A.; Lei, Y.; Huang, Y.-H.; Rao, A.; et al. DNMT3A and TET2 Compete and Cooperate to Repress Lineage-Specific Transcription Factors in Hematopoietic Stem Cells. Nat. Genet. 2016, 48, 1014–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.W.K.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-Gene Stemness Score for Rapid Determination of Risk in Acute Leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef]
- Bachas, C.; Schuurhuis, G.J.; Assaraf, Y.G.; Kwidama, Z.J.; Kelder, A.; Wouters, F.; Snel, A.N.; Kaspers, G.J.L.; Cloos, J. The Role of Minor Subpopulations within the Leukemic Blast Compartment of AML Patients at Initial Diagnosis in the Development of Relapse. Leukemia 2012, 26, 1313–1320. [Google Scholar] [CrossRef]
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.L.; et al. Distinct Evolution and Dynamics of Epigenetic and Genetic Heterogeneity in Acute Myeloid Leukemia. Nat. Med. 2016, 22, 792–799. [Google Scholar] [CrossRef] [Green Version]
- Fric, J.; Lim, C.X.; Mertes, A.; Lee, B.T.; Vigano, E.; Chen, J.; Zolezzi, F.; Poidinger, M.; Larbi, A.; Strobl, H.; et al. Calcium and Calcineurin-Nfat Signaling Regulate Granulocyte-Monocyte Progenitor Cell Cycle Via Flt3-L. Stem Cells 2014. [Google Scholar] [CrossRef] [Green Version]
- Metzelder, S.K.; Michel, C.; von Bonin, M.; Rehberger, M.; Hessmann, E.; Inselmann, S.; Solovey, M.; Wang, Y.; Sohlbach, K.; Brendel, C.; et al. NFATc1 as a Therapeutic Target in FLT3-ITD-Positive AML. Leukemia 2015, 29, 1470–1477. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic Inhibition of Fatty Acid Oxidation Sensitizes Human Leukemia Cells to Apoptosis Induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gherardi, G.; Monticelli, H.; Rizzuto, R.; Mammucari, C. The Mitochondrial Ca(2+) Uptake and the Fine-Tuning of Aerobic Metabolism. Front. Physiol. 2020, 11, 554904. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, M.T.; King, K.Y.; Boles, N.C.; Weksberg, D.C.; Goodell, M.A. Quiescent Haematopoietic Stem Cells Are Activated by IFN-Gamma in Response to Chronic Infection. Nature 2010, 465, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.A.; Gibb, A.A.; Arif, E.; Kolmetzky, D.W.; Tomar, D.; Luongo, T.S.; Jadiya, P.; Murray, E.K.; Lorkiewicz, P.K.; Hajnóczky, G.; et al. Mitochondrial Calcium Exchange Links Metabolism with the Epigenome to Control Cellular Differentiation. Nat. Commun. 2019, 10, 4509. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Kao, Y.-R.; Sun, D.; Todorova, T.I.; Reynolds, D.; Narayanagari, S.-R.; Montagna, C.; Will, B.; Verma, A.; Steidl, U. Myelodysplastic Syndrome Progression to Acute Myeloid Leukemia at the Stem Cell Level. Nat. Med. 2019, 25, 103–110. [Google Scholar] [CrossRef]
- Corces-Zimmerman, M.R.; Hong, W.-J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic Mutations in Human Acute Myeloid Leukemia Affect Epigenetic Regulators and Persist in Remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553. [Google Scholar] [CrossRef] [Green Version]
- Koeffler, H.P.; Leong, G. Preleukemia: One Name, Many Meanings. Leukemia 2017, 31, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Saeed, B.R.; Manta, L.; Raffel, S.; Pyl, P.T.; Buss, E.C.; Wang, W.; Eckstein, V.; Jauch, A.; Trumpp, A.; Huber, W.; et al. Analysis of Nonleukemic Cellular Subcompartments Reconstructs Clonal Evolution of Acute Myeloid Leukemia and Identifies Therapy-Resistant Preleukemic Clones. Int. J. Cancer 2021, 148, 2825–2838. [Google Scholar] [CrossRef]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of Pre-Leukaemic Haematopoietic Stem Cells in Acute Leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Bencomo-Alvarez, A.E.; Rubio, A.J.; Gonzalez, M.A.; Eiring, A.M. Energy Metabolism and Drug Response in Myeloid Leukaemic Stem Cells. Br. J. Haematol. 2019, 186, 524–537. [Google Scholar] [CrossRef]
- Gilliland, D.G.; Jordan, C.T.; Felix, C.A. The Molecular Basis of Leukemia. Hematol. Am. Soc. Hematol. Educ. Program. 2004, 80–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapuis, N.; Poulain, L.; Birsen, R.; Tamburini, J.; Bouscary, D. Rationale for Targeting Deregulated Metabolic Pathways as a Therapeutic Strategy in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, M.; Diesch, J.; Casquero, R.; Buschbeck, M. Epigenetic-Transcriptional Regulation of Fatty Acid Metabolism and Its Alterations in Leukaemia. Front. Genet. 2018, 9, 405. [Google Scholar] [CrossRef]
- Tabe, Y.; Konopleva, M.; Andreeff, M. Fatty Acid Metabolism, Bone Marrow Adipocytes, and AML. Front. Oncol. 2020, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Tcheng, M.; Roma, A.; Ahmed, N.; Smith, R.W.; Jayanth, P.; Minden, M.D.; Schimmer, A.D.; Hess, D.A.; Hope, K.; Rea, K.A.; et al. Very Long Chain Fatty Acid Metabolism Is Required in Acute Myeloid Leukemia. Blood 2021, 137, 3518–3532. [Google Scholar] [CrossRef]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty Acid Metabolism Underlies Venetoclax Resistance in Acute Myeloid Leukemia Stem Cells. Nat. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef]
- Zhong, W.; Xu, M.; Li, C.; Zhu, B.; Cao, X.; Li, D.; Chen, H.; Hu, C.; Li, R.; Luo, C.; et al. ORP4L Extracts and Presents PIP(2) from Plasma Membrane for PLCβ3 Catalysis: Targeting It Eradicates Leukemia Stem Cells. Cell Rep. 2019, 26, 2166–2177. [Google Scholar] [CrossRef] [Green Version]
- Csanády, L.; Törocsik, B. Four Ca2+ Ions Activate TRPM2 Channels by Binding in Deep Crevices near the Pore but Intracellularly of the Gate. J. Gen. Physiol. 2009, 133, 189–203. [Google Scholar] [CrossRef]
- Gutierrez, S.; Alvarado-Vázquez, P.A.; Eisenach, J.C.; Romero-Sandoval, E.A.; Boada, M.D. Tachykinins Modulate Nociceptive Responsiveness and Sensitization: In Vivo Electrical Characterization of Primary Sensory Neurons in Tachykinin Knockout (Tac1 KO) Mice. Mol. Pain 2019, 15, 1744806919845750. [Google Scholar] [CrossRef] [Green Version]
- Steinhoff, M.S.; von Mentzer, B.; Geppetti, P.; Pothoulakis, C.; Bunnett, N.W. Tachykinins and Their Receptors: Contributions to Physiological Control and the Mechanisms of Disease. Physiol. Rev. 2014, 94, 265–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, C.; Huang, H.; Huang, F.; Yang, T.; Zhang, T.; Wu, H.; Zhou, H.; Chen, Q.; Shi, Y.; Sun, Y.; et al. Neurokinin-1 Receptor Is an Effective Target for Treating Leukemia by Inducing Oxidative Stress through Mitochondrial Calcium Overload. Proc. Natl. Acad. Sci. USA 2019, 116, 19635–19645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, C.; Wuertzer, C.; Cui, X.; Bi, Y.; Davuluri, R.; Xiao, Y.-Y.; Wilson, M.; Owens, K.; Zhang, Y.; Perkins, A. Global Identification of EVI1 Target Genes in Acute Myeloid Leukemia. PLoS ONE 2013, 8, e67134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, W.; Yang, X.; Wang, L.; Wang, R.; Yang, F.; Wang, H.; Liu, X.; Ren, Q.; Zhang, Y.; Zhu, X.; et al. P2X7 Promotes the Progression of MLL-AF9 Induced Acute Myeloid Leukemia by Upregulation of Pbx3. Haematologica 2021, 106, 1278–1289. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Marinello, M.; Bononi, A.; Bonora, M.; Giorgi, C.; Rimessi, A.; Pinton, P. Selective Modulation of Subtype III IP3R by Akt Regulates ER Ca2+ Release and Apoptosis. Cell Death Dis. 2012, 3, e304. [Google Scholar] [CrossRef] [Green Version]
- Barazzuol, L.; Giamogante, F.; Calì, T. Mitochondria Associated Membranes (MAMs): Architecture and Physiopathological Role. Cell Calcium 2021, 94, 102343. [Google Scholar] [CrossRef]
- Kuchay, S.; Giorgi, C.; Simoneschi, D.; Pagan, J.; Missiroli, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Collazo-Lorduy, A.; Castillo-Martin, M.; et al. PTEN Counteracts FBXL2 to Promote IP3R3- and Ca(2+)-Mediated Apoptosis Limiting Tumour Growth. Nature 2017, 546, 554–558. [Google Scholar] [CrossRef]
- Høyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of Macroautophagy by Calcium, Calmodulin-Dependent Kinase Kinase-Beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, J.; Xu, Z.; Peng, B.; Huang, Q.; Arnold, E.; Ding, J. Structures of Human Cytosolic NADP-Dependent Isocitrate Dehydrogenase Reveal a Novel Self-Regulatory Mechanism of Activity. J. Biol. Chem. 2004, 279, 33946–33957. [Google Scholar] [CrossRef] [Green Version]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate Dehydrogenase Mutations in Gliomas. Neuro. Oncol. 2016, 18, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The Consensus Coding Sequences of Human Breast and Colorectal Cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Yen, K.E.; Bittinger, M.A.; Su, S.M.; Fantin, V.R. Cancer-Associated IDH Mutations: Biomarker and Therapeutic Opportunities. Oncogene 2010, 29, 6409–6417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-Associated Metabolite 2-Hydroxyglutarate Accumulates in Acute Myelogenous Leukemia with Isocitrate Dehydrogenase 1 and 2 Mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef]
- Marcucci, G.; Maharry, K.; Wu, Y.-Z.; Radmacher, M.D.; Mrózek, K.; Margeson, D.; Holland, K.B.; Whitman, S.P.; Becker, H.; Schwind, S.; et al. IDH1 and IDH2 Gene Mutations Identify Novel Molecular Subsets within de Novo Cytogenetically Normal Acute Myeloid Leukemia: A Cancer and Leukemia Group B Study. J. Clin. Oncol. 2010, 28, 2348–2355. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Cross, J.R.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.A.; Thompson, C.B. Identification of Additional IDH Mutations Associated with Oncometabolite R(-)-2-Hydroxyglutarate Production. Oncogene 2012, 31, 2491–2498. [Google Scholar] [CrossRef] [Green Version]
- McKenney, A.S.; Levine, R.L. Isocitrate Dehydrogenase Mutations in Leukemia. J. Clin. Investig. 2013, 123, 3672–3677. [Google Scholar] [CrossRef] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-Associated IDH1 Mutations Produce 2-Hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yang, J.; Sun, X.; Wang, Z.; Cheng, X.; Lu, W.; Cai, X.; Hu, C.; Shen, X.; Cao, P. Allosteric Inhibitor Remotely Modulates the Conformation of the Orthestric Pockets in Mutant IDH2/R140Q. Sci. Rep. 2017, 7, 16458. [Google Scholar] [CrossRef] [Green Version]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Mims, A.S.; Pratz, K.W.; Savona, M.R.; Stein, A.S.; Stone, R.M.; Winer, E.S.; Seet, C.S.; et al. Ivosidenib or Enasidenib Combined with Intensive Chemotherapy in Patients with Newly Diagnosed AML: A Phase 1 Study. Blood 2021, 137, 1792–1803. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Chinnaiyan, A.M. Metabolism Unhinged: IDH Mutations in Cancer. Nat. Med. 2011, 17, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Shafat, M.S.; Gnaneswaran, B.; Bowles, K.M.; Rushworth, S.A. The Bone Marrow Microenvironment – Home of the Leukemic Blasts. Blood Rev. 2017, 31, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinisch, A.; Chan, S.M.; Thomas, D.; Majeti, R. Biology and Clinical Relevance of Acute Myeloid Leukemia Stem Cells. Semin. Hematol. 2015, 52, 150–164. [Google Scholar] [CrossRef] [Green Version]
- Adams, G.B.; Chabner, K.T.; Alley, I.R.; Olson, D.P.; Szczepiorkowski, Z.M.; Poznansky, M.C.; Kos, C.H.; Pollak, M.R.; Brown, E.M.; Scadden, D.T. Stem Cell Engraftment at the Endosteal Niche Is Specified by the Calcium-Sensing Receptor. Nature 2006, 439, 599–603. [Google Scholar] [CrossRef]
- Mansour, A.; Abou-Ezzi, G.; Sitnicka, E.; Jacobsen, S.E.W.; Wakkach, A.; Blin-Wakkach, C. Osteoclasts Promote the Formation of Hematopoietic Stem Cell Niches in the Bone Marrow. J. Exp. Med. 2012, 209, 537–549. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Lévesque, J.-P.; Helwani, F.M.; Winkler, I.G. The Endosteal ‘Osteoblastic’ Niche and Its Role in Hematopoietic Stem Cell Homing and Mobilization. Leukemia 2010, 24, 1979–1992. [Google Scholar] [CrossRef] [Green Version]
- Le, P.M.; Andreeff, M.; Battula, V.L. Osteogenic Niche in the Regulation of Normal Hematopoiesis and Leukemogenesis. Haematologica 2018, 103, 1945–1955. [Google Scholar] [CrossRef] [Green Version]
- Nervi, B.; Ramirez, P.; Rettig, M.P.; Uy, G.L.; Holt, M.S.; Ritchey, J.K.; Prior, J.L.; Piwnica-Worms, D.; Bridger, G.; Ley, T.J.; et al. Chemosensitization of Acute Myeloid Leukemia (AML) Following Mobilization by the CXCR4 Antagonist AMD3100. Blood 2009, 113, 10. [Google Scholar] [CrossRef] [Green Version]
- Spoo, A.C.; Lübbert, M.; Wierda, W.G.; Burger, J.A. CXCR4 Is a Prognostic Marker in Acute Myelogenous Leukemia. Blood 2007, 109, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Z.; Xi Shi, Y.; Samudio, I.J.; Wang, R.-Y.; Ling, X.; Frolova, O.; Levis, M.; Rubin, J.B.; Negrin, R.R.; Estey, E.H.; et al. Targeting the Leukemia Microenvironment by CXCR4 Inhibition Overcomes Resistance to Kinase Inhibitors and Chemotherapy in AML. Blood 2009, 113, 6215–6224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uy, G.L.; Rettig, M.P.; Motabi, I.H.; McFarland, K.; Trinkaus, K.M.; Hladnik, L.M.; Kulkarni, S.; Abboud, C.N.; Cashen, A.F.; Stockerl-Goldstein, K.E.; et al. A Phase 1/2 Study of Chemosensitization with the CXCR4 Antagonist Plerixafor in Relapsed or Refractory Acute Myeloid Leukemia. Blood 2012, 119, 3917–3924. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.M.; MacLeod, R.J. Extracellular Calcium Sensing and Extracellular Calcium Signaling. Physiol. Rev. 2001, 81, 239–297. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.S.; Cunningham, C.; Adams, G.B. Pharmacologic Modulation of the Calcium-Sensing Receptor Enhances Hematopoietic Stem Cell Lodgment in the Adult Bone Marrow. Blood 2011, 117, 1167–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.M. Extracellular Ca2+ Sensing, Regulation of Parathyroid Cell Function, and Role of Ca2+ and Other Ions as Extracellular (First) Messengers. Physiol. Rev. 1991, 71, 371–411. [Google Scholar] [CrossRef]
- Linde, N.; Fluegen, G.; Aguirre-Ghiso, J.A. The Relationship Between Dormant Cancer Cells and Their Microenvironment. Adv. Cancer Res. 2016, 132, 45–71. [Google Scholar] [CrossRef] [Green Version]
- Purton, L.E.; Dworkin, S.; Olsen, G.H.; Walkley, C.R.; Fabb, S.A.; Collins, S.J.; Chambon, P. RARγ Is Critical for Maintaining a Balance between Hematopoietic Stem Cell Self-Renewal and Differentiation. J. Exp. Med. 2006, 203, 1283–1293. [Google Scholar] [CrossRef]
- Ghiaur, G.; Yegnasubramanian, S.; Perkins, B.; Gucwa, J.L.; Gerber, J.M.; Jones, R.J. Regulation of Human Hematopoietic Stem Cell Self-Renewal by the Microenvironment’s Control of Retinoic Acid Signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 16121–16126. [Google Scholar] [CrossRef] [Green Version]
- Nghiem, P.; Ollick, T.; Gardner, P.; Schulman, H. Interleukin-2 Transcriptional Block by Multifunctional Ca2+/Calmodulin Kinase. Nature 1994, 371, 347–350. [Google Scholar] [CrossRef]
- Anderson, K.A.; Means, A.R. Defective Signaling in a Subpopulation of CD4+ T Cells in the Absence of Ca2+/Calmodulin-Dependent Protein Kinase IV. Mol. Cell. Biol. 2002, 22, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, J.; Mueller, L.; Collins, S.J. CaMKII Regulates Retinoic Acid Receptor Transcriptional Activity and the Differentiation of Myeloid Leukemia Cells. J. Clin. Investig. 2007, 117, 1412–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Camacho, L.H.; Mehta, K. Retinoic Acid-Induced CD38 Antigen Promotes Leukemia Cells Attachment and Interferon-γ/Interleukin-1β-Dependent Apoptosis of Endothelial Cells: Implications in the Etiology of Retinoic Acid Syndrome. Leuk. Res. 2007, 31, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Bertagnolo, V.; Neri, L.M.; Marchisio, M.; Mischiati, C.; Capitani, S. Phosphoinositide 3-Kinase Activity Is Essential for All-Trans-Retinoic Acid-Induced Granulocytic Differentiation of HL-60 Cells. Cancer Res. 1999, 59, 542–546. [Google Scholar]
- Wu, X.; Shao, G.; Chen, S.; Wang, X.; Wang, Z.-Y. Studies on the Relationship between Protein Kinase C and Differentiation of Human Promyelocytic Leukemia Cells Induced by Retinoic Acid. Leuk. Res. 1989, 13, 869–874. [Google Scholar] [CrossRef]
- Yen, A.; Roberson, M.S.; Varvayanis, S.; Lee, A.T. Retinoic Acid Induced Mitogen-Activated Protein (MAP)/Extracellular Signal- Regulated Kinase (ERK) Kinase-Dependent MAP Kinase Activation Needed to Elicit HL-60 Cell Differentiation and Growth Arrest. Cancer Res. 1998, 58, 3163–3172. [Google Scholar]
- Su, M.; Alonso, S.; Jones, J.W.; Yu, J.; Kane, M.A.; Jones, R.J.; Ghiaur, G. All-Trans Retinoic Acid Activity in Acute Myeloid Leukemia: Role of Cytochrome P450 Enzyme Expression by the Microenvironment. PLoS ONE 2015, 10, e0127790. [Google Scholar] [CrossRef] [Green Version]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-Β2 Dictates Disseminated Tumour Cell Fate in Target Organs through TGF-β-RIII and P38α/β Signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [Green Version]
- Bruserud, Ø.; Tsykunova, G.; Hernandez-Valladares, M.; Reikvam, H.; Tvedt, T.H.A. Therapeutic Use of Valproic Acid and All-Trans Retinoic Acid in Acute Myeloid Leukemia—Literature Review and Discussion of Possible Use in Relapse after Allogeneic Stem Cell Transplantation. Pharmaceuticals 2021, 14, 423. [Google Scholar] [CrossRef]
- Kim, Y.; Jeung, H.-K.; Cheong, J.-W.; Song, J.; Bae, S.H.; Lee, J.I.; Min, Y.H. All-Trans Retinoic Acid Synergizes with Enasidenib to Induce Differentiation of IDH2-Mutant Acute Myeloid Leukemia Cells. Yonsei Med. J. 2020, 61, 762. [Google Scholar] [CrossRef]
- Ghiaur, G.; Wroblewski, M.; Loges, S. Acute Myelogenous Leukemia and Its Microenvironment: A Molecular Conversation. Semin. Hematol. 2015, 52, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Ciciarello, M.; Corradi, G.; Loscocco, F.; Visani, G.; Monaco, F.; Cavo, M.; Curti, A.; Isidori, A. The Yin and Yang of the Bone Marrow Microenvironment: Pros and Cons of Mesenchymal Stromal Cells in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benito, J.; Ramirez, M.S.; Millward, N.Z.; Velez, J.; Harutyunyan, K.G.; Lu, H.; Shi, Y.-X.; Matre, P.; Jacamo, R.; Ma, H.; et al. Hypoxia-Activated Prodrug TH-302 Targets Hypoxic Bone Marrow Niches in Preclinical Leukemia Models. Clin. Cancer Res. 2016, 22, 1687–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Oosterwijk, J.G.; Buelow, D.R.; Drenberg, C.D.; Vasilyeva, A.; Li, L.; Shi, L.; Wang, Y.-D.; Finkelstein, D.; Shurtleff, S.A.; Janke, L.J.; et al. Hypoxia-Induced Upregulation of BMX Kinase Mediates Therapeutic Resistance in Acute Myeloid Leukemia. J. Clin. Investig. 2018, 128, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.O.; Mortensen, B.T.; Hodgkiss, R.J.; Iversen, P.O.; Christensen, I.J.; Helledie, N.; Larsen, J.K. Increased Cellular Hypoxia and Reduced Proliferation of Both Normal and Leukaemic Cells during Progression of Acute Myeloid Leukaemia in Rats. Cell Prolif. 2000, 33, 381–395. [Google Scholar] [CrossRef]
- Portwood, S.; Lal, D.; Hsu, Y.-C.; Vargas, R.; Johnson, M.K.; Wetzler, M.; Hart, C.P.; Wang, E.S. Activity of the Hypoxia-Activated Prodrug, TH-302, in Preclinical Human Acute Myeloid Leukemia Models. Clin. Cancer Res. 2013, 19, 6506–6519. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 Is an Essential Pore Subunit of the CRAC Channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Yeromin, A.V.; Zhang, S.L.; Jiang, W.; Yu, Y.; Safrina, O.; Cahalan, M.D. Molecular Identification of the CRAC Channel by Altered Ion Selectivity in a Mutant of Orai. Nature 2006, 443, 226–229. [Google Scholar] [CrossRef]
- Sipkins, D.A.; Wei, X.; Wu, J.W.; Runnels, J.M.; Côté, D.; Means, T.K.; Luster, A.D.; Scadden, D.T.; Lin, C.P. In Vivo Imaging of Specialized Bone Marrow Endothelial Microdomains for Tumour Engraftment. Nature 2005, 435, 969–973. [Google Scholar] [CrossRef]
- Eyles, J.; Puaux, A.-L.; Wang, X.; Toh, B.; Prakash, C.; Hong, M.; Tan, T.G.; Zheng, L.; Ong, L.C.; Jin, Y.; et al. Tumor Cells Disseminate Early, but Immunosurveillance Limits Metastatic Outgrowth, in a Mouse Model of Melanoma. J. Clin. Investig. 2010, 120, 2030–2039. [Google Scholar] [CrossRef]
- Feske, S. Calcium Signalling in Lymphocyte Activation and Disease. Nat. Rev. Immunol. 2007, 7, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Wolf, I.M.A.; Guse, A.H. Ca2+ Microdomains in T-Lymphocytes. Front. Oncol. 2017, 7, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, K.; Gabriel, S.S.; Liao, Y.; Gloury, R.; Preston, S.; Henstridge, D.C.; Pellegrini, M.; Zehn, D.; Berberich-Siebelt, F.; Febbraio, M.A.; et al. Transcription Factor IRF4 Promotes CD8+ T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 2017, 47, 1129–1141.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8 + T Cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oestreich, K.J.; Yoon, H.; Ahmed, R.; Boss, J.M. NFATc1 Regulates PD-1 Expression upon T Cell Activation. J. Immunol. 2008, 181, 4832–4839. [Google Scholar] [CrossRef]
- Ritprajak, P.; Azuma, M. Intrinsic and Extrinsic Control of Expression of the Immunoregulatory Molecule PD-L1 in Epithelial Cells and Squamous Cell Carcinoma. Oral Oncol. 2015, 51, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Saudemont, A.; Quesnel, B. In a Model of Tumor Dormancy, Long-Term Persistent Leukemic Cells Have Increased B7-H1 and B7.1 Expression and Resist CTL-Mediated Lysis. Blood 2004, 104, 2124–2133. [Google Scholar] [CrossRef]
- Schneider, H.; Smith, X.; Liu, H.; Bismuth, G.; Rudd, C.E. CTLA-4 Disrupts ZAP70 Microcluster Formation with Reduced T Cell/APC Dwell Times and Calcium Mobilization. Eur. J. Immunol. 2008, 38, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Cherian, S. Acute Myeloid Leukemia Immunophenotyping by Flow Cytometric Analysis. Clin. Lab. Med. 2017, 37, 753–769. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Wu, P.; Chen, C.; Xu, Z.P.; Gu, W. Increased PD-L1 Expression in Breast and Colon Cancer Stem Cells. Clin. Exp. Pharmacol. Physiol. 2017, 44, 602–604. [Google Scholar] [CrossRef]
- Nair, J.S.; DaFonseca, C.J.; Tjernberg, A.; Sun, W.; Darnell, J.E.; Chait, B.T.; Zhang, J.J. Requirement of Ca2+ and CaMKII for Stat1 Ser-727 Phosphorylation in Response to IFN-. Proc. Natl. Acad. Sci. USA 2002, 99, 5971–5976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthon, C.; Driss, V.; Liu, J.; Kuranda, K.; Leleu, X.; Jouy, N.; Hetuin, D.; Quesnel, B. In Acute Myeloid Leukemia, B7-H1 (PD-L1) Protection of Blasts from Cytotoxic T Cells Is Induced by TLR Ligands and Interferon-Gamma and Can Be Reversed Using MEK Inhibitors. Cancer Immunol. Immunother. 2010, 59, 1839–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, L.K.; Fonseca, B.P.F.; Vieira-de-Abreu, A.; Barboza, B.A.; Robbs, B.K.; Bozza, P.T.; Viola, J.P.B. IFN- Production by CD8+ T Cells Depends on NFAT1 Transcription Factor and Regulates Th Differentiation. J. Immunol. 2005, 175, 5931–5939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, M.; Goldberg, A.D. Immune Checkpoint Inhibitors in Acute Myeloid Leukemia: Novel Combinations and Therapeutic Targets. Curr. Oncol. Rep. 2019, 21, 37. [Google Scholar] [CrossRef]
- Sehgal, A.; Whiteside, T.L.; Boyiadzis, M. Programmed Death-1 Checkpoint Blockade in Acute Myeloid Leukemia. Expert Opin. Biol. Ther. 2015, 15, 1191–1203. [Google Scholar] [CrossRef] [Green Version]
- Payne, K.K.; Keim, R.C.; Graham, L.; Idowu, M.O.; Wan, W.; Wang, X.-Y.; Toor, A.A.; Bear, H.D.; Manjili, M.H. Tumor-Reactive Immune Cells Protect against Metastatic Tumor and Induce Immunoediting of Indolent but Not Quiescent Tumor Cells. J. Leukoc. Biol. 2016, 100, 625–635. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Gajewski, T.F.; Kline, J. PD-1/PD-L1 Interactions Inhibit Antitumor Immune Responses in a Murine Acute Myeloid Leukemia Model. Blood 2009, 114, 1545–1552. [Google Scholar] [CrossRef] [Green Version]
- Segovia, M.; Russo, S.; Jeldres, M.; Mahmoud, Y.D.; Perez, V.; Duhalde, M.; Charnet, P.; Rousset, M.; Victoria, S.; Veigas, F.; et al. Targeting TMEM176B Enhances Antitumor Immunity and Augments the Efficacy of Immune Checkpoint Blockers by Unleashing Inflammasome Activation. Cancer Cell 2019, 35, 767–781.e6. [Google Scholar] [CrossRef] [Green Version]
- Rashidi, A.; Uy, G.L. Targeting the Microenvironment in Acute Myeloid Leukemia. Curr. Hematol. Malig. Rep. 2015, 10, 126–131. [Google Scholar] [CrossRef]
- Behrmann, L.; Wellbrock, J.; Fiedler, W. Acute Myeloid Leukemia and the Bone Marrow Niche—Take a Closer Look. Front. Oncol. 2018, 8, 444. [Google Scholar] [CrossRef] [Green Version]
- Karantanou, C.; Godavarthy, P.S.; Krause, D.S. Targeting the Bone Marrow Microenvironment in Acute Leukemia. Leuk. Lymphoma 2018, 59, 2535–2545. [Google Scholar] [CrossRef] [PubMed]
- Isidori, A.; Salvestrini, V.; Ciciarello, M.; Loscocco, F.; Visani, G.; Parisi, S.; Lecciso, M.; Ocadlikova, D.; Rossi, L.; Gabucci, E.; et al. The Role of the Immunosuppressive Microenvironment in Acute Myeloid Leukemia Development and Treatment. Expert Rev. Hematol. 2014, 7, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.K.; Dimou, A.; Pierce, S.; Kantarjian, H.; Andreeff, M. The Effect of Calcium Channel Blockers on the Outcome of Acute Myeloid Leukemia. Leuk. Lymphoma 2014, 55, 2822–2829. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yu, P.; Lin, H.; Jin, Z.; Zhao, S.; Zhang, Y.; Xu, Q.; Jin, H.; Liu, Z.; Yang, W.; et al. The Discovery of Novel ACA Derivatives as Specific TRPM2 Inhibitors That Reduce Ischemic Injury Both In Vitro and In Vivo. J. Med. Chem. 2021, 64, 3976–3996. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-X.; Zhang, L.; Huang, Z.-W.; Zhang, X.-N.; Jiang, Y.-Y.; Liu, F.-J.; Long, L.; Xue, M.-J.; Lu, G.; Liu, Q.; et al. Aurora Kinase Inhibitor Restrains STAT5-Activated Leukemic Cell Proliferation by Inducing Mitochondrial Impairment. J. Cell Physiol. 2020, 235, 8358–8370. [Google Scholar] [CrossRef] [PubMed]
- Löwenberg, B.; Muus, P.; Ossenkoppele, G.; Rousselot, P.; Cahn, J.-Y.; Ifrah, N.; Martinelli, G.; Amadori, S.; Berman, E.; Sonneveld, P.; et al. Phase 1/2 Study to Assess the Safety, Efficacy, and Pharmacokinetics of Barasertib (AZD1152) in Patients with Advanced Acute Myeloid Leukemia. Blood 2011, 118, 6030–6036. [Google Scholar] [CrossRef]
- Yang, J.; Ikezoe, T.; Nishioka, C.; Tasaka, T.; Taniguchi, A.; Kuwayama, Y.; Komatsu, N.; Bandobashi, K.; Togitani, K.; Koeffler, H.P.; et al. AZD1152, a Novel and Selective Aurora B Kinase Inhibitor, Induces Growth Arrest, Apoptosis, and Sensitization for Tubulin Depolymerizing Agent or Topoisomerase II Inhibitor in Human Acute Leukemia Cells in Vitro and in Vivo. Blood 2007, 110, 2034–2040. [Google Scholar] [CrossRef] [Green Version]
- Birkenkamp, K.U.; Geugien, M.; Lemmink, H.H.; Kruijer, W.; Vellenga, E. Regulation of Constitutive STAT5 Phosphorylation in Acute Myeloid Leukemia Blasts. Leukemia 2001, 15, 1923–1931. [Google Scholar] [CrossRef] [Green Version]
- Warsch, W.; Kollmann, K.; Eckelhart, E.; Fajmann, S.; Cerny-Reiterer, S.; Hölbl, A.; Gleixner, K.V.; Dworzak, M.; Mayerhofer, M.; Hoermann, G.; et al. High STAT5 Levels Mediate Imatinib Resistance and Indicate Disease Progression in Chronic Myeloid Leukemia. Blood 2011, 117, 3409–3420. [Google Scholar] [CrossRef]
- Hung, L.-Y.; Tseng, J.T.; Lee, Y.-C.; Xia, W.; Wang, Y.-N.; Wu, M.-L.; Chuang, Y.-H.; Lai, C.-H.; Chang, W.-C. Nuclear Epidermal Growth Factor Receptor (EGFR) Interacts with Signal Transducer and Activator of Transcription 5 (STAT5) in Activating Aurora-A Gene Expression. Nucleic Acids Res. 2008, 36, 4337–4351. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Mi, T.; Bradley, H.L.; Metts, J.; Sabnis, H.; Zhu, W.; Arbiser, J.; Bunting, K.D. Pimozide and Imipramine Blue Exploit Mitochondrial Vulnerabilities and Reactive Oxygen Species to Cooperatively Target High Risk Acute Myeloid Leukemia. Antioxidants 2021, 10, 956. [Google Scholar] [CrossRef]
- Chen, Y.; Hui, H.; Yang, H.; Zhao, K.; Qin, Y.; Gu, C.; Wang, X.; Lu, N.; Guo, Q. Wogonoside Induces Cell Cycle Arrest and Differentiation by Affecting Expression and Subcellular Localization of PLSCR1 in AML Cells. Blood 2013, 121, 3682–3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Xiao, J.; Adachi, M.; Liu, Z.; Zhou, J. 4-Aminopyridine Induces Apoptosis of Human Acute Myeloid Leukemia Cells via Increasing [Ca2+]i through P2X7 Receptor Pathway. Cell Physiol. Biochem. 2011, 28, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Angka, L.; Lee, E.A.; Rota, S.G.; Hanlon, T.; Sukhai, M.; Minden, M.; McMillan, E.M.; Quadrilatero, J.; Spagnuolo, P.A. Glucopsychosine Increases Cytosolic Calcium to Induce Calpain-Mediated Apoptosis of Acute Myeloid Leukemia Cells. Cancer Lett. 2014, 348, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanamandra, N.; Buzzeo, R.W.; Gabriel, M.; Hazlehurst, L.A.; Mari, Y.; Beaupre, D.M.; Cuevas, J. Tipifarnib-Induced Apoptosis in Acute Myeloid Leukemia and Multiple Myeloma Cells Depends on Ca2+ Influx through Plasma Membrane Ca2+ Channels. J. Pharm. Exp. Ther. 2011, 337, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Diez-Bello, R.; Jardin, I.; Salido, G.M.; Rosado, J.A. Orai1 and Orai2 Mediate Store-Operated Calcium Entry That Regulates HL60 Cell Migration and FAK Phosphorylation. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1064–1070. [Google Scholar] [CrossRef]
- Manteniotis, S.; Wojcik, S.; Göthert, J.R.; Dürig, J.; Dührsen, U.; Gisselmann, G.; Hatt, H. Deorphanization and Characterization of the Ectopically Expressed Olfactory Receptor OR51B5 in Myelogenous Leukemia Cells. Cell Death Discov. 2016, 2, 16010. [Google Scholar] [CrossRef] [Green Version]
- Yeh, Y.-C.; Parekh, A.B. CRAC Channels and Ca(2+)-Dependent Gene Expression. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W.J., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2018; pp. 93–106. ISBN 978-1-315-15259-2. [Google Scholar]
- He, X.; Dou, A.; Feng, S.; Roman-Rivera, A.; Hawkins, C.; Lawley, L.; Zhang, J.; Wunderlich, M.; Mizukawa, B.; Halene, S.; et al. Cyclosporine Enhances the Sensitivity to Lenalidomide in MDS/AML In Vitro. Exp. Hematol. 2020, 86, 21–27.e2. [Google Scholar] [CrossRef]
- Borella, G.; Da Ros, A.; Borile, G.; Porcù, E.; Tregnago, C.; Benetton, M.; Marchetti, A.; Bisio, V.; Montini, B.; Michielotto, B.; et al. Targeting the Plasticity of Mesenchymal Stromal Cells to Reroute the Course of Acute Myeloid Leukemia. Blood 2021, 138, 557–570. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sub-Sections | Targets | Mechanisms | Biological Effect | Biological Sources | References |
|---|---|---|---|---|---|
| Ca2+ Signaling and Cell Cycle Regulation | CaM | -Increased cytosolic CaM -Transition G1 to S | Increased proliferation | HL60 promyelocytic AML cell line | [17] |
| CaMKII | -Decreased Cdk inhibitors p27 (kip1) and p16 (ink4a) -Increased cyclin A, B1, D1 | -Increased proliferation -Cell cycle progression | AML cell lines | [19] | |
| CaMKIV | -Increased p27, p16 -Decreased cyclin A, B1, D1 | -Decreased proliferation | AML cell lines | [19] | |
| CaMKIV | -Phosphorylation Rb | Increased proliferation | Primary AML cells | [18] | |
| Calcineurin | -Transition G1 to S -Cyclin A, D1? -Cyclin E, E2? | Proliferation? Decreased calcineurin activity (−85%) | Sera from AML patient | [24] | |
| Ca2+ Channels and Proliferation in AML | TRPM2 | ATF4, CREB | Increased proliferation | Primary AML cells | [25] |
| Cav.1.2, L-type calcium channel | -Ca2+ entry | Increased proliferation | Primary AML cells | [26] | |
| ITPR2 | SERCA pumps | Cell cycle progression | Primary AML cells | [27] | |
| Notch and Ca2+ Signaling in AML Proliferation | Notch/Delta ligand | -Calcium sensor receptor -SOCE | Increased proliferation | TMD7 AML cell line | [29] |
| Notch/Delta1 ligand | -Calcium sensor receptor -SOCE | Increased proliferation | HL60 promyelocytic cell line | [30] | |
| Notch/Jagged1 ligand | -Calcium sensor receptor -SOCE | Decreased proliferation | Primary AML cells, AML cell lines | [28,33] | |
| Calcium Involvement in AML Differentiation | SERCA pumps | -Ca2+ | Increased differentiation | Primary AML cells | [34] |
| S100A9/TLR4 | -p38, ERK1/2, JNK | Increased differentiation | Primary AML cells | [35] | |
| Ca2+ concentration | Increased differentiation | Primary AML cells | [36] | ||
| IP3R1 | -Decreased c-myc expression | Increased AML cells | AML cell lines | [37] |
| Molecules | Targets | Clinical Use | Clinical Impact | Mechanism | Ref |
|---|---|---|---|---|---|
| Amlodipine/ Diltiazem | L-type calcium channels | Yes (heart disease, hypertension) | Decreased AML patient survival | L-type calcium channels inhibitors | [154] |
| A23 | TRPM2 channel | No | - | TRPM2 inhibitor makes AML cells more sensitive to chemotherapies in vitro (increases ROS production) | [25] |
| AKI604 | Aurora kinase A (AURKA) | No | - | AURKA inhibitor impairs mitochondrial activity, increases ROS production and cytoplasmic calcium concentration, and decreases tumor growth in xenograft models | [156] |
| Pimozide | Voltage-gated calcium channels | No | - | In combination with ABT-263 and AZD 8055, pimozide impairs mitochondrial functions and induce resistant AML cell lines apoptosis | [162] |
| Wogonoside | IP3R1 | No | - | Inhibits proliferation through PLSCR1 activation, IP3R1 upregulation, and the resulting increase in cytoplasmic calcium concentration leading to AML cell differentiation | [37,163] |
| 4-AP | Voltage-gated potassium channel | No | - | Inhibition of voltage-gated potassium channels by 4-AP leads to plasma membrane. depolarization, calcium entry into AML cells via ionotropic P2X7 receptor, and induction of apoptosis | [164] |
| Glucopsychosine | Unknown | No | - | Induces apoptosis in AML cells, but not in normal hematopoietic cells, via a calcium entry through unknown calcium channels | [165] |
| Tipifarnib | Farnesyltransferase | No | - | Tipifarnib inhibits farnesyltransferase and increases intracellular calcium concentration through SOC channels activation, leading to AML cell apoptosis | [166] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewuillon, C.; Laguillaumie, M.-O.; Quesnel, B.; Idziorek, T.; Touil, Y.; Lemonnier, L. Put in a “Ca2+ll” to Acute Myeloid Leukemia. Cells 2022, 11, 543. https://doi.org/10.3390/cells11030543
Lewuillon C, Laguillaumie M-O, Quesnel B, Idziorek T, Touil Y, Lemonnier L. Put in a “Ca2+ll” to Acute Myeloid Leukemia. Cells. 2022; 11(3):543. https://doi.org/10.3390/cells11030543
Chicago/Turabian StyleLewuillon, Clara, Marie-Océane Laguillaumie, Bruno Quesnel, Thierry Idziorek, Yasmine Touil, and Loïc Lemonnier. 2022. "Put in a “Ca2+ll” to Acute Myeloid Leukemia" Cells 11, no. 3: 543. https://doi.org/10.3390/cells11030543
APA StyleLewuillon, C., Laguillaumie, M. -O., Quesnel, B., Idziorek, T., Touil, Y., & Lemonnier, L. (2022). Put in a “Ca2+ll” to Acute Myeloid Leukemia. Cells, 11(3), 543. https://doi.org/10.3390/cells11030543