
Annexin-A1-Derived Peptide Ac2-26 Suppresses Allergic Airway Inflammation and Remodelling in Mice


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Animals
2.2. Sensitization, Allergen Challenge, and Treatment
2.3. Respiratory Mechanics
2.4. Bronchoalveolar Lavage Fluid
2.5. Histological Analyses
2.6. ELISA Analysis
2.7. Statistical Analysis
3. Results
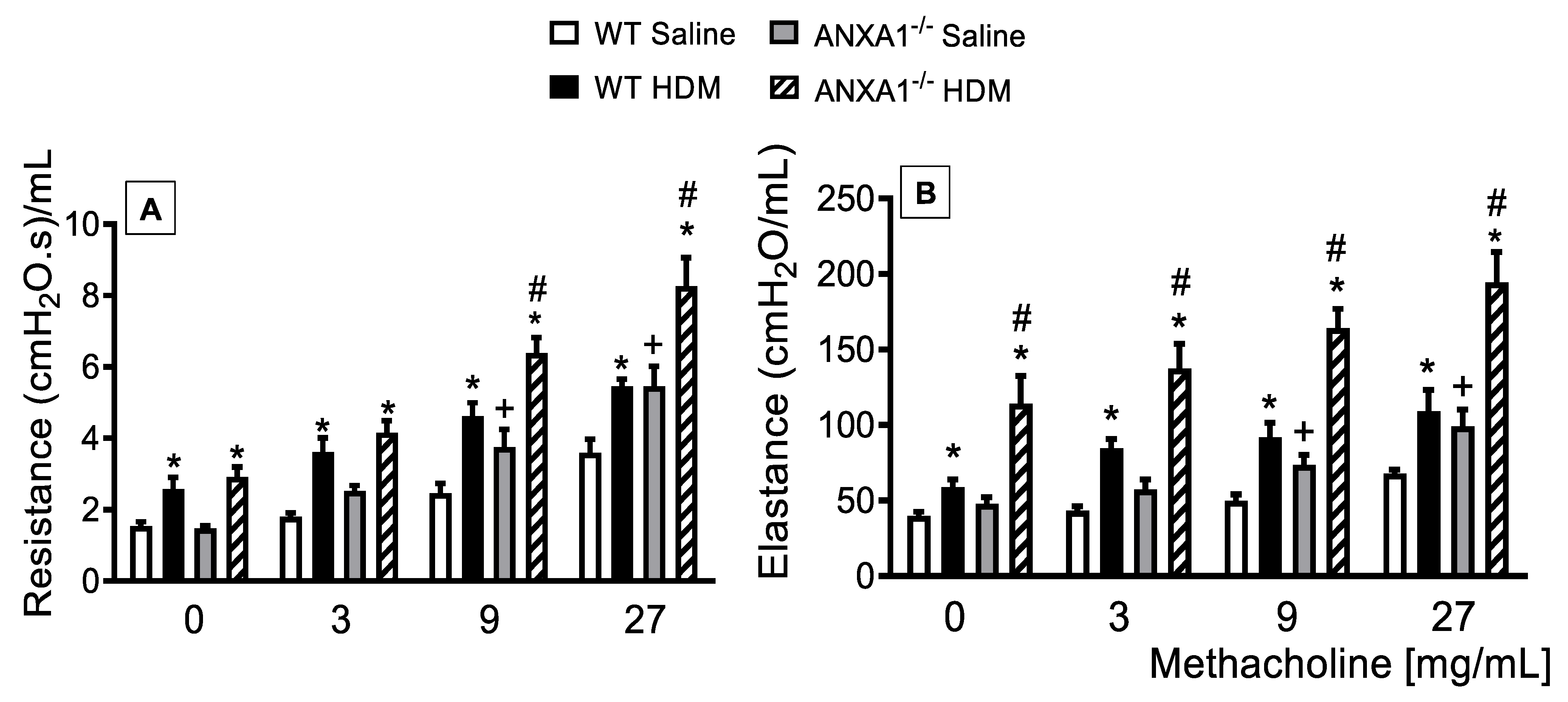
3.1. Lack of AnxA1 Expression Exacerbates Lung Function in HDM-Exposed Mice
3.2. Lack of AnxA1 Expression Exacerbates Airways Inflammation, Histological Changes, and Mediator Production in HDM-Exposed Mice
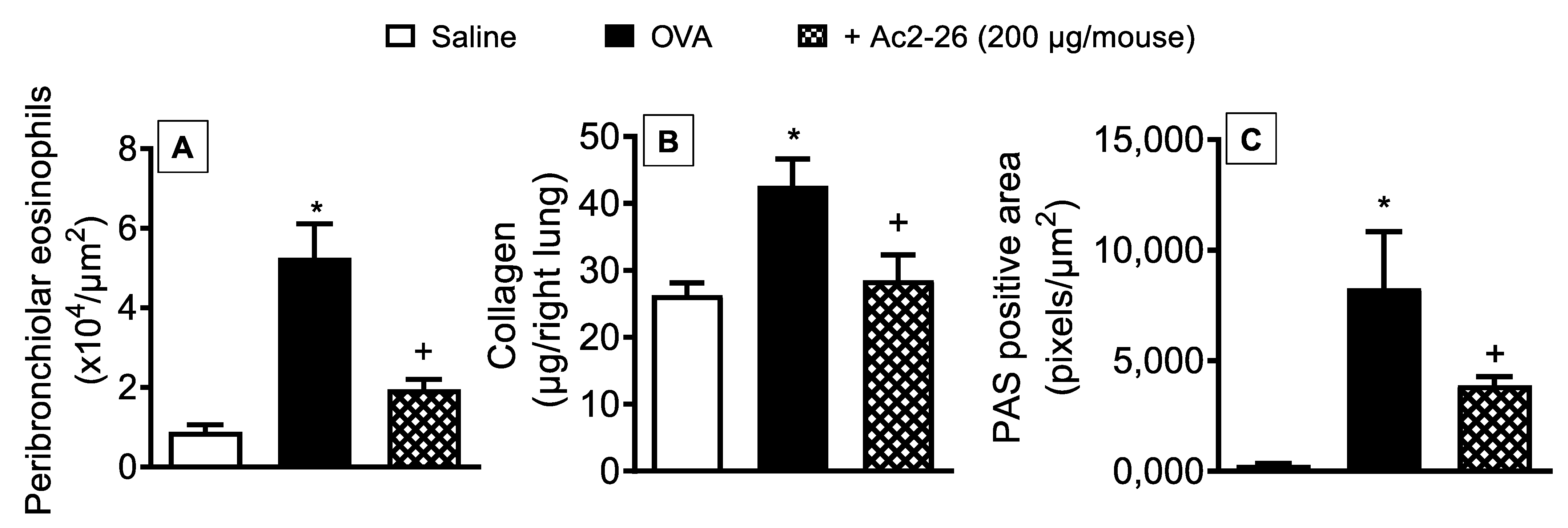
3.3. Effect of Peptide Ac2-26 on Lung Remodelling and Mucus Hypersecretion in OVA-Challenged Mice
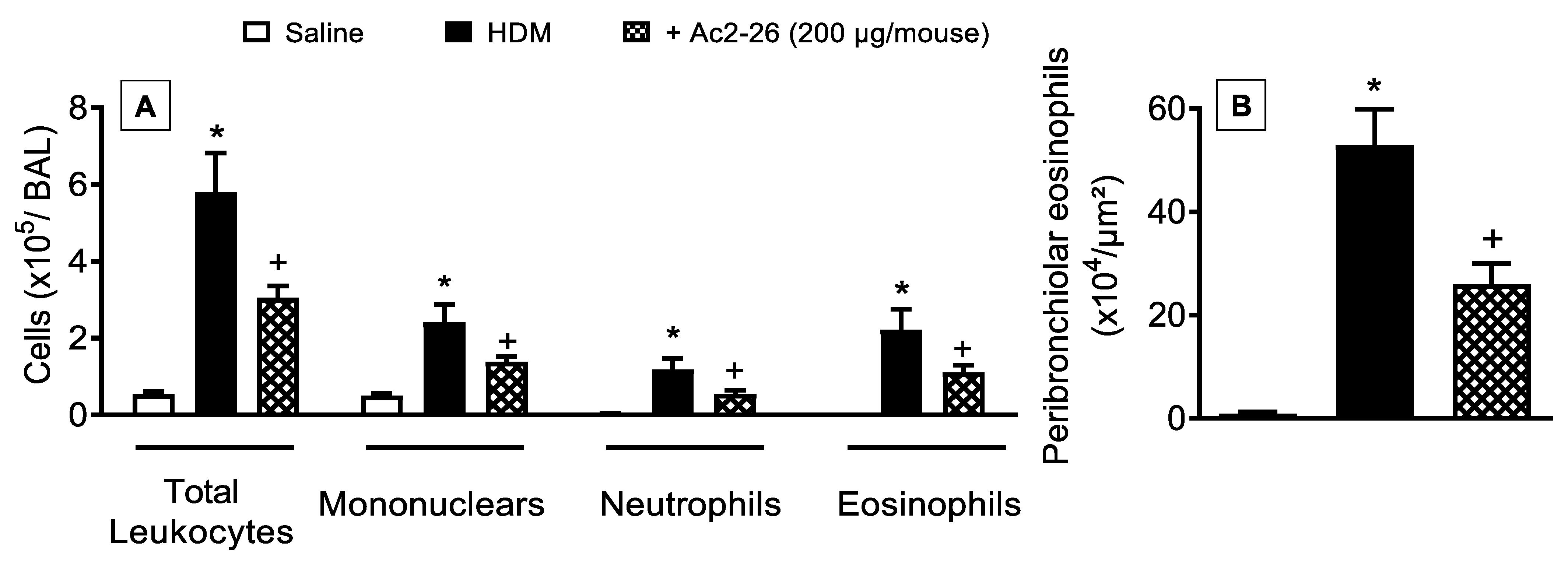
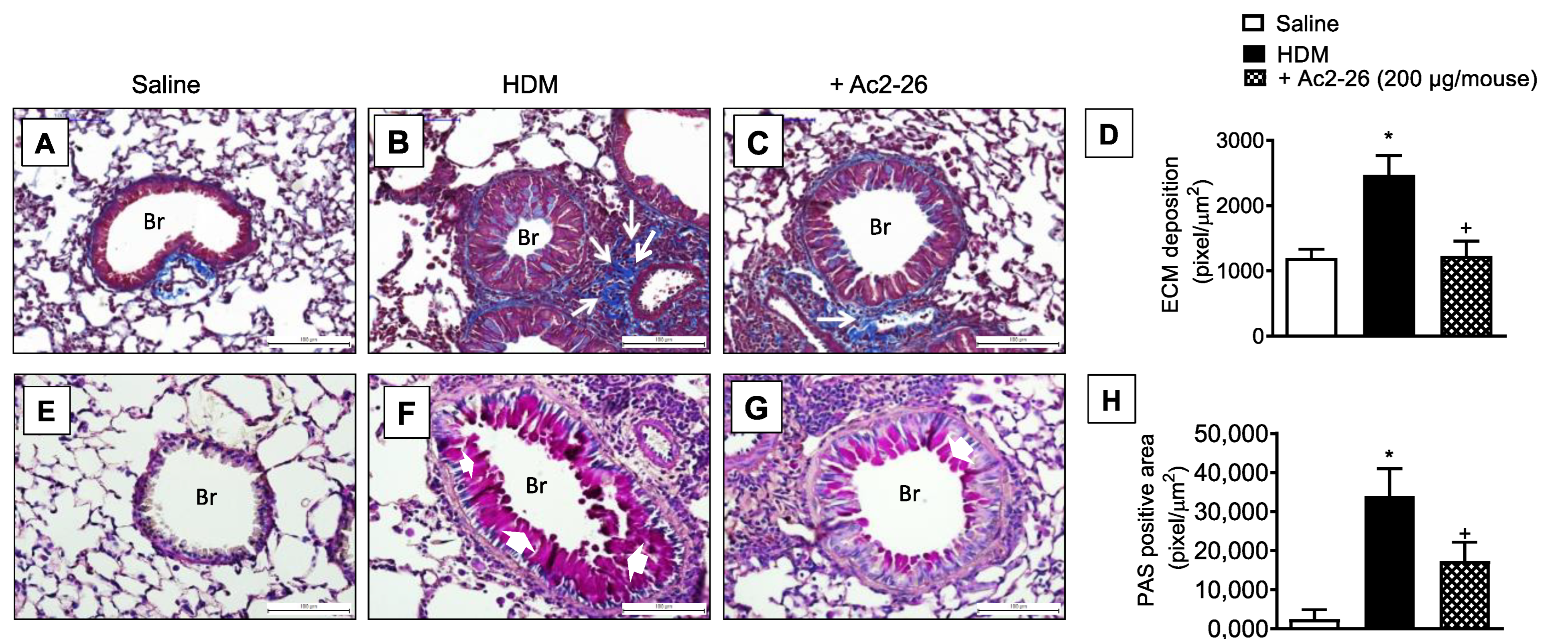
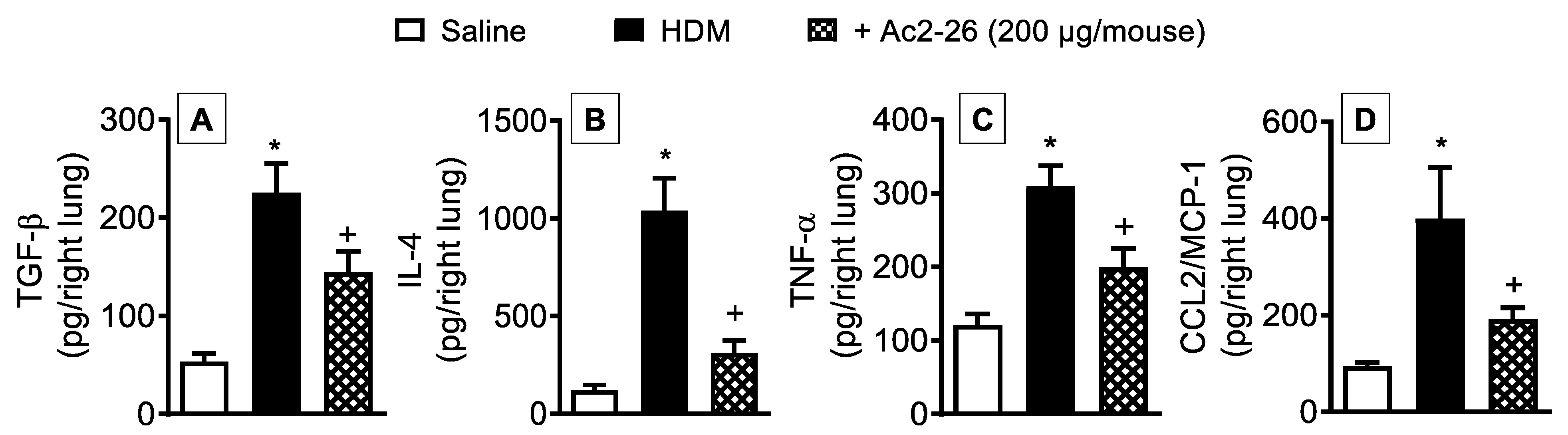
3.4. Effect of Peptide Ac2-26 on Lung Changes in HDM-Exposed Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| α-SMA | α-smooth muscle actin |
| AHR- | airway hyperreactivity |
| AnxA1 | annexin-A1 |
| CCL2/MCP-1 | CC ligand-2/ monocyte chemoattractant protein-1 |
| CCL11/eotaxin1 | CC ligand-11/eotaxin-1 |
| HDM | house dust mite |
| ECM | extracellular matrix |
| EDTA | ethylenediaminetetraacetic acid |
| GR | glucocorticoid receptor |
| GRE | glucocorticoid response element |
| GT | Gömöri trichrome |
| MMP-1 | matrix metalloproteinase-1 |
| PAS | periodic acid–Schiff |
| PBS | phosphate-buffered saline |
| TGF-β | transforming growth factor-β1 |
| TNF-α | tumour necrosis factor-α |
| WT | wildtype |
References
- Global Asthma Network. The Global Asthma Report 2018; Global Asthma Network: Auckland, New Zealand, 2018. [Google Scholar]
- GINA. Global Strategy for Asthma Management and Prevention. 2021. Available online: https://ginasthma.org/gina-reports/ (accessed on 29 December 2021).
- Lemanske, R.F., Jr.; Busse, W.W. Asthma. J. Allergy Clin. Immunol. 2003, 111, S502–S519. [Google Scholar] [CrossRef] [PubMed]
- Keglowich, L.; Borger, P. The three A’s in asthma—Airway smooth muscle, airway remodeling & angiogenesis. Open Respir. Med. J. 2015, 9, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Perretti, M.; D’Acquisto, F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 2009, 9, 62–70. [Google Scholar] [CrossRef] [PubMed]
- D’Acquisto, F.; Merghani, A.; Lecona, E.; Rosignoli, G.; Raza, K.; Buckley, C.D.; Flower, R.J.; Perretti, M. Annexin-1 modulates T-cell activation and differentiation. Blood 2007, 109, 1095–1102. [Google Scholar] [CrossRef] [Green Version]
- Strehl, C.; Ehlers, L.; Gaber, T.; Buttgereit, F. Glucocorticoids—All-rounders tackling the versatile players of the immune system. Front. Immunol. 2019, 10, 1744. [Google Scholar] [CrossRef] [Green Version]
- Perretti, M.; Flower, R.J. Annexin 1 and the biology of the neutrophil. J. Leukoc. Biol. 2004, 76, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Oliani, S.M.; Christian, H.C.; Manston, J.; Flower, R.J.; Perretti, M. An immunocytochemical and in situ hybridization analysis of Annexin 1 expression in rat mast cells: Modulation by inflammation and dexamethasone. Lab. Investig. 2000, 80, 1429–1438. [Google Scholar] [CrossRef] [Green Version]
- Hayhoe, R.P.G.; Kamal, A.M.; Solito, E.; Flower, R.J.; Cooper, D.; Perretti, M. Annexin 1 and its bioactive peptide inhibit neutrophil-endothelium interactions under flow: Indication of distinct receptor involvement. Blood 2006, 107, 2123–2130. [Google Scholar] [CrossRef] [Green Version]
- Cuzzocrea, S.; Tailor, A.; Zingarelli, B.; Salzman, A.L.; Flower, R.J.; Szabó, C.; Perretti, M. Lipocortin 1 protects against splanchnic artery occlusion and reperfusion injury by affecting neutrophil migration. J. Immunol. 1997, 159, 5089–5097. [Google Scholar]
- Luo, Z.; Wang, H.; Fang, S.; Li, L.; Li, X.; Shi, J.; Zhu, M.; Tan, Z.; Lu, Z. Annexin-1 mimetic peptide Ac2-26 suppresses inflammatory mediators in LPS-induced astrocytes and ameliorates pain hypersensitivity in a rat model of inflammatory pain. Cell. Mol. Neurobiol. 2020, 40, 569–585. [Google Scholar] [CrossRef]
- Liao, W.-I.; Wu, S.-Y.; Wu, G.-C.; Pao, H.-P.; Tang, S.-E.; Huang, K.-L.; Chu, S.-J. Ac2-26, an Annexin A1 peptide, attenuates ischemia-reperfusion-induced acute lung injury. Int. J. Mol. Sci. 2017, 18, 1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, M.G.; Tavares, L.P.; Souza, G.V.S.; Queiroz-Junior, C.M.; Ascenção, F.R.; Lopes, M.E.; Garcia, C.; Menezes, G.B.; Perretti, M.; Russo, R.C.; et al. The Annexin A1/FPR2 pathway controls the inflammatory response and bacterial dissemination in experimental pneumococcal pneumonia. FASEB J. 2020, 34, 2749–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandeira-Melo, C.; Bonavita, A.G.C.; Diaz, B.L.; e Silva, P.M.R.; Carvalho, V.F.; Jose, P.J.; Flower, R.J.; Perretti, M.; Martins, M.A. A novel effect for Annexin 1-derived peptide Ac2-26: Reduction of allergic inflammation in the rat. J. Pharmacol. Exp. Ther. 2005, 313, 1416–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, F.S.P.; Wong, K.Y.; Guan, S.P.; Mustafa, F.B.; Kajiji, T.S.; Bist, P.; Biswas, S.K.; Wong, W.S.F.; Lim, L.H.K. Annexin-1-deficient mice exhibit spontaneous airway hyperresponsiveness and exacerbated allergen-specific antibody responses in a mouse model of asthma. Clin. Exp. Allergy 2011, 41, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.K.; Herbert, C.; Foster, P.S. The “classical” ovalbumin challenge model of asthma in mice. Curr. Drug Targets 2008, 9, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Arshad, S.H. Does exposure to indoor allergens contribute to the development of asthma and allergy? Curr. Allergy Asthma Rep. 2010, 10, 49–55. [Google Scholar] [CrossRef]
- Hannon, R.; Croxtall, J.D.; Getting, S.J.; Roviezzo, F.; Yona, S.; Paul-Clark, M.J.; Gavins, F.N.E.; Perretti, M.; Morris, J.F.; Buckingham, J.C.; et al. Aberrant inflammation and resistance to glucocorticoids in Annexin 1−/− mouse. FASEB J. 2003, 17, 253–255. [Google Scholar] [CrossRef] [Green Version]
- Gregory, L.G.; Jones, C.P.; Walker, S.A.; Sawant, D.; Gowers, K.H.C.; Campbell, G.A.; McKenzie, A.N.J.; Lloyd, C.M. IL-25 drives remodelling in allergic airways disease induced by house dust mite. Thorax 2013, 68, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Olsen, P.; Ferreira, T.P.T.; Serra, M.F.; Farias-Filho, F.A.; Fonseca, B.D.P.; Viola, J.P.B.; Cordeiro, R.S.B.; Silva, P.M.R.; Costa, J.C.S.; Martins, M.A. Lidocaine-derivative JMF2-1 prevents ovalbumin-induced airway inflammation by regulating the function and survival of T cells. Clin. Exp. Allergy 2011, 41, 250–259. [Google Scholar] [CrossRef]
- Trentin, P.G.; Ferreira, T.P.T.; Arantes, A.C.S.; Ciambarella, B.T.; Cordeiro, R.S.B.; Flower, R.J.; Perretti, M.; Martins, M.A.; Silva, P.M.R. Annexin A1 mimetic peptide controls the inflammatory and fibrotic effects of silica particles in mice. Br. J. Pharmacol. 2015, 172, 3058–3071. [Google Scholar] [CrossRef] [Green Version]
- Serra, M.F.; Anjos-Valotta, E.A.; Olsen, P.; Couto, G.C.; Jurgilas, P.B.; Cotias, A.C.; Pão, C.R.; Ferreira, T.P.T.; Arantes, A.C.S.; Pires, A.L.A.; et al. Nebulized lidocaine prevents airway inflammation, peribronchial fibrosis, and mucus production in a murine model of asthma. Anesthesiology 2012, 117, 580–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids—New mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vago, J.P.; Nogueira, C.R.C.; Tavares, L.P.; Soriani, F.M.; Lopes, F.; Russo, R.C.; Pinho, V.; Teixeira, M.M.; Sousa, L.P. Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J. Leukoc. Biol. 2012, 92, 249–258. [Google Scholar] [CrossRef]
- Lee, S.-H.; Lee, P.-H.; Kim, B.-G.; Seo, H.-J.; Baek, A.-R.; Park, J.-S.; Lee, J.-H.; Park, S.-W.; Kim, D.-J.; Park, C.-S.; et al. Annexin A1 in plasma from patients with bronchial asthma: Its association with lung function. BMC Pulm. Med. 2018, 18, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.F.; Tetley, T.D.; Guz, A.; Flower, R.J. Detection of lipocortin 1 in human lung lavage fluid: Lipocortin degradation as a possible proteolytic mechanism in the control of inflammatory mediators and inflammation. Environ. Health Perspect. 1990, 85, 135–144. [Google Scholar] [CrossRef]
- Vishwanatha, J.; Davis, R.G.; Rubinstein, I.; Floreani, A. Annexin I degradation in bronchoalveolar lavage fluids from healthy smokers: A possible mechanism of inflammation. Clin. Cancer Res. 1998, 4, 2559–2564. [Google Scholar]
- Smith, S.F.; Tetley, T.D.; Datta, A.K.; Smith, T.; Guz, A.; Flower, R.J. Lipocortin-1 distribution in bronchoalveolar lavage from healthy human lung: Effect of prednisolone. J. Appl. Physiol. 1995, 79, 121–128. [Google Scholar] [CrossRef]
- D’Acquisto, F.; Paschalidis, N.; Sampaio, A.L.F.; Merghani, A.; Flower, R.J.; Perretti, M. Impaired T cell activation and increased Th2 lineage commitment in Annexin-1-deficient T cells. Eur. J. Immunol. 2007, 37, 3131–3142. [Google Scholar] [CrossRef]
- Damazo, A.S.; Sampaio, A.L.; Nakata, C.M.; Flower, R.J.; Perretti, M.; Oliani, S.M. Endogenous annexin A1 counter-regulates bleomycin-induced lung fibrosis. BMC Immunol. 2011, 12, 59. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Morand, E.F.; Song, W.; Cheng, Q.; Stewart, A.; Yang, Y.H. Regulation of lung fibroblast activation by annexin A1. J. Cell. Physiol. 2013, 228, 476–484. [Google Scholar] [CrossRef]
- Perretti, M.; Ahluwalia, A.; Harris, J.G.; Goulding, N.J.; Flower, R.J. Lipocortin-1 fragments inhibit neutrophil accumulation and neutrophil-dependent edema in the mouse: A qualitative comparison with an anti-CD11b monoclonal antibody. J. Immunol. 1993, 151, 4306–4314. [Google Scholar] [PubMed]
- Lee, S.H.; Kim, D.W.; Kim, H.R.; Woo, S.J.; Kim, S.M.; Jo, H.S.; Jeon, S.G.; Cho, S.-W.; Park, J.H.; Won, M.H.; et al. Anti-inflammatory effects of Tat-Annexin protein on ovalbumin-induced airway inflammation in a mouse model of asthma. Biochem. Biophys. Res. Commun. 2012, 417, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-M.; Li, W.-H.; Xu, Y.-C.; Wei, Q.; Zhao, H.; Jiang, X.-F. Annexin 1-derived peptide Ac2-26 inhibits eosinophil recruitment in vivo via decreasing prostaglandin (D2). Int. Arch. Allergy Immunol. 2011, 154, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Walther, A.; Riehemann, K.; Gerke, V. A novel ligand of the formyl peptide receptor: Annexin I regulates neutrophil extravasation by interacting with the FPR. Mol. Cell 2000, 5, 831–840. [Google Scholar] [CrossRef]
- Perretti, M.; Chiang, N.; La, M.; Fierro, I.M.; Marullo, S.; Getting, S.J.; Solito, E.; Serhan, C.N. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat. Med. 2002, 8, 1296–1302. [Google Scholar] [CrossRef]
- Cooray, S.N.; Gobbetti, T.; Montero-Melendez, T.; McArthur, S.; Thompson, D.; Clark, A.J.L.; Flower, R.J.; Perretti, M. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. USA 2013, 110, 18232–18237. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.G.; Sawatzky, D.A.; Walker, A.; Ward, C.; Sheldrake, T.A.; Riley, N.A.; Caldicott, A.; Martinez-Losa, M.; Walker, T.R.; Duffin, R.; et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 2006, 12, 1056–1064. [Google Scholar] [CrossRef]
- McArthur, S.; Juban, G.; Gobbetti, T.; Desgeorges, T.; Theret, M.; Gondin, J.; Toller-Kawahisa, J.E.; Reutelingsperger, C.P.; Chazaud, B.; Perretti, M.; et al. Annexin A1 drives macrophage skewing to accelerate muscle regeneration through AMPK activation. J. Clin. Investig. 2020, 130, 1156–1167. [Google Scholar] [CrossRef] [Green Version]
- Neymeyer, H.; Labes, R.; Reverte, V.; Saez, F.; Stroh, T.; Dathe, C.; Hohberger, S.; Zeisberg, M.; Müller, G.A.; Salazar, J.; et al. Activation of annexin A1 signalling in renal fibroblasts exerts antifibrotic effects. Acta Physiol. 2015, 215, 144–158. [Google Scholar] [CrossRef]
- Craig, V.J.; Zhang, L.; Hagood, J.S.; Owen, C.A. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 585–600. [Google Scholar] [CrossRef] [Green Version]
- Tagoe, C.E.; Marjanovic, N.; Park, J.Y.; Chan, E.S.; Abeles, A.; Attur, M.; Abramson, S.; Pillinger, M. Annexin-1 mediates TNF-α-stimulated matrix metalloproteinase secretion from rheumatoid arthritis synovial fibroblasts. J. Immunol. 2008, 181, 2813–2820. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, T.P.T.; Guimarães, F.V.; Sá, Y.A.P.J.; da Silva Ribeiro, N.B.; de Arantes, A.C.S.; de Frias Carvalho, V.; Sousa, L.P.; Perretti, M.; Martins, M.A.; e Silva, P.M.R. Annexin-A1-Derived Peptide Ac2-26 Suppresses Allergic Airway Inflammation and Remodelling in Mice. Cells 2022, 11, 759. https://doi.org/10.3390/cells11050759
Ferreira TPT, Guimarães FV, Sá YAPJ, da Silva Ribeiro NB, de Arantes ACS, de Frias Carvalho V, Sousa LP, Perretti M, Martins MA, e Silva PMR. Annexin-A1-Derived Peptide Ac2-26 Suppresses Allergic Airway Inflammation and Remodelling in Mice. Cells. 2022; 11(5):759. https://doi.org/10.3390/cells11050759
Chicago/Turabian StyleFerreira, Tatiana Paula Teixeira, Fernanda Verdini Guimarães, Yago Amigo Pinho Jannini Sá, Natalia Barreto da Silva Ribeiro, Ana Carolina Santos de Arantes, Vinicius de Frias Carvalho, Lirlândia Pires Sousa, Mauro Perretti, Marco Aurélio Martins, and Patrícia Machado Rodrigues e Silva. 2022. "Annexin-A1-Derived Peptide Ac2-26 Suppresses Allergic Airway Inflammation and Remodelling in Mice" Cells 11, no. 5: 759. https://doi.org/10.3390/cells11050759
APA StyleFerreira, T. P. T., Guimarães, F. V., Sá, Y. A. P. J., da Silva Ribeiro, N. B., de Arantes, A. C. S., de Frias Carvalho, V., Sousa, L. P., Perretti, M., Martins, M. A., & e Silva, P. M. R. (2022). Annexin-A1-Derived Peptide Ac2-26 Suppresses Allergic Airway Inflammation and Remodelling in Mice. Cells, 11(5), 759. https://doi.org/10.3390/cells11050759