
Maintaining Golgi Homeostasis: A Balancing Act of Two Proteolytic Pathways


{kind=link}
{kind=link}
Abstract
:1. Introduction
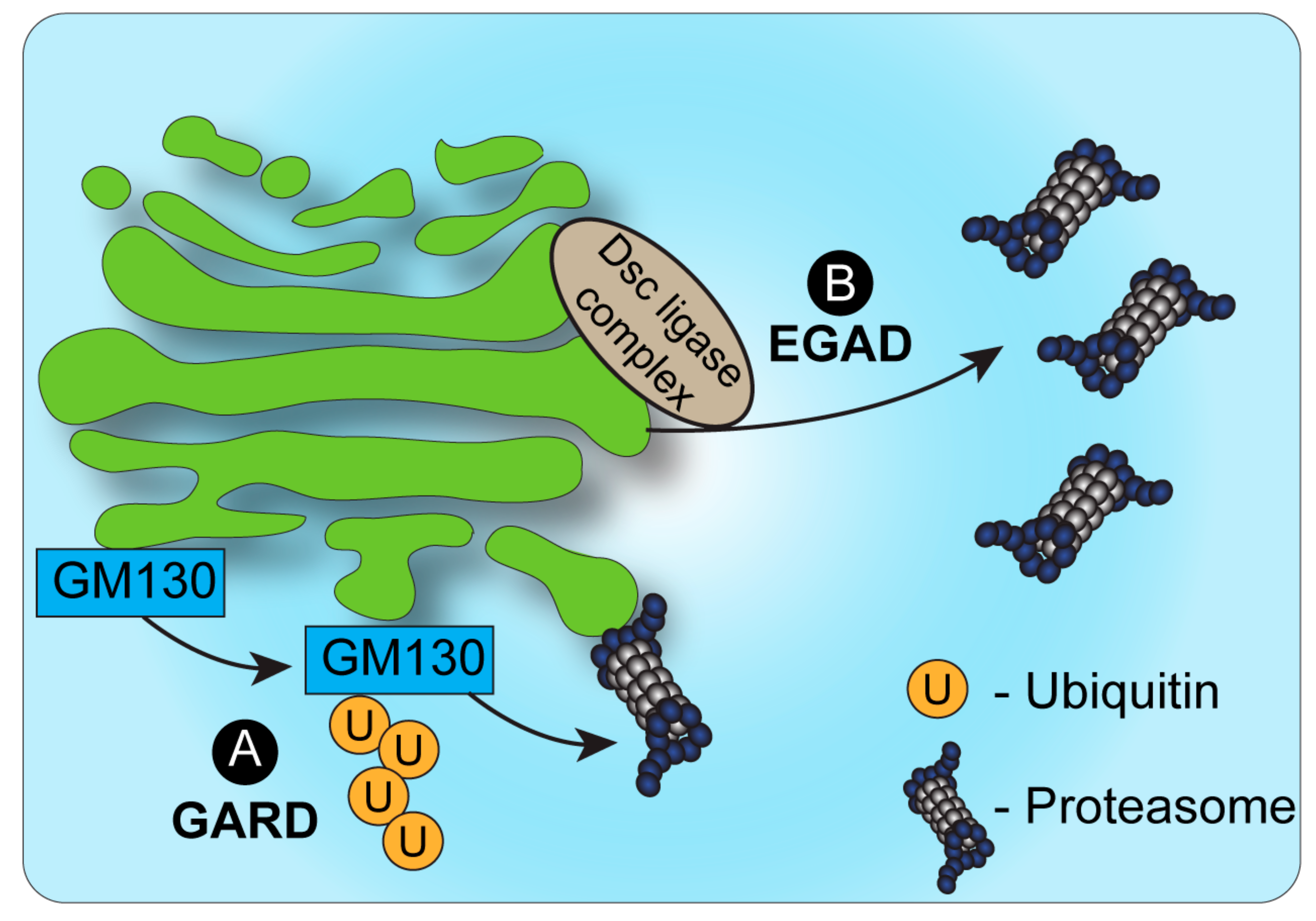
2. Proteasomal Degradation and the Golgi
Quality Control and Stress Response at the Golgi
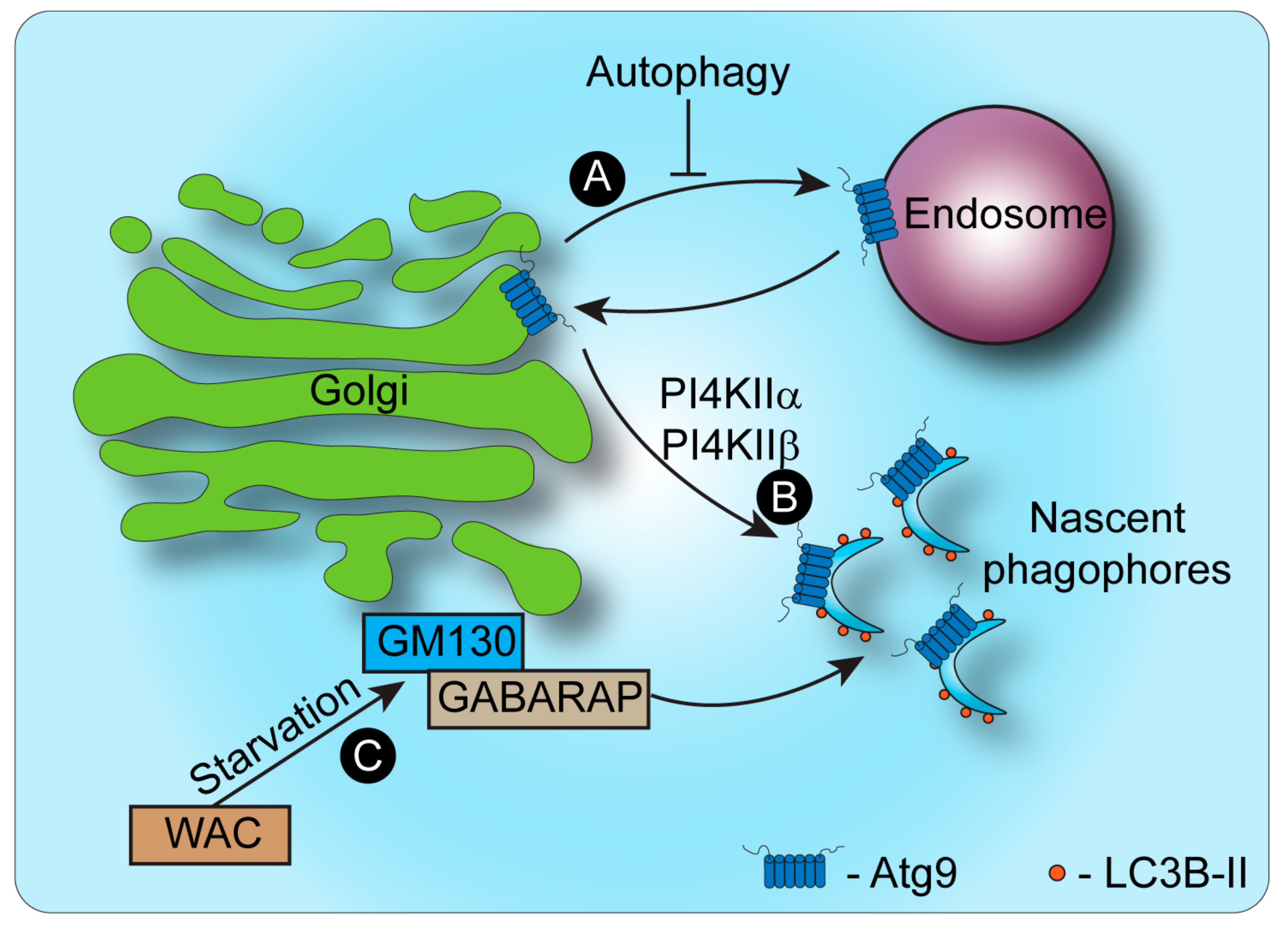
3. Autophagy and the Golgi Apparatus
Maintenance of Golgi Morphology and Trafficking Are Linked to Autophagy
4. Discussion and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Viotti, C. ER to Golgi-Dependent Protein Secretion: The Conventional Pathway. Methods Mol. Biol. 2016, 1459, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Stanley, P. Golgi glycosylation. Cold Spring Harb. Perspect. Biol. 2011, 3, a005199. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Struwe, W.B.; Harvey, D.J.; Ferguson, M.A.J.; Robinson, C.V. N-glycan microheterogeneity regulates interactions of plasma proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 8763–8768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joazeiro, C.A.P. Mechanisms and functions of ribosome-associated protein quality control. Nat. Rev. Mol. Cell Biol. 2019, 20, 368–383. [Google Scholar] [CrossRef] [PubMed]
- Guna, A.; Hegde, R.S. Transmembrane Domain Recognition during Membrane Protein Biogenesis and Quality Control. Curr. Biol. 2018, 28, R498–R511. [Google Scholar] [CrossRef] [Green Version]
- Juszkiewicz, S.; Hegde, R.S. Quality Control of Orphaned Proteins. Mol. Cell 2018, 71, 443–457. [Google Scholar] [CrossRef] [Green Version]
- Benyair, R.; Ron, E.; Lederkremer, G.Z. Protein quality control, retention, and degradation at the endoplasmic reticulum. Int. Rev. Cell Mol. Biol. 2011, 292, 197–280. [Google Scholar] [CrossRef]
- Adams, B.M.; Oster, M.E.; Hebert, D.N. Protein Quality Control in the Endoplasmic Reticulum. Protein J. 2019, 38, 317–329. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Chino, H.; Mizushima, N. ER-Phagy: Quality Control and Turnover of Endoplasmic Reticulum. Trends Cell Biol. 2020, 30, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, J.L. Cleaning up: ER-associated degradation to the rescue. Cell 2012, 151, 1163–1167. [Google Scholar] [CrossRef] [Green Version]
- Spear, E.D.; Ng, D.T. Stress tolerance of misfolded carboxypeptidase Y requires maintenance of protein trafficking and degradative pathways. Mol. Biol. Cell 2003, 14, 2756–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, C.M.; Caldwell, S.; Cooper, A.A. An HRD/DER-independent ER quality control mechanism involves Rsp5p-dependent ubiquitination and ER-Golgi transport. J. Cell Biol. 2002, 158, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Vashist, S.; Kim, W.; Belden, W.J.; Spear, E.D.; Barlowe, C.; Ng, D.T. Distinct retrieval and retention mechanisms are required for the quality control of endoplasmic reticulum protein folding. J. Cell Biol. 2001, 155, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, S.R.; Hill, K.J.; Cooper, A.A. Degradation of endoplasmic reticulum (ER) quality control substrates requires transport between the ER and Golgi. J. Biol. Chem. 2001, 276, 23296–23303. [Google Scholar] [CrossRef] [Green Version]
- Vashist, S.; Ng, D.T. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol. 2004, 165, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Kincaid, M.M.; Cooper, A.A. Misfolded proteins traffic from the endoplasmic reticulum (ER) due to ER export signals. Mol. Biol. Cell 2007, 18, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Hsu, V.W.; Yuan, L.C.; Nuchtern, J.G.; Lippincott-Schwartz, J.; Hammerling, G.J.; Klausner, R.D. A recycling pathway between the endoplasmic reticulum and the Golgi apparatus for retention of unassembled MHC class I molecules. Nature 1991, 352, 441–444. [Google Scholar] [CrossRef]
- Elkabetz, Y.; Kerem, A.; Tencer, L.; Winitz, D.; Kopito, R.R.; Bar-Nun, S. Immunoglobulin light chains dictate vesicular transport-dependent and -independent routes for IgM degradation by the ubiquitin-proteasome pathway. J. Biol. Chem. 2003, 278, 18922–18929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amitay, R.; Bar-Nun, S.; Haimovich, J.; Rabinovich, E.; Shachar, I. Post-translational regulation of IgM expression in B lymphocytes. Selective nonlysosomal degradation of assembled secretory IgM is temperature-dependent and occurs prior to the trans-Golgi. J. Biol. Chem. 1991, 266, 12568–12573. [Google Scholar] [CrossRef]
- Winitz, D.; Shachar, I.; Elkabetz, Y.; Amitay, R.; Samuelov, M.; Bar-Nun, S. Degradation of distinct assembly forms of immunoglobulin M occurs in multiple sites in permeabilized B cells. J. Biol. Chem. 1996, 271, 27645–27651. [Google Scholar] [CrossRef] [Green Version]
- Letourneur, F.; Cosson, P. Targeting to the endoplasmic reticulum in yeast cells by determinants present in transmembrane domains. J. Biol. Chem. 1998, 273, 33273–33278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puccia, R.; Grondin, B.; Herscovics, A. Disruption of the processing alpha-mannosidase gene does not prevent outer chain synthesis in Saccharomyces cerevisiae. Biochem. J. 1993, 290, 21–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvan, P.; Zhao, X.; Ramos-Castaneda, J.; Chang, A. Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic 2002, 3, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Pelham, H.R. A transmembrane ubiquitin ligase required to sort membrane proteins into multivesicular bodies. Nat. Cell Biol. 2002, 4, 117–123. [Google Scholar] [CrossRef]
- Hwang, J.; Ribbens, D.; Raychaudhuri, S.; Cairns, L.; Gu, H.; Frost, A.; Urban, S.; Espenshade, P.J. A Golgi rhomboid protease Rbd2 recruits Cdc48 to cleave yeast SREBP. EMBO J. 2016, 35, 2332–2349. [Google Scholar] [CrossRef] [Green Version]
- Stewart, E.V.; Nwosu, C.C.; Tong, Z.; Roguev, A.; Cummins, T.D.; Kim, D.U.; Hayles, J.; Park, H.O.; Hoe, K.L.; Powell, D.W.; et al. Yeast SREBP cleavage activation requires the Golgi Dsc E3 ligase complex. Mol. Cell 2011, 42, 160–171. [Google Scholar] [CrossRef] [Green Version]
- Kober, D.L.; Xu, S.; Li, S.; Bajaj, B.; Liang, G.; Rosenbaum, D.M.; Radhakrishnan, A. Identification of a degradation signal at the carboxy terminus of SREBP2: A new role for this domain in cholesterol homeostasis. Proc. Natl. Acad. Sci. USA 2020, 117, 28080–28091. [Google Scholar] [CrossRef]
- Schmidt, O.; Weyer, Y.; Baumann, V.; Widerin, M.A.; Eising, S.; Angelova, M.; Schleiffer, A.; Kremser, L.; Lindner, H.; Peter, M.; et al. Endosome and Golgi-associated degradation (EGAD) of membrane proteins regulates sphingolipid metabolism. EMBO J. 2019, 38, e101433. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Lerner, A.; Benyair, R.; Hizkiahou, N.; Nudel, N.; Maor, R.; Kramer, M.P.; Shmueli, M.D.; Zigdon, I.; Cherniavsky Lev, M.; Ulman, A.; et al. Golgi organization is regulated by proteasomal degradation. Nat. Commun. 2020, 11, 409. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni-Gosavi, P.; Makhoul, C.; Gleeson, P.A. Form and function of the Golgi apparatus: Scaffolds, cytoskeleton and signalling. FEBS Lett. 2019, 593, 2289–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litterman, N.; Ikeuchi, Y.; Gallardo, G.; O’Connell, B.C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W.; Bonni, A. An OBSL1-Cul7Fbxw8 ubiquitin ligase signaling mechanism regulates Golgi morphology and dendrite patterning. PLoS Biol. 2011, 9, e1001060. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Apaja, P.M.; Lukacs, G.L. Protein quality control at the plasma membrane. Curr. Opin. Cell Biol. 2011, 23, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Jadiya, P.; Tomar, D. Mitochondrial Protein Quality Control Mechanisms. Genes 2020, 11, 563. [Google Scholar] [CrossRef]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef]
- Coughlan, C.M.; Walker, J.L.; Cochran, J.C.; Wittrup, K.D.; Brodsky, J.L. Degradation of mutated bovine pancreatic trypsin inhibitor in the yeast vacuole suggests post-endoplasmic reticulum protein quality control. J. Biol. Chem. 2004, 279, 15289–15297. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ng, D.T. Evasion of endoplasmic reticulum surveillance makes Wsc1p an obligate substrate of Golgi quality control. Mol. Biol. Cell 2010, 21, 1153–1165. [Google Scholar] [CrossRef] [Green Version]
- Briant, K.; Johnson, N.; Swanton, E. Transmembrane domain quality control systems operate at the endoplasmic reticulum and Golgi apparatus. PLoS ONE 2017, 12, e0173924. [Google Scholar] [CrossRef]
- Hellerschmied, D.; Serebrenik, Y.V.; Shao, L.; Burslem, G.M.; Crews, C.M. Protein folding state-dependent sorting at the Golgi apparatus. Mol. Biol. Cell 2019, 30, 2296–2308. [Google Scholar] [CrossRef] [PubMed]
- Benyair, R.; Ogen-Shtern, N.; Lederkremer, G.Z. Glycan regulation of ER-associated degradation through compartmentalization. Semin. Cell Dev. Biol. 2015, 41, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Chan, E.Y.; Hu, X.W.; Kochl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reggiori, F.; Tucker, K.A.; Stromhaug, P.E.; Klionsky, D.J. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev. Cell 2004, 6, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Torii, S.; Yamaguchi, H.; Nakanishi, A.; Arakawa, S.; Honda, S.; Moriwaki, K.; Nakano, H.; Shimizu, S. Identification of a phosphorylation site on Ulk1 required for genotoxic stress-induced alternative autophagy. Nat. Commun. 2020, 11, 1754. [Google Scholar] [CrossRef]
- Popovic, D.; Dikic, I. TBC1D5 and the AP2 complex regulate ATG9 trafficking and initiation of autophagy. EMBO Rep. 2014, 15, 392–401. [Google Scholar] [CrossRef]
- Imai, K.; Hao, F.; Fujita, N.; Tsuji, Y.; Oe, Y.; Araki, Y.; Hamasaki, M.; Noda, T.; Yoshimori, T. Atg9A trafficking through the recycling endosomes is required for autophagosome formation. J. Cell Sci. 2016, 129, 3781–3791. [Google Scholar] [CrossRef] [Green Version]
- Mari, M.; Griffith, J.; Rieter, E.; Krishnappa, L.; Klionsky, D.J.; Reggiori, F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 2010, 190, 1005–1022. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Ma, K.; Gao, R.; Mu, C.; Chen, L.; Liu, Q.; Luo, Q.; Feng, D.; Zhu, Y.; Chen, Q. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 2017, 27, 184–201. [Google Scholar] [CrossRef] [Green Version]
- Judith, D.; Jefferies, H.B.J.; Boeing, S.; Frith, D.; Snijders, A.P.; Tooze, S.A. ATG9A shapes the forming autophagosome through Arfaptin 2 and phosphatidylinositol 4-kinase IIIbeta. J. Cell Biol. 2019, 218, 1634–1652. [Google Scholar] [CrossRef] [Green Version]
- Slobodkin, M.R.; Elazar, Z. The Atg8 family: Multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem. 2013, 55, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joachim, J.; Tooze, S.A. Control of GABARAP-mediated autophagy by the Golgi complex, centrosome and centriolar satellites. Biol. Cell 2018, 110, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Joachim, J.; Jefferies, H.B.; Razi, M.; Frith, D.; Snijders, A.P.; Chakravarty, P.; Judith, D.; Tooze, S.A. Activation of ULK Kinase and Autophagy by GABARAP Trafficking from the Centrosome Is Regulated by WAC and GM130. Mol. Cell 2015, 60, 899–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Hamasaki, M.; Noda, T.; Ohsumi, Y. The early secretory pathway contributes to autophagy in yeast. Cell Struct. Funct. 2003, 28, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Chang, C.; Huang, R.; Liu, B.; Bao, L.; Liu, W. AP1 is essential for generation of autophagosomes from the trans-Golgi network. J. Cell Sci. 2012, 125, 1706–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Yang, Z.; Vollmer, L.L.; Gao, Y.; Fu, Y.; Liu, C.; Chen, X.; Liu, P.; Vogt, A.; Yin, X.M. AMDE-1 is a dual function chemical for autophagy activation and inhibition. PLoS ONE 2015, 10, e0122083. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Liu, Y.; Hong, L.; Yang, Z.; Cai, X.; Chen, X.; Fu, Y.; Lin, Y.; Wen, W.; Li, S.; et al. Golgi-associated LC3 lipidation requires V-ATPase in noncanonical autophagy. Cell Death Dis. 2016, 7, e2330. [Google Scholar] [CrossRef] [Green Version]
- Shorter, J.; Watson, R.; Giannakou, M.E.; Clarke, M.; Warren, G.; Barr, F.A. GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. EMBO J. 1999, 18, 4949–4960. [Google Scholar] [CrossRef] [Green Version]
- Barr, F.A.; Puype, M.; Vandekerckhove, J.; Warren, G. GRASP65, a protein involved in the stacking of Golgi cisternae. Cell 1997, 91, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Wang, Y. GRASP55 and GRASP65 play complementary and essential roles in Golgi cisternal stacking. J. Cell Biol. 2010, 188, 237–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Seemann, J. Rapid degradation of GRASP55 and GRASP65 reveals their immediate impact on the Golgi structure. J. Cell Biol. 2021, 220, e202007052. [Google Scholar] [CrossRef] [PubMed]
- Grond, R.; Veenendaal, T.; Duran, J.M.; Raote, I.; van Es, J.H.; Corstjens, S.; Delfgou, L.; El Haddouti, B.; Malhotra, V.; Rabouille, C. The function of GORASPs in Golgi apparatus organization in vivo. J. Cell Biol. 2020, 219, e202004191. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, L.; Lak, B.; Li, J.; Jokitalo, E.; Wang, Y. GRASP55 Senses Glucose Deprivation through O-GlcNAcylation to Promote Autophagosome-Lysosome Fusion. Dev. Cell 2018, 45, 245–261e246. [Google Scholar] [CrossRef] [Green Version]
- Nuchel, J.; Tauber, M.; Nolte, J.L.; Morgelin, M.; Turk, C.; Eckes, B.; Demetriades, C.; Plomann, M. An mTORC1-GRASP55 signaling axis controls unconventional secretion to reshape the extracellular proteome upon stress. Mol. Cell 2021, 81, 3275–3293.e12. [Google Scholar] [CrossRef]
- Nakamura, N.; Lowe, M.; Levine, T.P.; Rabouille, C.; Warren, G. The vesicle docking protein p115 binds GM130, a cis-Golgi matrix protein, in a mitotically regulated manner. Cell 1997, 89, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Mei, M.; Li, Q.; Roboti, P.; Pang, Q.; Ying, Z.; Gao, F.; Lowe, M.; Bao, S. Loss of the golgin GM130 causes Golgi disruption, Purkinje neuron loss, and ataxia in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 346–351. [Google Scholar] [CrossRef] [Green Version]
- Luke, M.R.; Kjer-Nielsen, L.; Brown, D.L.; Stow, J.L.; Gleeson, P.A. GRIP domain-mediated targeting of two new coiled-coil proteins, GCC88 and GCC185, to subcompartments of the trans-Golgi network. J. Biol. Chem. 2003, 278, 4216–4226. [Google Scholar] [CrossRef] [Green Version]
- Gosavi, P.; Houghton, F.J.; McMillan, P.J.; Hanssen, E.; Gleeson, P.A. The Golgi ribbon in mammalian cells negatively regulates autophagy by modulating mTOR activity. J. Cell Sci. 2018, 131, jcs211987. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Pluhackova, K.; Bockmann, R.A. The Multifaceted Role of SNARE Proteins in Membrane Fusion. Front. Physiol. 2017, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Nichols, B.J.; Pelham, H.R. SNAREs and membrane fusion in the Golgi apparatus. Biochim. Biophys. Acta 1998, 1404, 9–31. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.Q.; Wang, Z.; Wang, C.Y.; Zhang, D.Y.; Wan, H.D.; Zhao, Z.L.; Gu, J.; Zhang, Y.X.; Li, Z.G.; Man, K.Y.; et al. PAQR3 controls autophagy by integrating AMPK signaling to enhance ATG14L-associated PI3K activity. EMBO J. 2016, 35, 496–514. [Google Scholar] [CrossRef] [PubMed]
- Naydenov, N.G.; Harris, G.; Morales, V.; Ivanov, A.I. Loss of a membrane trafficking protein alphaSNAP induces non-canonical autophagy in human epithelia. Cell Cycle 2012, 11, 4613–4625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dascher, C.; Matteson, J.; Balch, W.E. Syntaxin 5 regulates endoplasmic reticulum to Golgi transport. J. Biol. Chem. 1994, 269, 29363–29366. [Google Scholar] [CrossRef]
- Renna, M.; Schaffner, C.; Winslow, A.R.; Menzies, F.M.; Peden, A.A.; Floto, R.A.; Rubinsztein, D.C. Autophagic substrate clearance requires activity of the syntaxin-5 SNARE complex. J. Cell Sci. 2011, 124, 469–482. [Google Scholar] [CrossRef] [Green Version]
- Suga, K.; Hattori, H.; Saito, A.; Akagawa, K. RNA interference-mediated silencing of the syntaxin 5 gene induces Golgi fragmentation but capable of transporting vesicles. FEBS Lett. 2005, 579, 4226–4234. [Google Scholar] [CrossRef] [Green Version]
- Muppirala, M.; Gupta, V.; Swarup, G. Syntaxin 17 cycles between the ER and ERGIC and is required to maintain the architecture of ERGIC and Golgi. Biol. Cell 2011, 103, 333–350. [Google Scholar] [CrossRef]
- Kumar, S.; Gu, Y.; Abudu, Y.P.; Bruun, J.A.; Jain, A.; Farzam, F.; Mudd, M.; Anonsen, J.H.; Rusten, T.E.; Kasof, G.; et al. Phosphorylation of Syntaxin 17 by TBK1 Controls Autophagy Initiation. Dev. Cell 2019, 49, 130–144. [Google Scholar] [CrossRef] [Green Version]
- Tam, R.C.; Li, M.W.; Gao, Y.P.; Pang, Y.T.; Yan, S.; Ge, W.; Lau, C.S.; Chan, V.S. Human CLEC16A regulates autophagy through modulating mTOR activity. Exp. Cell Res. 2017, 352, 304–312. [Google Scholar] [CrossRef]
- Hewavitharana, T.; Wedegaertner, P.B. PAQR3 regulates Golgi vesicle fission and transport via the Gbetagamma-PKD signaling pathway. Cell. Signal. 2015, 27, 2444–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Wang, Z.; Wan, H.; Xu, L.; You, X.; Liao, L.; Chen, Y. PAQR3 Regulates Endoplasmic Reticulum-to-Golgi Trafficking of COPII Vesicle via Interaction with Sec13/Sec31 Coat Proteins. iScience 2018, 9, 382–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Pan, Y.; Huang, M.; You, X.; Guo, F.; Chen, Y. PAQR3 augments amino acid deprivation-induced autophagy by inhibiting mTORC1 signaling. Cell. Signal. 2017, 33, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Redmann, V.; Lamb, C.A.; Hwang, S.; Orchard, R.C.; Kim, S.; Razi, M.; Milam, A.; Park, S.; Yokoyama, C.C.; Kambal, A.; et al. Clec16a is Critical for Autolysosome Function and Purkinje Cell Survival. Sci. Rep. 2016, 6, 23326. [Google Scholar] [CrossRef] [Green Version]
- Kuna, R.S.; Field, S.J. GOLPH3: A Golgi phosphatidylinositol(4)phosphate effector that directs vesicle trafficking and drives cancer. J. Lipid Res. 2019, 60, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Welch, L.G.; Peak-Chew, S.Y.; Begum, F.; Stevens, T.J.; Munro, S. GOLPH3 and GOLPH3L are broad-spectrum COPI adaptors for sorting into intra-Golgi transport vesicles. J. Cell Biol. 2021, 220, e202106115. [Google Scholar] [CrossRef]
- Li, T.; You, H.; Mo, X.; He, W.; Tang, X.; Jiang, Z.; Chen, S.; Chen, Y.; Zhang, J.; Hu, Z. GOLPH3 Mediated Golgi Stress Response in Modulating N2A Cell Death upon Oxygen-Glucose Deprivation and Reoxygenation Injury. Mol. Neurobiol. 2016, 53, 1377–1385. [Google Scholar] [CrossRef]
- Lu, L.Q.; Tang, M.Z.; Qi, Z.H.; Huang, S.F.; He, Y.Q.; Li, D.K.; Li, L.F.; Chen, L.X. Regulation of the Golgi apparatus via GOLPH3-mediated new selective autophagy. Life Sci. 2020, 253, 117700. [Google Scholar] [CrossRef]
- Dippold, H.C.; Ng, M.M.; Farber-Katz, S.E.; Lee, S.K.; Kerr, M.L.; Peterman, M.C.; Sim, R.; Wiharto, P.A.; Galbraith, K.A.; Madhavarapu, S.; et al. GOLPH3 bridges phosphatidylinositol-4- phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 2009, 139, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Scott, K.L.; Kabbarah, O.; Liang, M.C.; Ivanova, E.; Anagnostou, V.; Wu, J.; Dhakal, S.; Wu, M.; Chen, S.; Feinberg, T.; et al. GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature 2009, 459, 1085–1090. [Google Scholar] [CrossRef] [Green Version]
- Dobzinski, N.; Chuartzman, S.G.; Kama, R.; Schuldiner, M.; Gerst, J.E. Starvation-Dependent Regulation of Golgi Quality Control Links the TOR Signaling and Vacuolar Protein Sorting Pathways. Cell Rep. 2015, 12, 1876–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coe, J.G.; Lim, A.C.; Xu, J.; Hong, W. A role for Tlg1p in the transport of proteins within the Golgi apparatus of Saccharomyces cerevisiae. Mol. Biol. Cell 1999, 10, 2407–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdez-Taubas, J.; Pelham, H. Swf1-dependent palmitoylation of the SNARE Tlg1 prevents its ubiquitination and degradation. EMBO J. 2005, 24, 2524–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerial, M.; McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhuin, T.; Roy, J.K. Rab proteins: The key regulators of intracellular vesicle transport. Exp. Cell Res. 2014, 328, 1–19. [Google Scholar] [CrossRef]
- Ao, X.; Zou, L.; Wu, Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014, 21, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Homma, Y.; Hiragi, S.; Fukuda, M. Rab family of small GTPases: An updated view on their regulation and functions. FEBS J. 2021, 288, 36–55. [Google Scholar] [CrossRef]
- Liu, S.; Storrie, B. How Rab proteins determine Golgi structure. Int. Rev. Cell Mol. Biol. 2015, 315, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Wang, P.S.; Yang, L. Golgi-associated Rab GTPases implicated in autophagy. Cell Biosci. 2021, 11, 35. [Google Scholar] [CrossRef]
- Morgan, N.E.; Cutrona, M.B.; Simpson, J.C. Multitasking Rab Proteins in Autophagy and Membrane Trafficking: A Focus on Rab33b. Int. J. Mol. Sci. 2019, 20, 3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraste, J. Spatial and Functional Aspects of ER-Golgi Rabs and Tethers. Front. Cell Dev. Biol. 2016, 4, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, A.; Wang, Y.; Malsam, J.; Beard, M.B.; Warren, G. Golgin-84 is a rab1 binding partner involved in Golgi structure. Traffic 2003, 4, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoppino, F.C.; Militello, R.D.; Slavin, I.; Alvarez, C.; Colombo, M.I. Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic 2010, 11, 1246–1261. [Google Scholar] [CrossRef]
- Lorincz, P.; Toth, S.; Benko, P.; Lakatos, Z.; Boda, A.; Glatz, G.; Zobel, M.; Bisi, S.; Hegedus, K.; Takats, S.; et al. Rab2 promotes autophagic and endocytic lysosomal degradation. J. Cell Biol. 2017, 216, 1937–1947. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Jiang, X.; Tian, R.; Zhao, P.; Li, L.; Wang, X.; Chen, S.; Zhu, Y.; Mei, M.; Bao, S.; et al. RAB2 regulates the formation of autophagosome and autolysosome in mammalian cells. Autophagy 2019, 15, 1774–1786. [Google Scholar] [CrossRef] [Green Version]
- Dickson, L.J.; Liu, S.; Storrie, B. Rab6 is required for rapid, cisternal-specific, intra-Golgi cargo transport. Sci. Rep. 2020, 10, 16604. [Google Scholar] [CrossRef]
- Ayala, C.I.; Kim, J.; Neufeld, T.P. Rab6 promotes insulin receptor and cathepsin trafficking to regulate autophagy induction and activity in Drosophila. J. Cell Sci. 2018, 131, jcs216127. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Rosenwald, A.G. Autophagy in Saccharomyces cerevisiae requires the monomeric GTP-binding proteins, Arl1 and Ypt6. Autophagy 2016, 12, 1721–1737. [Google Scholar] [CrossRef] [Green Version]
- Ganley, I.G.; Carroll, K.; Bittova, L.; Pfeffer, S. Rab9 GTPase regulates late endosome size and requires effector interaction for its stability. Mol. Biol. Cell 2004, 15, 5420–5430. [Google Scholar] [CrossRef] [Green Version]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, S.; Honda, S.; Yamaguchi, H.; Shimizu, S. Molecular mechanisms and physiological roles of Atg5/Atg7-independent alternative autophagy. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, D.; Soldati, T.; Riederer, M.A.; Goda, Y.; Zerial, M.; Pfeffer, S.R. Rab9 functions in transport between late endosomes and the trans Golgi network. EMBO J. 1993, 12, 677–682. [Google Scholar] [CrossRef] [Green Version]
- Riederer, M.A.; Soldati, T.; Shapiro, A.D.; Lin, J.; Pfeffer, S.R. Lysosome biogenesis requires Rab9 function and receptor recycling from endosomes to the trans-Golgi network. J. Cell Biol. 1994, 125, 573–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, T.; Fujita, N.; Kanno, E.; Yamamoto, A.; Yoshimori, T.; Fukuda, M. Golgi-resident small GTPase Rab33B interacts with Atg16L and modulates autophagosome formation. Mol. Biol. Cell 2008, 19, 2916–2925. [Google Scholar] [CrossRef] [Green Version]
- Pantoom, S.; Konstantinidis, G.; Voss, S.; Han, H.; Hofnagel, O.; Li, Z.; Wu, Y.W. RAB33B recruits the ATG16L1 complex to the phagophore via a noncanonical RAB binding protein. Autophagy 2021, 17, 2290–2304. [Google Scholar] [CrossRef]
- Sekito, T.; Kawamata, T.; Ichikawa, R.; Suzuki, K.; Ohsumi, Y. Atg17 recruits Atg9 to organize the pre-autophagosomal structure. Genes Cells 2009, 14, 525–538. [Google Scholar] [CrossRef]
- Nishimura, T.; Tooze, S.A. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 2020, 6, 32. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846. [Google Scholar] [CrossRef] [Green Version]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Mattera, R.; Park, S.Y.; De Pace, R.; Guardia, C.M.; Bonifacino, J.S. AP-4 mediates export of ATG9A from the trans-Golgi network to promote autophagosome formation. Proc. Natl. Acad. Sci. USA 2017, 114, E10697–E10706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Li, C.M.; Luo, X.M.; Siu, G.K.; Gan, W.J.; Zhang, L.; Wu, W.K.; Chan, H.C.; Yu, S. Mammalian TRAPPIII Complex positively modulates the recruitment of Sec13/31 onto COPII vesicles. Sci. Rep. 2017, 7, 43207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, C.A.; Tooze, S.A. TBC1D14 sets the TRAPP for ATG9. Autophagy 2016, 12, 1212–1213. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Nuhlen, S.; Judith, D.; Frith, D.; Snijders, A.P.; Behrends, C.; Tooze, S.A. TBC1D14 regulates autophagy via the TRAPP complex and ATG9 traffic. EMBO J. 2016, 35, 281–301. [Google Scholar] [CrossRef]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Ramirez-Peinado, S.; Ignashkova, T.I.; van Raam, B.J.; Baumann, J.; Sennott, E.L.; Gendarme, M.; Lindemann, R.K.; Starnbach, M.N.; Reiling, J.H. TRAPPC13 modulates autophagy and the response to Golgi stress. J. Cell Sci. 2017, 130, 2251–2265. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, J.B.; D’Souza, Z.; Lupashin, V.V. Maintaining order: COG complex controls Golgi trafficking, processing, and sorting. FEBS Lett. 2019, 593, 2466–2487. [Google Scholar] [CrossRef] [Green Version]
- Ungar, D.; Oka, T.; Brittle, E.E.; Vasile, E.; Lupashin, V.V.; Chatterton, J.E.; Heuser, J.E.; Krieger, M.; Waters, M.G. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J. Cell Biol. 2002, 157, 405–415. [Google Scholar] [CrossRef]
- Yen, W.L.; Shintani, T.; Nair, U.; Cao, Y.; Richardson, B.C.; Li, Z.; Hughson, F.M.; Baba, M.; Klionsky, D.J. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J. Cell Biol. 2010, 188, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Sohda, M.; Misumi, Y.; Ogata, S.; Sakisaka, S.; Hirose, S.; Ikehara, Y.; Oda, K. Trans-Golgi protein p230/golgin-245 is involved in phagophore formation. Biochem. Biophys. Res. Commun. 2015, 456, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Bremnes, B.; Ronning, E.; Aasland, R.; Stenmark, H. Syntaxin-16, a putative Golgi t-SNARE. Eur. J. Cell Biol. 1998, 75, 223–231. [Google Scholar] [CrossRef]
- Tang, B.L.; Low, D.Y.; Lee, S.S.; Tan, A.E.; Hong, W. Molecular cloning and localization of human syntaxin 16, a member of the syntaxin family of SNARE proteins. Biochem. Biophys. Res. Commun. 1998, 242, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Syntaxin 16’s Newly Deciphered Roles in Autophagy. Cells 2019, 8, 1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyagi, K.; Itakura, M.; Fukutomi, T.; Nishiwaki, C.; Nakamichi, Y.; Torii, S.; Makiyama, T.; Harada, A.; Ohara-Imaizumi, M. VAMP7 Regulates Autophagosome Formation by Supporting Atg9a Functions in Pancreatic beta-Cells From Male Mice. Endocrinology 2018, 159, 3674–3688. [Google Scholar] [CrossRef] [Green Version]
- Simunovic, M.; Evergren, E.; Callan-Jones, A.; Bassereau, P. Curving Cells Inside and Out: Roles of BAR Domain Proteins in Membrane Shaping and Its Cellular Implications. Annu. Rev. Cell Dev. Biol. 2019, 35, 111–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Meyerkord, C.L.; Hori, T.; Runkle, K.; Fox, T.E.; Kester, M.; Loughran, T.P.; Wang, H.G. Bif-1 regulates Atg9 trafficking by mediating the fission of Golgi membranes during autophagy. Autophagy 2011, 7, 61–73. [Google Scholar] [CrossRef]
- Knaevelsrud, H.; Soreng, K.; Raiborg, C.; Haberg, K.; Rasmuson, F.; Brech, A.; Liestol, K.; Rusten, T.E.; Stenmark, H.; Neufeld, T.P.; et al. Membrane remodeling by the PX-BAR protein SNX18 promotes autophagosome formation. J. Cell Biol. 2013, 202, 331–349. [Google Scholar] [CrossRef]
- Soreng, K.; Munson, M.J.; Lamb, C.A.; Bjorndal, G.T.; Pankiv, S.; Carlsson, S.R.; Tooze, S.A.; Simonsen, A. SNX18 regulates ATG9A trafficking from recycling endosomes by recruiting Dynamin-2. EMBO Rep. 2018, 19, e44837. [Google Scholar] [CrossRef]
- Wang, H.; Sun, H.Q.; Zhu, X.; Zhang, L.; Albanesi, J.; Levine, B.; Yin, H. GABARAPs regulate PI4P-dependent autophagosome:lysosome fusion. Proc. Natl. Acad. Sci. USA 2015, 112, 7015–7020. [Google Scholar] [CrossRef] [Green Version]
- Waugh, M.G. The Great Escape: How phosphatidylinositol 4-kinases and PI4P promote vesicle exit from the Golgi (and drive cancer). Biochem. J. 2019, 476, 2321–2346. [Google Scholar] [CrossRef] [PubMed]
- Rahajeng, J.; Kuna, R.S.; Makowski, S.L.; Tran, T.T.T.; Buschman, M.D.; Li, S.; Cheng, N.; Ng, M.M.; Field, S.J. Efficient Golgi Forward Trafficking Requires GOLPH3-Driven, PI4P-Dependent Membrane Curvature. Dev. Cell 2019, 50, 573–585.e5. [Google Scholar] [CrossRef] [PubMed]
- Miao, G.; Zhang, Y.; Chen, D.; Zhang, H. The ER-Localized Transmembrane Protein TMEM39A/SUSR2 Regulates Autophagy by Controlling the Trafficking of the PtdIns(4)P Phosphatase SAC1. Mol. Cell 2020, 77, 618–632.e5. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Arakawa, S.; Kanaseki, T.; Miyatsuka, T.; Fujitani, Y.; Watada, H.; Tsujimoto, Y.; Shimizu, S. Golgi membrane-associated degradation pathway in yeast and mammals. EMBO J. 2016, 35, 1991–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longatti, A.; Tooze, S.A. Vesicular trafficking and autophagosome formation. Cell Death Differ. 2009, 16, 956–965. [Google Scholar] [CrossRef] [PubMed]
- De Tito, S.; Hervas, J.H.; van Vliet, A.R.; Tooze, S.A. The Golgi as an Assembly Line to the Autophagosome. Trends Biochem. Sci. 2020, 45, 484–496. [Google Scholar] [CrossRef]
- Schultz, M.J.; Swindall, A.F.; Bellis, S.L. Regulation of the metastatic cell phenotype by sialylated glycans. Cancer Metastasis Rev. 2012, 31, 501–518. [Google Scholar] [CrossRef] [Green Version]
- Petrosyan, A. Onco-Golgi: Is Fragmentation a Gate to Cancer Progression? Biochem. Mol. Biol. J. 2015, 1, 16. [Google Scholar] [CrossRef] [Green Version]
- Sausville, E.A.; Duncan, K.L.; Senderowicz, A.; Plowman, J.; Randazzo, P.A.; Kahn, R.; Malspeis, L.; Grever, M.R. Antiproliferative effect in vitro and antitumor activity in vivo of brefeldin A. Cancer J. Sci. Am. 1996, 2, 52–58. [Google Scholar]
- Martinez-Menarguez, J.A.; Tomas, M.; Martinez-Martinez, N.; Martinez-Alonso, E. Golgi Fragmentation in Neurodegenerative Diseases: Is There a Common Cause? Cells 2019, 8, 748. [Google Scholar] [CrossRef] [Green Version]
- Ayala, I.; Colanzi, A. Alterations of Golgi organization in Alzheimer’s disease: A cause or a consequence? Tissue Cell 2017, 49, 133–140. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benyair, R.; Eisenberg-Lerner, A.; Merbl, Y. Maintaining Golgi Homeostasis: A Balancing Act of Two Proteolytic Pathways. Cells 2022, 11, 780. https://doi.org/10.3390/cells11050780
Benyair R, Eisenberg-Lerner A, Merbl Y. Maintaining Golgi Homeostasis: A Balancing Act of Two Proteolytic Pathways. Cells. 2022; 11(5):780. https://doi.org/10.3390/cells11050780
Chicago/Turabian StyleBenyair, Ron, Avital Eisenberg-Lerner, and Yifat Merbl. 2022. "Maintaining Golgi Homeostasis: A Balancing Act of Two Proteolytic Pathways" Cells 11, no. 5: 780. https://doi.org/10.3390/cells11050780
APA StyleBenyair, R., Eisenberg-Lerner, A., & Merbl, Y. (2022). Maintaining Golgi Homeostasis: A Balancing Act of Two Proteolytic Pathways. Cells, 11(5), 780. https://doi.org/10.3390/cells11050780