
Transcriptome Differences in Normal Human Bronchial Epithelial Cells in Response to Influenza A pdmH1N1 or H7N9 Virus Infection


Abstract
:1. Introduction
2. Materials and Methods
2.1. NHBE Cells
2.2. Virus Isolation and Preparation
2.3. Virus Infection
2.4. RNA Extraction
2.5. Influenza A Virus Quantification by Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction
2.6. Affymetrix Analysis
2.7. Bio-Plex Cytokine Analysis
3. Results
3.1. Viral RNA Quantities Comparison of Influenza A pdmH1N1 and H7N9 in NHBE Cells
3.2. Global Overview of RNA-Seq Data of pdmH1N1- and H7N9-Infected NHBE Cells
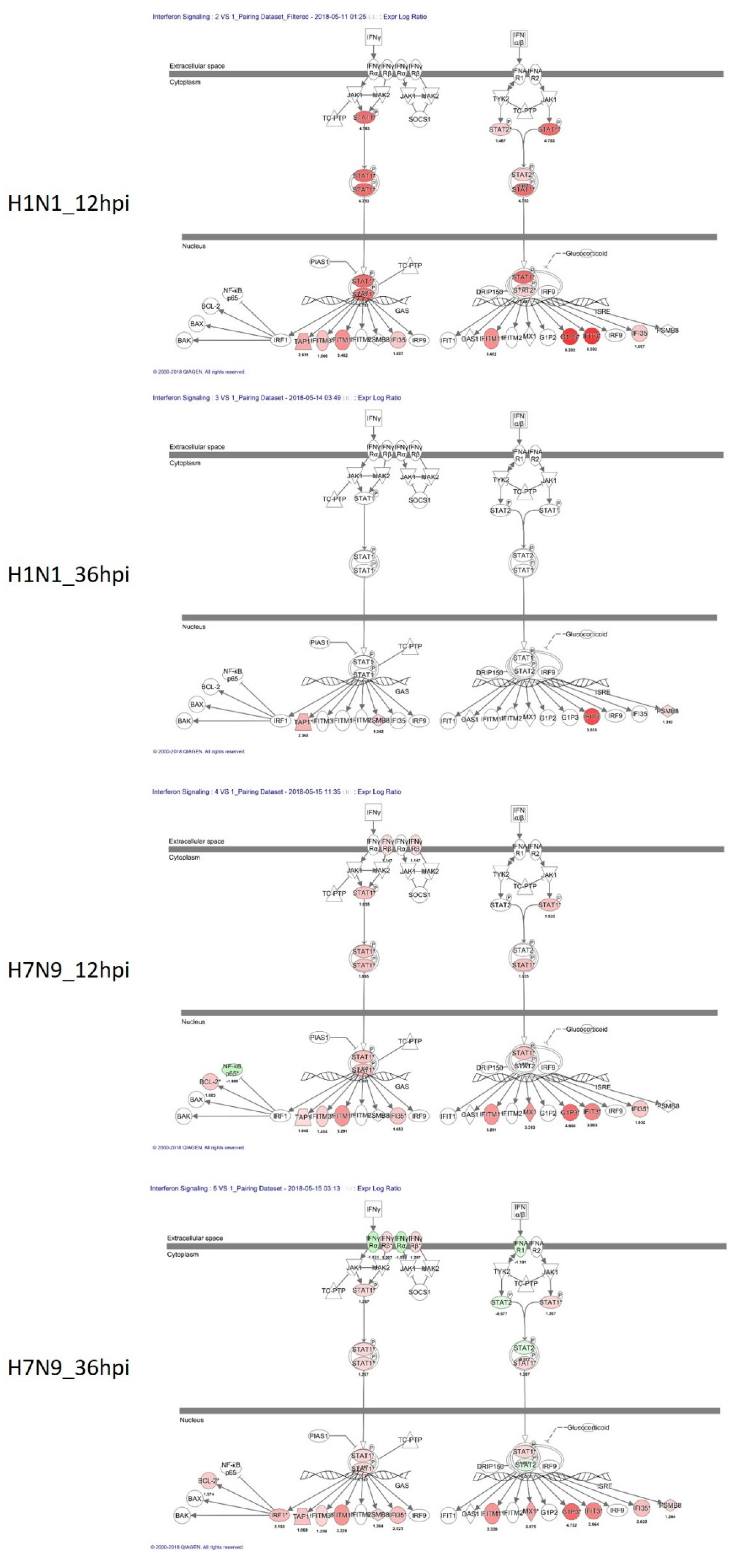
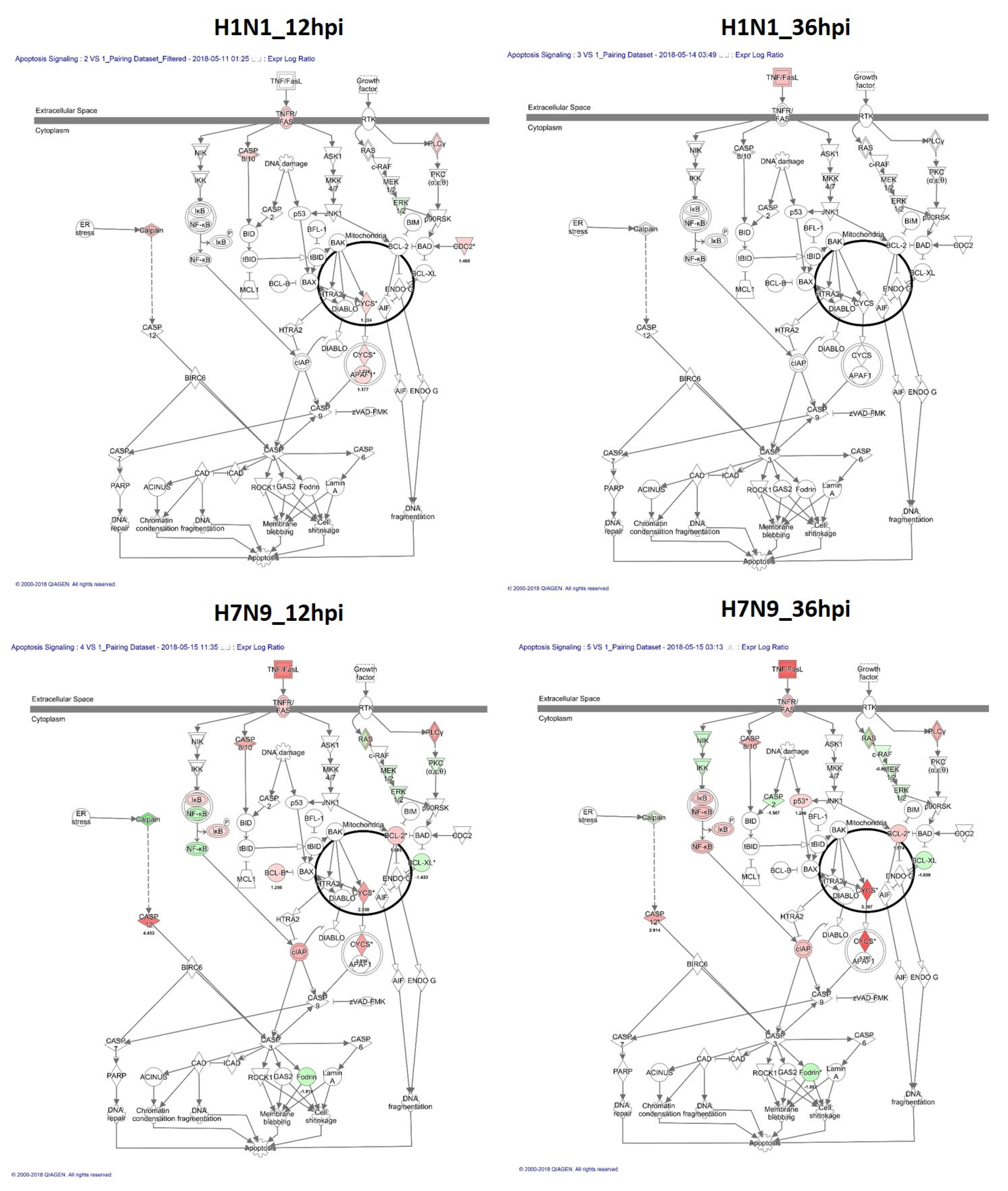
3.3. Canonical Signaling Pathways Analysis Based on DEGs
3.4. Differential Cytokine Expression in NHBE Cells Challenged by pdmH1N1 or H7N9
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Steel, J.; Lowen, A.C. Influenza A Virus Reassortment. Influenza Pathog. Control-Vol. I 2014, 385, 377–401. [Google Scholar] [CrossRef]
- Wu, J.T.; Ma, E.S.K.; Lee, C.K.; Chu, D.K.W.; Ho, P.; Shen, A.L.; Ho, A.; Hung, I.F.N.; Riley, S.; Ho, L.M.; et al. The Infection Attack Rate and Severity of 2009 Pandemic H1N1 Influenza in Hong Kong. Clin. Infect. Dis. 2010, 51, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention, 2009 H1N1 Pandemic (H1N1pdm09 Virus), June 2019. Available online: https://www.cdc.gov/flu/pandemic-resources/2009-h1n1-pandemic.html (accessed on 9 January 2022).
- Dai, J.; Zhou, X.; Dong, D.; Liu, Y.; Gu, Q.; Zhu, B.; Wu, C.; Cai, H. Human infection with a novel avian-origin influenza A (H7N9) virus: Serial chest radiographic and CT findings. Chin. Med. J. 2014, 127, 2206–2211. [Google Scholar] [PubMed]
- Chen, Y.; Liang, W.; Yang, S.; Wu, N.; Gao, H.; Sheng, J.; Yao, H.; Wo, J.; Fang, Q.; Cui, D.; et al. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: Clinical analysis and characterisation of viral genome. Lancet 2013, 381, 1916–1925. [Google Scholar] [CrossRef]
- Wiwanitkit, V. H7N9 influenza: The emerging infectious disease. N. Am. J. Med. Sci. 2013, 5, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Shi, W.; Shi, Y.; Wang, D.; Xiao, H.; Li, W.; Bi, Y.; Wu, Y.; Li, X.; Yan, J.; et al. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: Phylogenetic, structural, and coalescent analyses. Lancet 2013, 381, 1926–1932. [Google Scholar] [CrossRef]
- Watanabe, T.; Kiso, M.; Fukuyama, S.; Nakajima, N.; Imai, M.; Yamada, S.; Murakami, S.; Yamayoshi, S.; Iwatsuki-Horimoto, K.; Sakoda, Y.; et al. Characterization of H7N9 influenza A viruses isolated from humans. Nature 2013, 501, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Cowling, B.; Feng, L.; Lau, E.; Liao, Q.; Tsang, T.K.L.; Peng, Z.; Wu, P.; Liu, F.; Fang, V.J.; et al. Human infection with avian influenza A H7N9 virus: An assessment of clinical severity. Lancet 2013, 382, 138–145. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Avian Influenza A(H7N9) Response: An Investment in Public Health Preparedness, December 2013. Available online: https://www.who.int/publications/i/item/avian-influenza-a(-h7n9)-response (accessed on 10 February 2022).
- Song, L.; Xiong, D.; Hu, M.; Jiao, X.; Pan, Z. Enhanced Th1/Th2 mixed immune responses elicited by polyethyleneimine adjuvanted influenza A (H7N9) antigen HA1-2 in chickens. Poult. Sci. 2018, 97, 4245–4251. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, K.; Liu, L.; Li, W.; Zhu, B.; Zhang, S.; Xu, P.; Liu, W.; Li, J. Global transcriptome analysis of H5N1 influenza virus-infected human cells. Hereditas 2019, 156, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-J.; Kumar, M.; Kwon, H.-I.; Seong, R.-K.; Han, K.; Song, J.-M.; Kim, C.-J.; Choi, Y.-K.; Shin, O.S. Dynamic changes in host gene expression associated with H5N8 avian influenza virus infection in mice. Sci. Rep. 2015, 5, 16512. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-G.; Lee, L.-A.; Wu, Y.-C.; Hsiao, M.-J.; Horng, J.-T.; Kuo, R.-L.; Huang, C.-H.; Lin, Y.-C.; Tsao, K.-C.; Chen, M.-C.; et al. A pilot study on primary cultures of human respiratory tract epithelial cells to predict patients’ responses to H7N9 infection. Oncotarget 2018, 9, 14492–14508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taiwan Centers for Disease Control Press Releases, April 2014. Available online: https://www.cdc.gov.tw/En/Bulletin/Detail/uKe3mLC55DB0E8TdfwXR2g?typeid=158 (accessed on 8 February 2022).
- Chen, G.-W.; Kuo, S.-M.; Yang, S.-L.; Gong, Y.-N.; Hsiao, M.-R.; Liu, Y.-C.; Shih, S.-R.; Tsao, K.-C. Genomic Signatures for Avian H7N9 Viruses Adapting to Humans. PLoS ONE 2016, 11, e0148432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, P.F.; de La Vega, M.-A.; Paradis, É.; Mendoza, E.; Coombs, K.M.; Kobasa, D.; Beauchemin, C. Avian influenza viruses that cause highly virulent infections in humans exhibit distinct replicative properties in contrast to human H1N1 viruses. Sci. Rep. 2016, 6, 24154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Weng, H.; Lan, C.; Zhang, H.; Wang, X.; Pan, J.; Chen, L.; Huang, J. Comparison of patients with avian influenza A (H7N9) and influenza A (H1N1) complicated by acute respiratory distress syndrome. Medicine 2018, 97, e0194. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yu, H.; Horby, P.; Cao, B.; Wu, P.; Yang, S.; Gao, H.; Li, H.; Tsang, T.K.L.; Liao, Q.; et al. Comparison of Patients Hospitalized with Influenza A Subtypes H7N9, H5N1, and 2009 Pandemic H1N1. Clin. Infect. Dis. 2014, 58, 1095–1103. [Google Scholar] [CrossRef]
- Barber, G.N. Host defense, viruses and apoptosis. Cell Death Differ. 2001, 8, 113–126. [Google Scholar] [CrossRef] [Green Version]
- Cameron, C.M.; Cameron, M.J.; Bermejo-Martin, J.F.; Ran, L.; Xu, L.; Turner, P.V.; Ran, R.; Danesh, A.; Fang, Y.; Chan, P.-K.M.; et al. Gene Expression Analysis of Host Innate Immune Responses during Lethal H5N1 Infection in Ferrets. J. Virol. 2008, 82, 11308–11317. [Google Scholar] [CrossRef] [Green Version]
- Maines, T.R.; Szretter, K.J.; Perrone, L.; Belser, J.A.; Bright, R.A.; Zeng, H.; Tumpey, T.M.; Katz, J.M. Pathogenesis of emerging avian influenza viruses in mammals and the host innate immune response. Immunol. Rev. 2008, 225, 68–84. [Google Scholar] [CrossRef]
- Nichols, J.E.; Niles, J.A.; Roberts, N.J. Human Lymphocyte Apoptosis after Exposure to Influenza A Virus. J. Virol. 2001, 75, 5921–5929. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.C.Y.; To, K.; Zhu, H.; Chu, H.; Li, C.; Mak, W.W.N.; Zhang, J.; Yuen, K.-Y. Avian influenza virus A H7N9 infects multiple mononuclear cell types in peripheral blood and induces dysregulated cytokine responses and apoptosis in infected monocytes. J. Gen. Virol. 2017, 98, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Cui, D.; Wang, X.; Zou, Q.; Zhao, D.; Zheng, S.; Yu, F.; Huang, L.; Dong, Y.; Yang, X.; et al. Decreased Frequencies of Th17 and Tc17 Cells in Patients Infected with Avian Influenza A (H7N9) Virus. J. Immunol. Res. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, L.; Li, X.; Zhou, D.; Geng, J.; Chen, L. Role of Hippo signaling in regulating immunity. Cell. Mol. Immunol. 2018, 15, 1003–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taye, B.; Chen, H.; Yeo, D.S.-Y.; Seah, S.G.-K.; Wong, M.S.-Y.; Sugrue, R.J.; Tan, B.-H. A System Based-Approach to Examine Host Response during Infection with Influenza A Virus Subtype H7N9 in Human and Avian Cells. Cells 2020, 9, 448. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Shi, D.; Fang, D.; Guo, F.; Guo, J.; Huang, F.; Chen, Y.; Lv, L.; Li, L. A new perspective on C-reactive protein in H7N9 infections. Int. J. Infect. Dis. 2016, 44, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Chen, Z.; Li, X.; Xu, L.; Guan, W.; Cao, Y.; Hu, Y.; Zhang, J. Host immunological response and factors associated with clinical outcome in patients with the novel influenza A H7N9 infection. Clin. Microbiol. Infect. 2014, 20, O493–O500. [Google Scholar] [CrossRef] [Green Version]
- Betakova, T.; Kostrabova, A.; Lachova, V.; Turianova, L. Cytokines Induced During Influenza Virus Infection. Curr. Pharm. Des. 2017, 23, 2616–2622. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Gene No. 67,528 | Upregulation | Downregulation | Significant Gene No. | % in Total Gene | ||
|---|---|---|---|---|---|---|
| Coding | Non-Coding | Coding | Non-Coding | |||
| H1N1_12hpi VS Mock | 1678 | 773 | 735 | 262 | 3448 | 5% |
| H1N1_36hpi VS Mock | 908 | 214 | 78 | 13 | 1213 | 2% |
| H7N9_12hpi VS Mock | 5559 | 2820 | 1566 | 408 | 10,353 | 15% |
| H7N9_36hpi VS Mock | 6727 | 3213 | 2155 | 574 | 12,669 | 19% |
| Upregulation | Downregulation | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| hpi | H1N1 | Fold Change | p-Value | H7N9 | Fold Change | p-Value | hpi | H1N1 | Fold Change | p-Value | H7N9 | Fold Change | p-Value |
| 12 hpi | IFI6 | 315.62 | 0.00854 | RP1-12G14.6 | 944.41 | 0.000383 | 12 hpi | CRNN | −293.19 | 0.003856 | ATP12A | −2305.39 | 0.000245 |
| IFI44L | 191.33 | 0.0036 | RNA5SP115 | 435.61 | 0.00153 | LCE3D | −273.6 | 0.017274 | CNFN | −1263.62 | 0.010624 | ||
| IFIT1 | 138.87 | 0.006031 | POLG2 | 372.61 | 0.000438 | CNFN | −194.27 | 0.024209 | KLK7 | −751.41 | 0.004461 | ||
| MX1 | 137.1 | 0.003247 | AMPD1 | 195.68 | 0.000091 | FLG | −149.21 | 0.012985 | CEACAM5 | −749.71 | 0.001844 | ||
| IFIT3 | 96.49 | 0.007147 | LINC00641 | 167.28 | 0.006579 | NCCRP1 | −140.35 | 0.001799 | SBSN | −719.43 | 0.009373 | ||
| IFI44 | 88.53 | 0.001676 | MSH4 | 164.85 | 0.000503 | KLK7 | −118.29 | 0.008553 | NCCRP1 | −642.08 | 0.001569 | ||
| RSAD2 | 86.66 | 0.015444 | MIR215 | 158.55 | 0.00367 | SBSN | −117.62 | 0.019107 | KRT4 | −598.76 | 0.000358 | ||
| DDX60L | 81.55 | 0.001921 | RNY5P3 | 152.08 | 0.004924 | LCE3E | −113.71 | 0.007499 | TMPRSS11E | −574.16 | 0.007775 | ||
| IFIT2 | 76.81 | 0.015337 | CKAP2L | 123.31 | 0.004605 | ATP12A | −110.6 | 0.003672 | IVL | −497.27 | 0.000799 | ||
| CXCL11 | 58.57 | 0.01463 | LDHC | 107.28 | 0.000013 | WFDC12 | −102.89 | 0.004885 | CRNN | −435.42 | 0.003444 | ||
| 36 hpi | VTRNA1-3 | 1663.32 | 0.017978 | RNA5SP115 | 1176.93 | 0.000935 | 36 hpi | CRNN | −21.31 | 0.03233 | ATP12A | −1273.8 | 0.000371 |
| IFI6 | 738.27 | 0.062186 | RP1-12G14.6 | 1164.64 | 0.000848 | WFDC12 | −10.05 | 0.040629 | KRT4 | −640.49 | 0.000352 | ||
| RNA5SP402 | 603.17 | 0.005802 | POLG2 | 953.23 | 0.00052 | IL36A | −7.45 | 0.027829 | CEACAM5 | −533.67 | 0.002502 | ||
| RNA5SP496 | 417.73 | 0.014153 | RP11-505P4.6 | 647.16 | 0.000201 | SPINK5 | −5.73 | 0.019001 | SBSN | −489.11 | 0.01055 | ||
| MIR4521 | 314.04 | 0.01979 | MIR4659A | 508.36 | 0.001933 | GCNT3 | −4.97 | 0.042463 | NCCRP1 | −486.72 | 0.001416 | ||
| RSAD2 | 267.72 | 0.045687 | HIST2H4B | 362.02 | 0.001517 | SCNN1B | −4.77 | 0.022182 | KLK7 | −410.65 | 0.005305 | ||
| TRNAI6 | 255.48 | 0.000532 | VTRNA1-3 | 326.31 | 0.024592 | IVL | −4.63 | 0.046796 | RPTN | −404.42 | 0.073667 | ||
| RNA5SP318 | 248.85 | 0.011112 | HCP5 | 324.61 | 0.001104 | KCNH5 | −4.59 | 0.032183 | CNFN | −401.92 | 0.015334 | ||
| TRNAI2 | 243.8 | 0.002995 | RP1-40E16.11 | 260.6 | 0.000742 | CLCA4 | −4.33 | 0.031389 | TMPRSS11E | −380.78 | 0.008919 | ||
| RSAD2 | 198.39 | 0.016356 | RGS2 | 257.86 | 0.00157 | SH3BGRL2 | −3.96 | 0.028623 | PPL | −376.61 | 0.010291 | ||
| Category | Name | H7N9 | pdmH1N1 | ||||
|---|---|---|---|---|---|---|---|
| 4 hpi | 8 hpi | 12 hpi | 4 hpi | 8 hpi | 12 hpi | ||
| Chemokines | IL-8 | 0.1 | 0.9 | 1.0 | −2.7 | −0.8 | −0.1 |
| IP-10 | 0.8 | 3.7 | 4.3 | −1.3 | 3.3 | 4.2 | |
| RANTES | −0.2 | 0.3 | 0.9 | 0.0 | 0.0 | 4.5 | |
| MIP-1β | −0.6 | −0.9 | −0.2 | 0.0 | 0.0 | 0.0 | |
| Eotaxin | −0.4 | −0.8 | −0.5 | 0.0 | 0.0 | 0.0 | |
| MCP-1(MCAF) | −0.5 | −1.1 | −0.8 | 0.0 | 0.0 | 0.0 | |
| MIP-1α | −0.1 | −0.1 | −0.3 | −0.5 | −0.5 | −0.1 | |
| Growth Factors | PDGF-BB | −0.6 | −1.1 | −1.6 | 0.0 | 0.0 | 0.0 |
| FGF basic | −0.1 | −0.2 | −0.4 | 1.3 | −2.9 | −0.1 | |
| GM-CSF | −0.1 | −0.4 | −0.3 | −0.2 | 0.01 | 0.05 | |
| VEGF | −0.1 | −0.1 | 0.2 | −1.9 | −1.4 | −0.6 | |
| G-CSF | 0.3 | 0.5 | 0.2 | −9.5 | −2.1 | −1.0 | |
| Pro inflammatoryFactors | IL-1β | 0.2 | 0.3 | 0.02 | −1.6 | −0.3 | −0.1 |
| IL-6 | 0.7 | 0.9 | 0.0 | −2.9 | −0.3 | −0.3 | |
| TNF-α | 0.7 | 1.6 | 2.9 | −4.1 | −1.1 | 0.2 | |
| T-helper cytokines | IL-12(p70) | −0.5 | −0.7 | −0.5 | −3.2 | −2.6 | −0.5 |
| IL-2 | −1.6 | −1.9 | −1.7 | 0.0 | 0.0 | 0.0 | |
| IL-5 | −0.2 | −1.0 | −0.4 | 0.0 | 0.0 | 0.0 | |
| IL-9 | −0.2 | −0.3 | −0.3 | 0.0 | 0.0 | 0.0 | |
| IL-17 | −0.5 | −0.6 | −0.7 | 0.0 | 0.0 | 0.0 | |
| IFN-γ | −0.4 | −0.5 | −0.6 | 0.0 | 1.8 | 1.1 | |
| IL-10 | −0.1 | −0.4 | −0.3 | 0.0 | 0.2 | 0.9 | |
| IL-4 | −0.3 | −0.6 | −0.4 | −0.9 | 1.7 | 2.0 | |
| IL-13 | −0.3 | −0.3 | −0.8 | 6.5 | 6.0 | 5.9 | |
| Others | IL-1Ra | 0.0 | −0.3 | −0.3 | 0.1 | 0.1 | 0.1 |
| IL-7 | −0.5 | −1.1 | −0.9 | 0.0 | 0.0 | 0.0 | |
| IL-15 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, T.-H.; Lin, Y.-J.; Hsioa, M.-J.; Wang, H.-J.; Chen, L.-T.; Yang, S.-L.; Huang, C.-G. Transcriptome Differences in Normal Human Bronchial Epithelial Cells in Response to Influenza A pdmH1N1 or H7N9 Virus Infection. Cells 2022, 11, 781. https://doi.org/10.3390/cells11050781
Hsieh T-H, Lin Y-J, Hsioa M-J, Wang H-J, Chen L-T, Yang S-L, Huang C-G. Transcriptome Differences in Normal Human Bronchial Epithelial Cells in Response to Influenza A pdmH1N1 or H7N9 Virus Infection. Cells. 2022; 11(5):781. https://doi.org/10.3390/cells11050781
Chicago/Turabian StyleHsieh, Tzu-Hsuan, Ya-Jhu Lin, Mei-Jen Hsioa, Hsin-Ju Wang, Lu-Ting Chen, Shu-Li Yang, and Chung-Guei Huang. 2022. "Transcriptome Differences in Normal Human Bronchial Epithelial Cells in Response to Influenza A pdmH1N1 or H7N9 Virus Infection" Cells 11, no. 5: 781. https://doi.org/10.3390/cells11050781
APA StyleHsieh, T.-H., Lin, Y.-J., Hsioa, M.-J., Wang, H.-J., Chen, L.-T., Yang, S.-L., & Huang, C.-G. (2022). Transcriptome Differences in Normal Human Bronchial Epithelial Cells in Response to Influenza A pdmH1N1 or H7N9 Virus Infection. Cells, 11(5), 781. https://doi.org/10.3390/cells11050781