
Cell-Type Resolved Insights into the Cis-Regulatory Genome of NAFLD


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
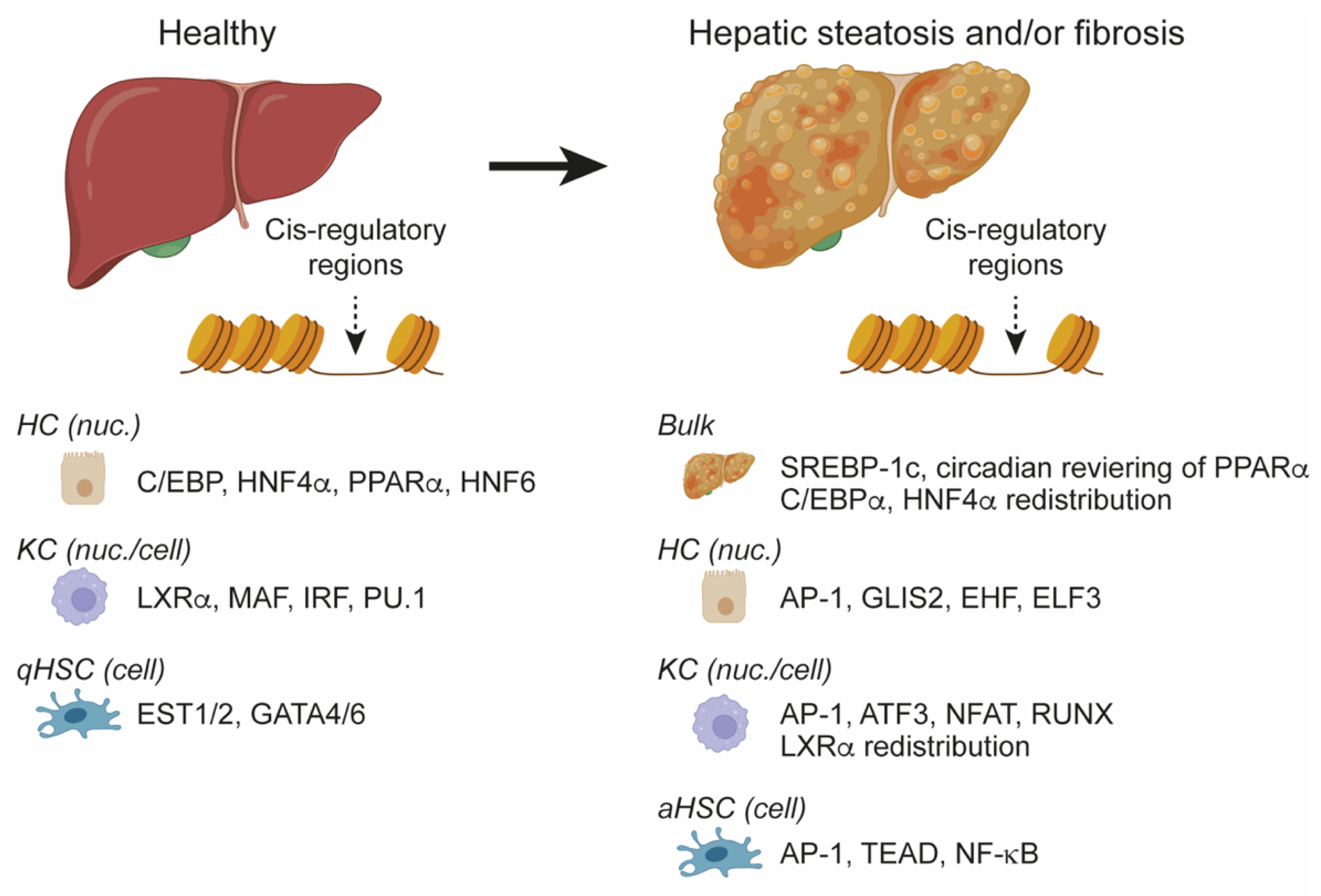
2. Identification of Cis-Regulatory Regions in the Diseased Liver
Hepatic Chromatin Remodeling Induced by NAFLD
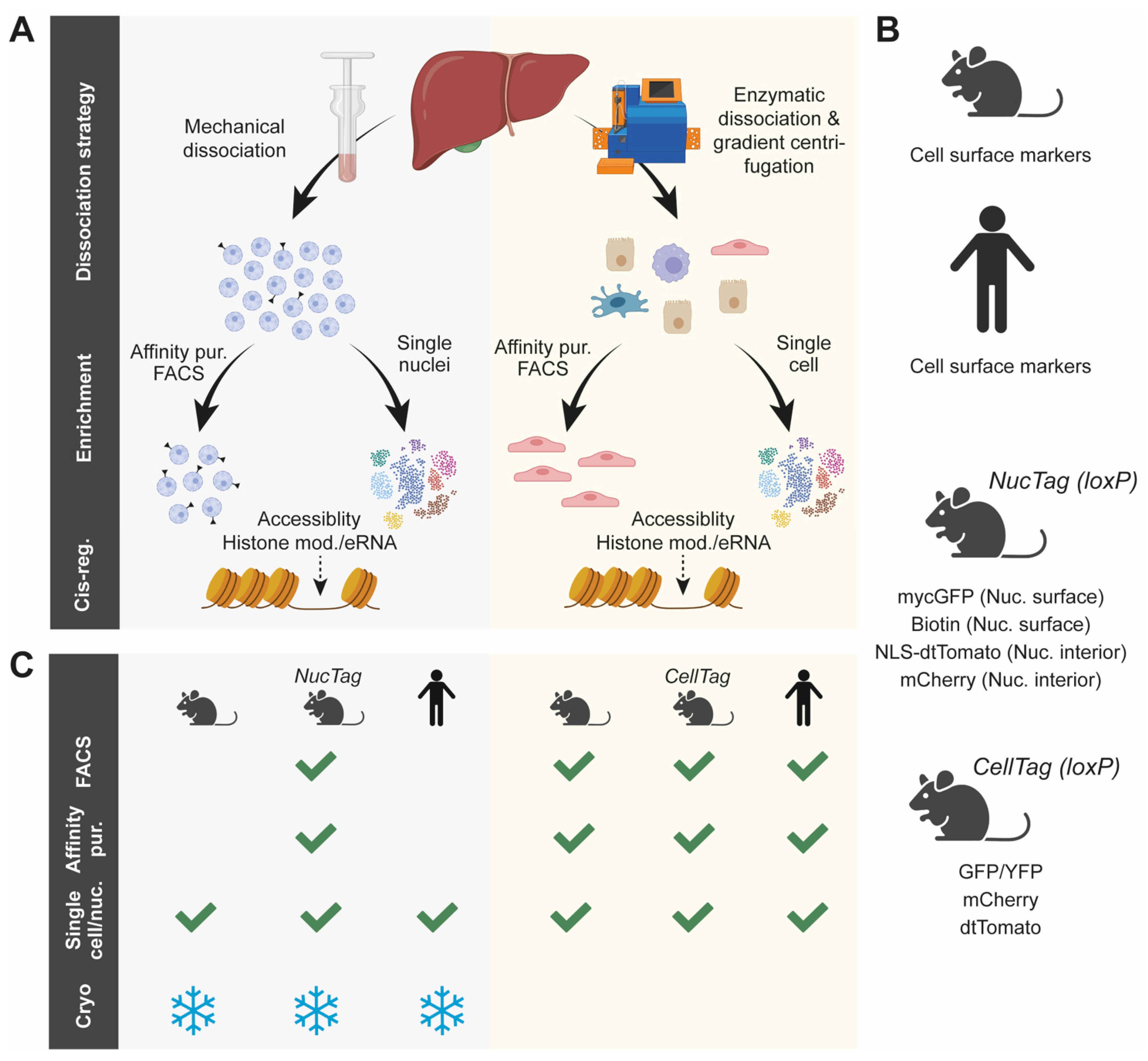
3. Strategies to Study Cis-Regulatory Regions in Individual Cell Populations of the Liver
4. The Cis-Regulatory Genome in Specific Cell Populations of the Liver
4.1. Hepatocytes
4.2. Kupffer Cells and Infiltrating Macrophages
4.3. Hepatic Stellate Cells
4.4. NAFLD/NASH Remodel Lineage-Determining Cis-Regulatory Regions
5. Emerging Technologies to Analyze the Cis-Regulatory Genome at Single-Cell Level
5.1. Mapping Chromatin Accessibility at Single-Cell Resolution
5.2. Mapping Histone Modifications and TF Binding at Single-Cell Resolution
5.3. Mapping Chromatin Interactions at Single-Cell Resolution
5.4. Multi-Omics Technologies for the Profiling of Different Regulatory Layers
5.5. Linking Gene Expression to Cellular Function: Single-Cell Proteomics and Single-Cell Metabolomics
6. Summary and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Remmerie, A.; Martens, L.; Thoné, T.; Castoldi, A.; Seurinck, R.; Pavie, B.; Roels, J.; Vanneste, B.; De Prijck, S.; Vanhockerhout, M.; et al. Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct from Kupffer Cells in the Fatty Liver. Immunity 2020, 53, 641–657.e14. [Google Scholar] [CrossRef] [PubMed]
- Miyao, M.; Kotani, H.; Ishida, T.; Kawai, C.; Manabe, S.; Abiru, H.; Tamaki, K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab. Investig. 2015, 95, 1130–1144. [Google Scholar] [CrossRef] [Green Version]
- Marcher, A.-B.; Bendixen, S.M.; Terkelsen, M.K.; Hohmann, S.S.; Hansen, M.H.; Larsen, B.D.; Mandrup, S.; Dimke, H.; Detlefsen, S.; Ravnskjaer, K. Transcriptional regulation of Hepatic Stellate Cell activation in NASH. Sci. Rep. 2019, 9, 2324. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Kundu, D.; Robles-Linares, J.; Meadows, V.; Sato, K.; Baiocchi, L.; Ekser, B.; Glaser, S.; Alpini, G.; Francis, H.; et al. Feedback Signaling between Cholangiopathies, Ductular Reaction, and Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 2072. [Google Scholar] [CrossRef]
- Sato, K.; Marzioni, M.; Meng, F.; Francis, H.; Glaser, S.; Alpini, G. Ductular reaction in liver diseases: Pathological mechanisms and translational significances. Hepatology 2019, 69, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Pantano, L.; Agyapong, G.; Shen, Y.; Zhuo, Z.; Fernandez-Albert, F.; Rust, W.; Knebel, D.; Hill, J.; Boustany-Kari, C.M.; Doerner, J.F.; et al. Molecular characterization and cell type composition deconvolution of fibrosis in NAFLD. Sci. Rep. 2021, 11, 18045. [Google Scholar] [CrossRef]
- Fan, Z.; Li, L.; Li, M.; Zhang, X.; Hao, C.; Yu, L.; Zeng, S.; Xu, H.; Fang, M.; Shen, A.; et al. The histone methyltransferase Suv39h2 contributes to nonalcoholic steatohepatitis in mice. Hepatology 2017, 65, 1904–1919. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Xu, J.; Rosenthal, S.; Zhang, L.-J.; McCubbin, R.; Meshgin, N.; Shang, L.; Koyama, Y.; Ma, H.-Y.; Sharma, S.; et al. Identification of Lineage-Specific Transcription Factors That Prevent Activation of Hepatic Stellate Cells and Promote Fibrosis Resolution. Gastroenterology 2020, 158, 1728–1744.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidman, J.S.; Troutman, T.D.; Sakai, M.; Gola, A.; Spann, N.J.; Bennett, H.; Bruni, C.M.; Ouyang, Z.; Li, R.Z.; Sun, X.; et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 2020, 52, 1057–1074.e7. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Masoodi, M.; Gastaldelli, A.; Hyötyläinen, T.; Arretxe, E.; Alonso, C.; Gaggini, M.; Brosnan, J.; Anstee, Q.M.; Millet, O.; Ortiz, P.; et al. Metabolomics and lipidomics in NAFLD: Biomarkers and non-invasive diagnostic tests. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 835–856. [Google Scholar] [CrossRef] [PubMed]
- Spitz, F.; Furlong, E.E.M. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Heinz, S.; Romanoski, C.E.; Benner, C.; Glass, C.K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015, 16, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550. [Google Scholar] [CrossRef]
- Lam, M.T.Y.; Li, W.; Rosenfeld, M.G.; Glass, C.K. Enhancer RNAs and regulated transcriptional programs. Trends Biochem. Sci. 2014, 39, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeva, V. Analysis of genomic sequence motifs for deciphering transcription factor binding and transcriptional regulation in eukaryotic cells. Front. Genet. 2016, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siersbæk, M.; Varticovski, L.; Yang, S.; Baek, S.; Nielsen, R.; Mandrup, S.; Hager, G.L.; Chung, J.H.; Grøntved, L. High fat diet-induced changes of mouse hepatic transcription and enhancer activity can be reversed by subsequent weight loss. Sci. Rep. 2017, 7, 40220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchiyama, H.; Komatsu, K.-I.; Nakata, A.; Sato, K.; Mihara, Y.; Takaguri, A.; Nagashima, T.; Wakame, K. Global Liver Gene Expression Analysis on a Murine Hepatic Steatosis Model Treated with Mulberry (Morus alba L.) Leaf Powder. Anticancer Res. 2018, 38, 4305–4311. [Google Scholar] [CrossRef]
- Ganguly, S.; Muench, G.A.; Shang, L.; Rosenthal, S.B.; Rahman, G.; Wang, R.; Wang, Y.; Kwon, H.C.; Diomino, A.M.; Kisseleva, T.; et al. Nonalcoholic steatohepatitis and HCC in a hyperphagic mouse accelerated by western diet. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 891–920. [Google Scholar] [CrossRef]
- Leung, A.; Parks, B.W.; Du, J.; Trac, C.; Setten, R.; Chen, Y.; Brown, K.; Lusis, A.J.; Natarajan, R.; Schones, D.E. Open chromatin profiling in mice livers reveals unique chromatin variations induced by high fat diet. J. Biol. Chem. 2014, 289, 23557–23567. [Google Scholar] [CrossRef] [Green Version]
- Leung, A.; Trac, C.; Du, J.; Natarajan, R.; Schones, D.E. Persistent chromatin modifications induced by high fat diet. J. Biol. Chem. 2016, 291, 10446–10455. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Grimm, S.A.; Roberts, J.D.; Chrysovergis, K.; Wade, P.A. Alterations in promoter interaction landscape and transcriptional network underlying metabolic adaptation to diet. Nat. Commun. 2020, 11, 962. [Google Scholar] [CrossRef] [Green Version]
- Guan, D.; Xiong, Y.; Borck, P.C.; Jang, C.; Doulias, P.-T.; Papazyan, R.; Fang, B.; Jiang, C.; Zhang, Y.; Briggs, E.R.; et al. Diet-Induced Circadian Enhancer Remodeling Synchronizes Opposing Hepatic Lipid Metabolic Processes. Cell 2018, 174, 831–842.e12. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zalzala, M.; Xu, J.; Li, Y.; Yin, L.; Zhang, Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat. Commun. 2015, 6, 7466. [Google Scholar] [CrossRef]
- Yang, T.; Poenisch, M.; Khanal, R.; Hu, Q.; Dai, Z.; Li, R.; Song, G.; Yuan, Q.; Yao, Q.; Shen, X.; et al. Therapeutic HNF4A mRNA attenuates liver fibrosis in a preclinical model. J. Hepatol. 2021, 75, 1420–1433. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.Y.; Yin, C.; Hou, J.L.; Zeng, X.; Chen, Y.X.; Zhong, W.; Hu, P.F.; Deng, X.; Tan, Y.X.; Zhang, J.P.; et al. Hepatocyte nuclear factor 4alpha attenuates hepatic fibrosis in rats. Gut 2010, 59, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Bell, A.; Brooks, J.M.; Setoyama, K.; Melis, M.; Han, B.; Fukumitsu, K.; Handa, K.; Tian, J.; Kaestner, K.H.; et al. Resetting the transcription factor network reverses terminal chronic hepatic failure. J. Clin. Investig. 2015, 125, 1533–1544. [Google Scholar] [CrossRef] [Green Version]
- Stefflova, K.; Thybert, D.; Wilson, M.D.; Streeter, I.; Aleksic, J.; Karagianni, P.; Brazma, A.; Adams, D.J.; Talianidis, I.; Marioni, J.C.; et al. Cooperativity and rapid evolution of cobound transcription factors in closely related mammals. Cell 2013, 154, 530–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, T.A.; Bereshchenko, O.; Garcia-Silva, S.; Ermakova, O.; Kurz, E.; Mandrup, S.; Porse, B.T.; Nerlov, C. Distinct C/EBPalpha motifs regulate lipogenic and gluconeogenic gene expression in vivo. EMBO J. 2007, 26, 1081–1093. [Google Scholar] [CrossRef] [Green Version]
- Millward, C.A.; Heaney, J.D.; Sinasac, D.S.; Chu, E.C.; Bederman, I.R.; Gilge, D.A.; Previs, S.F.; Croniger, C.M. Mice with a deletion in the gene for CCAAT/enhancer-binding protein beta are protected against diet-induced obesity. Diabetes 2007, 56, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Iakova, P.; Breaux, M.; Sullivan, E.; Jawanmardi, N.; Chen, D.; Jiang, Y.; Medrano, E.M.; Timchenko, N.A. Increased expression of enzymes of triglyceride synthesis is essential for the development of hepatic steatosis. Cell Rep. 2013, 3, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; MacLean, P.S.; You, H.; Schaack, J.; Shao, J. knocking down liver ccaat/enhancer-binding protein alpha by adenovirus-transduced silent interfering ribonucleic acid improves hepatic gluconeogenesis and lipid homeostasis in db/db mice. Endocrinology 2006, 147, 3060–3069. [Google Scholar] [CrossRef] [Green Version]
- Matsusue, K.; Gavrilova, O.; Lambert, G.; Brewer, H.B.; Ward, J.M.; Inoue, Y.; LeRoith, D.; Gonzalez, F.J. Hepatic CCAAT/enhancer binding protein alpha mediates induction of lipogenesis and regulation of glucose homeostasis in leptin-deficient mice. Mol. Endocrinol. 2004, 18, 2751–2764. [Google Scholar] [CrossRef]
- Wang, Y.; Viscarra, J.; Kim, S.-J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Guichard, C.; Ferré, P.; Foufelle, F. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc. Natl. Acad. Sci. USA 1999, 96, 12737–12742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foretz, M.; Pacot, C.; Dugail, I.; Lemarchand, P.; Guichard, C.; Le Lièpvre, X.; Berthelier-Lubrano, C.; Spiegelman, B.; Kim, J.B.; Ferré, P.; et al. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol. Cell. Biol. 1999, 19, 3760–3768. [Google Scholar] [CrossRef] [Green Version]
- Ponugoti, B.; Kim, D.-H.; Xiao, Z.; Smith, Z.; Miao, J.; Zang, M.; Wu, S.-Y.; Chiang, C.-M.; Veenstra, T.D.; Kemper, J.K. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 2010, 285, 33959–33970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohjima, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; Enjoji, M.; et al. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2008, 21, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.-X.; Shen, W.; Sun, H. Effects of nuclear receptor FXR on the regulation of liver lipid metabolism in patients with non-alcoholic fatty liver disease. Hepatol Int. 2010, 4, 741–748. [Google Scholar] [CrossRef] [Green Version]
- Knebel, B.; Haas, J.; Hartwig, S.; Jacob, S.; Köllmer, C.; Nitzgen, U.; Muller-Wieland, D.; Kotzka, J. Liver-specific expression of transcriptionally active SREBP-1c is associated with fatty liver and increased visceral fat mass. PLoS ONE 2012, 7, e31812. [Google Scholar] [CrossRef] [Green Version]
- Hua, X.; Nohturfft, A.; Goldstein, J.L.; Brown, M.S. Sterol resistance in CHO cells traced to point mutation in SREBP cleavage-activating protein. Cell 1996, 87, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Baker, S.S.; Liu, W.; Tao, M.-H.; Patel, R.; Nowak, N.J.; Baker, R.D. Lipid in the livers of adolescents with nonalcoholic steatohepatitis: Combined effects of pathways on steatosis. Metab. Clin. Exp. 2011, 60, 1001–1011. [Google Scholar] [CrossRef]
- Patsouris, D.; Reddy, J.K.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology 2006, 147, 1508–1516. [Google Scholar] [CrossRef] [Green Version]
- Régnier, M.; Polizzi, A.; Smati, S.; Lukowicz, C.; Fougerat, A.; Lippi, Y.; Fouché, E.; Lasserre, F.; Naylies, C.; Bétoulières, C.; et al. Hepatocyte-specific deletion of Pparα promotes NAFLD in the context of obesity. Sci. Rep. 2020, 10, 6489. [Google Scholar] [CrossRef]
- Zhang, N.; Lu, Y.; Shen, X.; Bao, Y.; Cheng, J.; Chen, L.; Li, B.; Zhang, Q. Fenofibrate treatment attenuated chronic endoplasmic reticulum stress in the liver of nonalcoholic fatty liver disease mice. Pharmacology 2015, 95, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Shiri-Sverdlov, R.; Wouters, K.; van Gorp, P.J.; Gijbels, M.J.; Noel, B.; Buffat, L.; Staels, B.; Maeda, N.; van Bilsen, M.; Hofker, M.H. Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J. Hepatol. 2006, 44, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Haleim, E.A.; Bahgat, A.K.; Saleh, S. Resveratrol and fenofibrate ameliorate fructose-induced nonalcoholic steatohepatitis by modulation of genes expression. World J. Gastroenterol. 2016, 22, 2931–2948. [Google Scholar] [CrossRef]
- Brosch, M.; Kattler, K.; Herrmann, A.; von Schönfels, W.; Nordström, K.; Seehofer, D.; Damm, G.; Becker, T.; Zeissig, S.; Nehring, S.; et al. Epigenomic map of human liver reveals principles of zonated morphogenic and metabolic control. Nat. Commun. 2018, 9, 4150. [Google Scholar] [CrossRef]
- Loft, A.; Alfaro, A.J.; Schmidt, S.F.; Pedersen, F.B.; Terkelsen, M.K.; Puglia, M.; Chow, K.K.; Feuchtinger, A.; Troullinaki, M.; Maida, A.; et al. Liver-fibrosis-activated transcriptional networks govern hepatocyte reprogramming and intra-hepatic communication. Cell Metab. 2021, 33, 1685–1700.e9. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Troutman, T.D.; Seidman, J.S.; Ouyang, Z.; Spann, N.J.; Abe, Y.; Ego, K.M.; Bruni, C.M.; Deng, Z.; Schlachetzki, J.C.M.; et al. Liver-Derived Signals Sequentially Reprogram Myeloid Enhancers to Initiate and Maintain Kupffer Cell Identity. Immunity 2019, 51, 655–670.e8. [Google Scholar] [CrossRef] [PubMed]
- Troutman, T.D.; Bennett, H.; Sakai, M.; Seidman, J.S.; Heinz, S.; Glass, C.K. Purification of mouse hepatic non-parenchymal cells or nuclei for use in ChIP-seq and other next-generation sequencing approaches. STAR Protoc. 2021, 2, 100363. [Google Scholar] [CrossRef]
- Denisenko, E.; Guo, B.B.; Jones, M.; Hou, R.; de Kock, L.; Lassmann, T.; Poppe, D.; Clément, O.; Simmons, R.K.; Lister, R.; et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 2020, 21, 130. [Google Scholar] [CrossRef]
- Binek, A.; Rojo, D.; Godzien, J.; Rupérez, F.J.; Nuñez, V.; Jorge, I.; Ricote, M.; Vázquez, J.; Barbas, C. Flow cytometry has a significant impact on the cellular metabolome. J. Proteome Res. 2019, 18, 169–181. [Google Scholar] [CrossRef]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.-Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell 2019, 75, 644–660.e5. [Google Scholar] [CrossRef]
- Mo, A.; Mukamel, E.A.; Davis, F.P.; Luo, C.; Henry, G.L.; Picard, S.; Urich, M.A.; Nery, J.R.; Sejnowski, T.J.; Lister, R.; et al. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 2015, 86, 1369–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, H.C.; Tsai, L.T.-Y.; Lyubetskaya, A.; Tenen, D.; Kumari, M.; Rosen, E.D. Simultaneous Transcriptional and Epigenomic Profiling from Specific Cell Types within Heterogeneous Tissues In Vivo. Cell Rep. 2017, 18, 1048–1061. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [Green Version]
- Hasenfuss, S.C.; Bakiri, L.; Thomsen, M.K.; Williams, E.G.; Auwerx, J.; Wagner, E.F. Regulation of steatohepatitis and PPARγ signaling by distinct AP-1 dimers. Cell Metab. 2014, 19, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.-I.; Hoshida, Y.; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef]
- Kita, Y.; Takamura, T.; Misu, H.; Ota, T.; Kurita, S.; Takeshita, Y.; Uno, M.; Matsuzawa-Nagata, N.; Kato, K.-I.; Ando, H.; et al. Metformin prevents and reverses inflammation in a non-diabetic mouse model of nonalcoholic steatohepatitis. PLoS ONE 2012, 7, e43056. [Google Scholar] [CrossRef] [PubMed]
- Frades, I.; Andreasson, E.; Mato, J.M.; Alexandersson, E.; Matthiesen, R.; Martínez-Chantar, M.L. Integrative genomic signatures of hepatocellular carcinoma derived from nonalcoholic Fatty liver disease. PLoS ONE 2015, 10, e0124544. [Google Scholar] [CrossRef]
- du Plessis, J.; van Pelt, J.; Korf, H.; Mathieu, C.; van der Schueren, B.; Lannoo, M.; Oyen, T.; Topal, B.; Fetter, G.; Nayler, S.; et al. Association of adipose tissue inflammation with histologic severity of nonalcoholic fatty liver disease. Gastroenterology 2015, 149, 635–648.e14. [Google Scholar] [CrossRef] [Green Version]
- Moylan, C.A.; Pang, H.; Dellinger, A.; Suzuki, A.; Garrett, M.E.; Guy, C.D.; Murphy, S.K.; Ashley-Koch, A.E.; Choi, S.S.; Michelotti, G.A.; et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014, 59, 471–482. [Google Scholar] [CrossRef]
- Ahrens, M.; Ammerpohl, O.; von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; Wang, Q.; Wang, S.; Zhang, J.; Liu, T.; Guo, L.; Yu, Y.; Lin, J.D. Mapping the molecular signatures of diet-induced NASH and its regulation by the hepatokine Tsukushi. Mol. Metab. 2019, 20, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Sladek, F.M. Orphan receptor HNF-4 and liver-specific gene expression. Receptor 1993, 3, 223–232. [Google Scholar] [PubMed]
- Wang, K.; Holterman, A.-X. Pathophysiologic role of hepatocyte nuclear factor 6. Cell Signal. 2012, 24, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Yoshida, Y.; Hughes, D.E.; Costa, R.H. Increased expression of hepatocyte nuclear factor 6 stimulates hepatocyte proliferation during mouse liver regeneration. Gastroenterology 2006, 130, 1283–1300. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Yu, J.; Wang, L.; Tang, Y.; Zhou, Q.; Ji, S.; Wang, Y.; Santos, L.; Haeusler, R.A.; Que, J.; et al. Inhibition of PU.1 ameliorates metabolic dysfunction and non-alcoholic steatohepatitis. J. Hepatol. 2020, 73, 361–370. [Google Scholar] [CrossRef]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [Green Version]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [Green Version]
- Bennett, H.; Troutman, T.D.; Sakai, M.; Glass, C.K. Epigenetic regulation of kupffer cell function in health and disease. Front. Immunol. 2020, 11, 609618. [Google Scholar] [CrossRef]
- Bahr, M.J.; Vincent, K.J.; Arthur, M.J.; Fowler, A.V.; Smart, D.E.; Wright, M.C.; Clark, I.M.; Benyon, R.C.; Iredale, J.P.; Mann, D.A. Control of the tissue inhibitor of metalloproteinases-1 promoter in culture-activated rat hepatic stellate cells: Regulation by activator protein-1 DNA binding proteins. Hepatology 1999, 29, 839–848. [Google Scholar] [CrossRef]
- Smart, D.E.; Vincent, K.J.; Arthur, M.J.; Eickelberg, O.; Castellazzi, M.; Mann, J.; Mann, D.A. JunD regulates transcription of the tissue inhibitor of metalloproteinases-1 and interleukin-6 genes in activated hepatic stellate cells. J. Biol. Chem. 2001, 276, 24414–24421. [Google Scholar] [CrossRef] [Green Version]
- Park, E.Y.; Shin, S.M.; Ma, C.J.; Kim, Y.C.; Kim, S.G. meso-dihydroguaiaretic acid from Machilus thunbergii down-regulates TGF-beta1 gene expression in activated hepatic stellate cells via inhibition of AP-1 activity. Planta Med. 2005, 71, 393–398. [Google Scholar] [CrossRef]
- Mann, J.; Mann, D.A. Transcriptional regulation of hepatic stellate cells. Adv. Drug Deliv. Rev. 2009, 61, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, K.; Pritchett, J.; Llewellyn, J.; Mullan, A.F.; Athwal, V.S.; Dobie, R.; Harvey, E.; Zeef, L.; Farrow, S.; Streuli, C.; et al. PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat. Commun. 2016, 7, 12502. [Google Scholar] [CrossRef]
- Mannaerts, I.; Leite, S.B.; Verhulst, S.; Claerhout, S.; Eysackers, N.; Thoen, L.F.R.; Hoorens, A.; Reynaert, H.; Halder, G.; van Grunsven, L.A. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 2015, 63, 679–688. [Google Scholar] [CrossRef]
- Halpern, K.B.; Shenhav, R.; Matcovitch-Natan, O.; Toth, B.; Lemze, D.; Golan, M.; Massasa, E.E.; Baydatch, S.; Landen, S.; Moor, A.E.; et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 2017, 542, 352–356. [Google Scholar] [CrossRef]
- Su, T.; Yang, Y.; Lai, S.; Jeong, J.; Jung, Y.; McConnell, M.; Utsumi, T.; Iwakiri, Y. Single-Cell Transcriptomics Reveals Zone-Specific Alterations of Liver Sinusoidal Endothelial Cells in Cirrhosis. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 1139–1161. [Google Scholar] [CrossRef]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef] [PubMed]
- Daemen, S.; Gainullina, A.; Kalugotla, G.; He, L.; Chan, M.M.; Beals, J.W.; Liss, K.H.; Klein, S.; Feldstein, A.E.; Finck, B.N.; et al. Dynamic shifts in the composition of resident and recruited macrophages influence tissue remodeling in NASH. Cell Rep. 2021, 34, 108626. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, B.A.; Zebroski, H.L.; Finkbeiner, C.R.; Chitsazan, A.D.; Beach, K.E.; Sen, N.; Zhang, R.C.; Bermingham-McDonogh, O. Novel cell types and developmental lineages revealed by single-cell RNA-seq analysis of the mouse crista ampullaris. eLife 2021, 10, e60108. [Google Scholar] [CrossRef]
- MacParland, S.A.; Liu, J.C.; Ma, X.-Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aizarani, N.; Saviano, A.; Sagar; Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grün, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Shema, E.; Bernstein, B.E.; Buenrostro, J.D. Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat. Genet. 2019, 51, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.L.; Schilling, J.D.; King, K.R.; Feldstein, A.E. The Power of Single-Cell Analysis for the Study of Liver Pathobiology. Hepatology 2021, 73, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Saviano, A.; Henderson, N.C.; Baumert, T.F. Single-cell genomics and spatial transcriptomics: Discovery of novel cell states and cellular interactions in liver physiology and disease biology. J. Hepatol. 2020, 73, 1219–1230. [Google Scholar] [CrossRef]
- Ramachandran, P.; Matchett, K.P.; Dobie, R.; Wilson-Kanamori, J.R.; Henderson, N.C. Single-cell technologies in hepatology: New insights into liver biology and disease pathogenesis. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Remmerie, A.; Martens, L.; Scott, C.L. Macrophage subsets in obesity, aligning the liver and adipose tissue. Front. Endocrinol. 2020, 11, 259. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Cusanovich, D.A.; Daza, R.; Adey, A.; Pliner, H.A.; Christiansen, L.; Gunderson, K.L.; Steemers, F.J.; Trapnell, C.; Shendure, J. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 2015, 348, 910–914. [Google Scholar] [CrossRef] [Green Version]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef]
- Pliner, H.A.; Packer, J.S.; McFaline-Figueroa, J.L.; Cusanovich, D.A.; Daza, R.M.; Aghamirzaie, D.; Srivatsan, S.; Qiu, X.; Jackson, D.; Minkina, A.; et al. Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol. Cell 2018, 71, 858–871.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, R.; Preissl, S.; Li, Y.; Hou, X.; Lucero, J.; Wang, X.; Motamedi, A.; Shiau, A.K.; Zhou, X.; Xie, F.; et al. Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat. Commun. 2021, 12, 1337. [Google Scholar] [CrossRef] [PubMed]
- Granja, J.M.; Corces, M.R.; Pierce, S.E.; Bagdatli, S.T.; Choudhry, H.; Chang, H.Y.; Greenleaf, W.J. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021, 53, 403–411. [Google Scholar] [CrossRef]
- Stuart, T.; Srivastava, A.; Madad, S.; Lareau, C.A.; Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 2021, 18, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Trevino, A.E.; Müller, F.; Andersen, J.; Sundaram, L.; Kathiria, A.; Shcherbina, A.; Farh, K.; Chang, H.Y.; Pașca, A.M.; Kundaje, A.; et al. Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell 2021, 184, 5053–5069.e23. [Google Scholar] [CrossRef]
- Allaway, K.C.; Gabitto, M.I.; Wapinski, O.; Saldi, G.; Wang, C.-Y.; Bandler, R.C.; Wu, S.J.; Bonneau, R.; Fishell, G. Genetic and epigenetic coordination of cortical interneuron development. Nature 2021, 597, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, M.; Giannese, F.; Lazarević, D.; Giansanti, V.; Rosano, D.; Monzani, S.; Catalano, I.; Grassi, E.; Zanella, E.R.; Botrugno, O.A.; et al. Chromatin Velocity reveals epigenetic dynamics by single-cell profiling of heterochromatin and euchromatin. Nat. Biotechnol. 2022, 40, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Rotem, A.; Ram, O.; Shoresh, N.; Sperling, R.A.; Goren, A.; Weitz, D.A.; Bernstein, B.E. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 2015, 33, 1165–1172. [Google Scholar] [CrossRef]
- Grosselin, K.; Durand, A.; Marsolier, J.; Poitou, A.; Marangoni, E.; Nemati, F.; Dahmani, A.; Lameiras, S.; Reyal, F.; Frenoy, O.; et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet. 2019, 51, 1060–1066. [Google Scholar] [CrossRef]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [CrossRef] [Green Version]
- Skene, P.J.; Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 2017, 6, e21856. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Henikoff, J.G.; Henikoff, S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 2018, 13, 1006–1019. [Google Scholar] [CrossRef] [PubMed]
- Gopalan, S.; Wang, Y.; Harper, N.W.; Garber, M.; Fazzio, T.G. Simultaneous profiling of multiple chromatin proteins in the same cells. Mol. Cell 2021, 81, 4736–4746.e5. [Google Scholar] [CrossRef] [PubMed]
- Bartosovic, M.; Kabbe, M.; Castelo-Branco, G. Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat. Biotechnol. 2021, 39, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flyamer, I.M.; Gassler, J.; Imakaev, M.; Brandão, H.B.; Ulianov, S.V.; Abdennur, N.; Razin, S.V.; Mirny, L.A.; Tachibana-Konwalski, K. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 2017, 544, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Nagano, T.; Lubling, Y.; Várnai, C.; Dudley, C.; Leung, W.; Baran, Y.; Mendelson Cohen, N.; Wingett, S.; Fraser, P.; Tanay, A. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 2017, 547, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Ramani, V.; Deng, X.; Qiu, R.; Gunderson, K.L.; Steemers, F.J.; Disteche, C.M.; Noble, W.S.; Duan, Z.; Shendure, J. Massively multiplex single-cell Hi-C. Nat. Methods 2017, 14, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Xing, D.; Chang, C.-H.; Li, H.; Xie, X.S. Three-dimensional genome structures of single diploid human cells. Science 2018, 361, 924–928. [Google Scholar] [CrossRef] [Green Version]
- Ramani, V.; Deng, X.; Qiu, R.; Lee, C.; Disteche, C.M.; Noble, W.S.; Shendure, J.; Duan, Z. Sci-Hi-C: A single-cell Hi-C method for mapping 3D genome organization in large number of single cells. Methods 2020, 170, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Galitsyna, A.A.; Gelfand, M.S. Single-cell Hi-C data analysis: Safety in numbers. Brief. Bioinform. 2021, 22, bbab316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhou, T.; Ma, J. Multiscale and integrative single-cell Hi-C analysis with Higashi. Nat. Biotechnol. 2022, 40, 254–261. [Google Scholar] [CrossRef]
- Kim, H.-J.; Yardımcı, G.G.; Bonora, G.; Ramani, V.; Liu, J.; Qiu, R.; Lee, C.; Hesson, J.; Ware, C.B.; Shendure, J.; et al. Capturing cell type-specific chromatin compartment patterns by applying topic modeling to single-cell Hi-C data. PLoS Comput. Biol. 2020, 16, e1008173. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lin, D.; Yardimci, G.G.; Noble, W.S. Unsupervised embedding of single-cell Hi-C data. Bioinformatics 2018, 34, i96–i104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Ma, J.; Chen, Y.; Cheng, C.; Bao, B.; Peng, J.; Sejnowski, T.J.; Dixon, J.R.; Ecker, J.R. Robust single-cell Hi-C clustering by convolution- and random-walk-based imputation. Proc. Natl. Acad. Sci. USA 2019, 116, 14011–14018. [Google Scholar] [CrossRef] [Green Version]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef] [Green Version]
- Peterson, V.M.; Zhang, K.X.; Kumar, N.; Wong, J.; Li, L.; Wilson, D.C.; Moore, R.; McClanahan, T.K.; Sadekova, S.; Klappenbach, J.A. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 2017, 35, 936–939. [Google Scholar] [CrossRef]
- Chen, X.; Litzenburger, U.M.; Wei, Y.; Schep, A.N.; LaGory, E.L.; Choudhry, H.; Giaccia, A.J.; Greenleaf, W.J.; Chang, H.Y. Joint single-cell DNA accessibility and protein epitope profiling reveals environmental regulation of epigenomic heterogeneity. Nat. Commun. 2018, 9, 4590. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.F.; Parks, B.; Kathiria, A.; Ober-Reynolds, B.; Goronzy, J.; Greenleaf, W. NEAT-seq: Simultaneous profiling of intra-nuclear proteins, chromatin accessibility, and gene expression in single cells. BioRxiv 2021. [Google Scholar] [CrossRef]
- Seydel, C. Single-cell metabolomics hits its stride. Nat. Methods 2021, 18, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Perkel, J.M. Single-cell proteomics takes centre stage. Nature 2021, 597, 580–582. [Google Scholar] [CrossRef] [PubMed]
- Rappez, L.; Stadler, M.; Triana, S.; Gathungu, R.M.; Ovchinnikova, K.; Phapale, P.; Heikenwalder, M.; Alexandrov, T. SpaceM reveals metabolic states of single cells. Nat. Methods 2021, 18, 799–805. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dam, T.V.; Toft, N.I.; Grøntved, L. Cell-Type Resolved Insights into the Cis-Regulatory Genome of NAFLD. Cells 2022, 11, 870. https://doi.org/10.3390/cells11050870
Dam TV, Toft NI, Grøntved L. Cell-Type Resolved Insights into the Cis-Regulatory Genome of NAFLD. Cells. 2022; 11(5):870. https://doi.org/10.3390/cells11050870
Chicago/Turabian StyleDam, Trine V., Nicolaj I. Toft, and Lars Grøntved. 2022. "Cell-Type Resolved Insights into the Cis-Regulatory Genome of NAFLD" Cells 11, no. 5: 870. https://doi.org/10.3390/cells11050870