
Failure of Alzheimer’s Mice Brain Resident Neural Precursor Cells in Supporting Microglia-Mediated Amyloid β Clearance


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Mice Genetic Screening
2.3. Isolation and Growth of Microglia
2.4. Isolation and Growth of Adult Mouse SVZ-Derived NPCs
2.5. Isolation and Expansion of Nestin-GFP SVZ-NPCs
2.6. Isolation and Growth of Newborn Mouse NPCs
2.7. Co-Culturing Microglia with NPCs
2.8. Time-Lapse
2.9. Phagocytosis and Degradation Assays
2.10. Histopathology
2.11. RNA Extraction and Real Time PCR
2.12. Analysis
3. Results
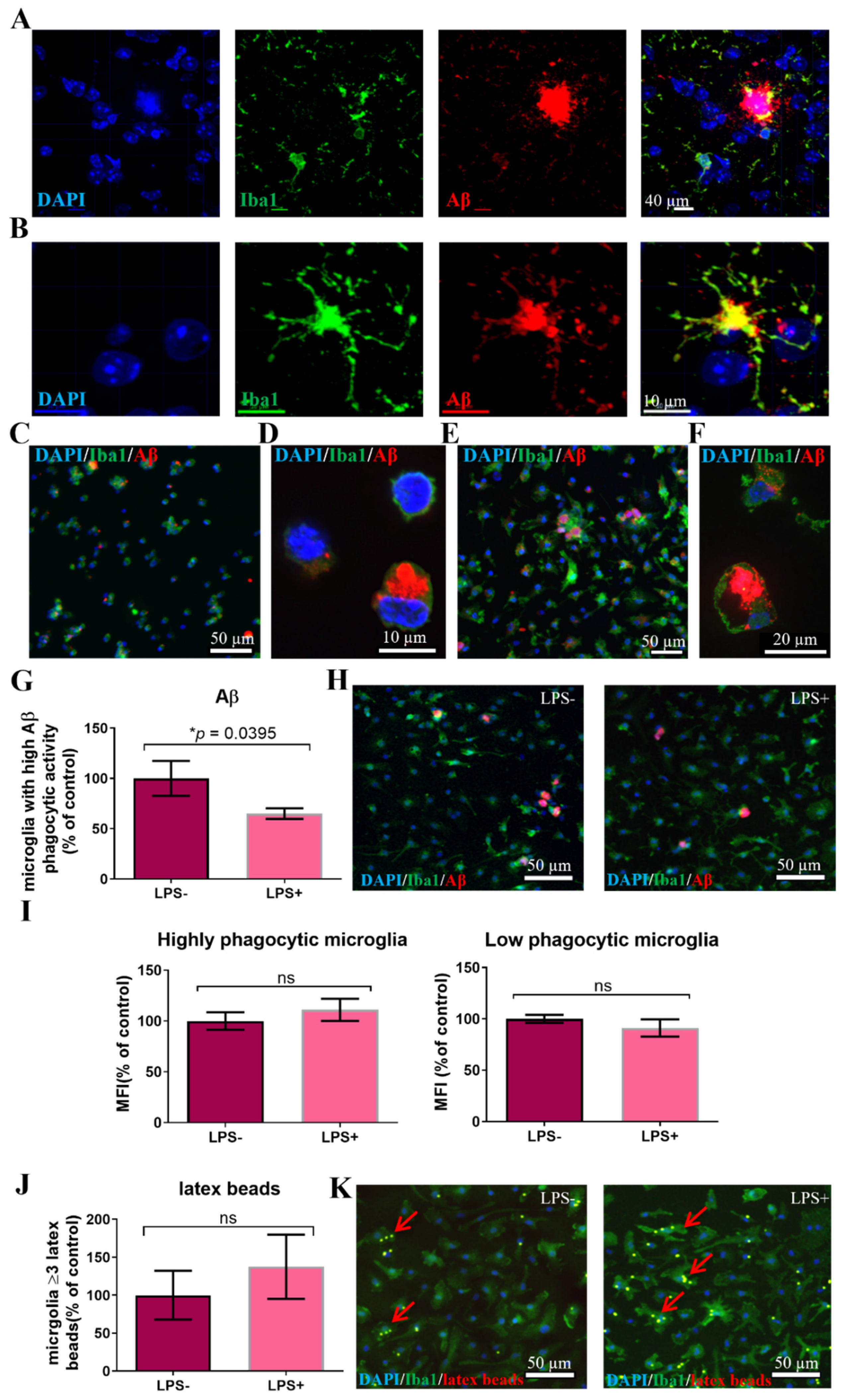
3.1. Microglia from Adult AD Brain Exhibit an Increased Latex Beads’ Phagocytosis
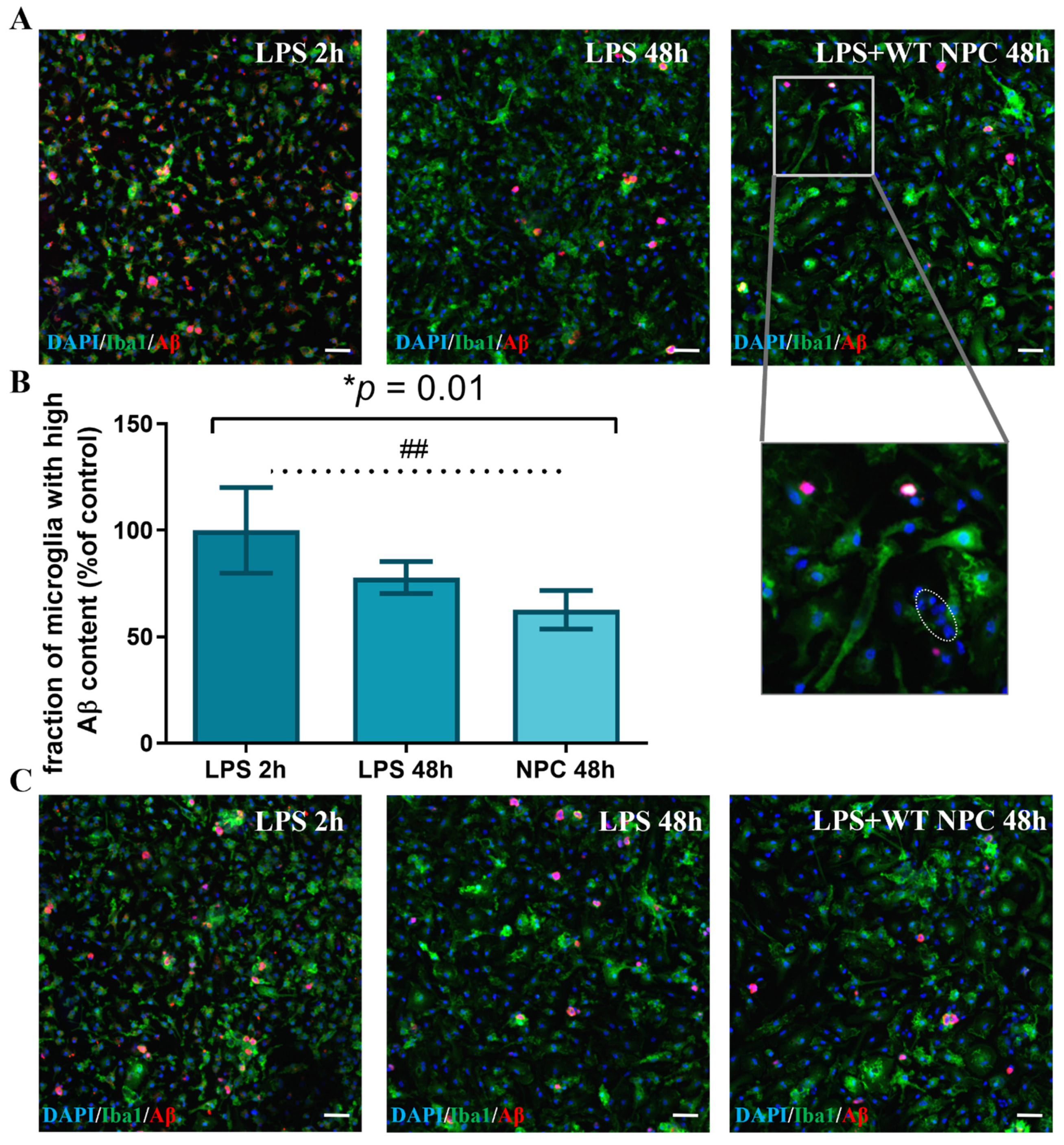
3.2. A Subpopulation of Microglia in the AD Brain Exhibits High Aβ Phagocytic Activity
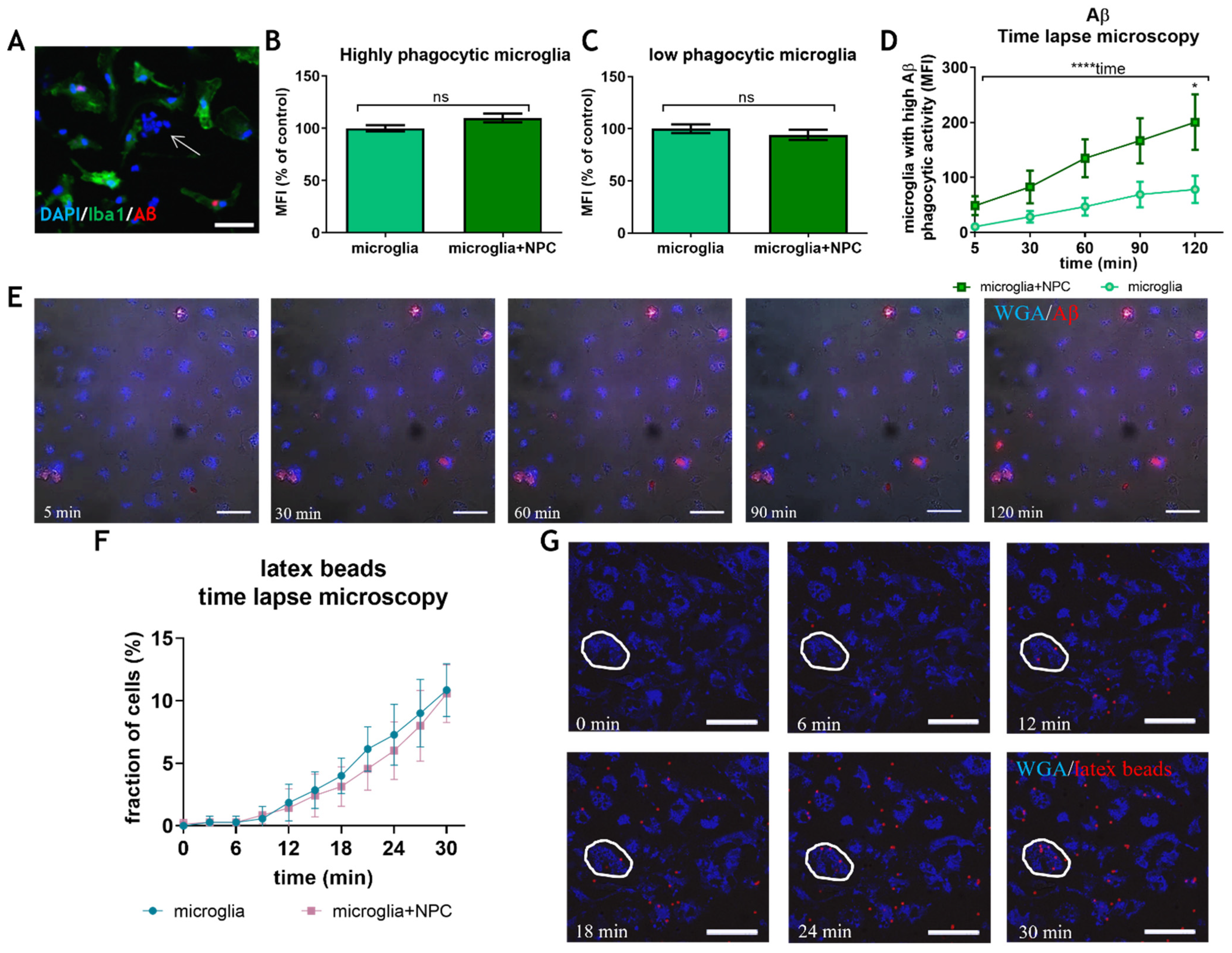
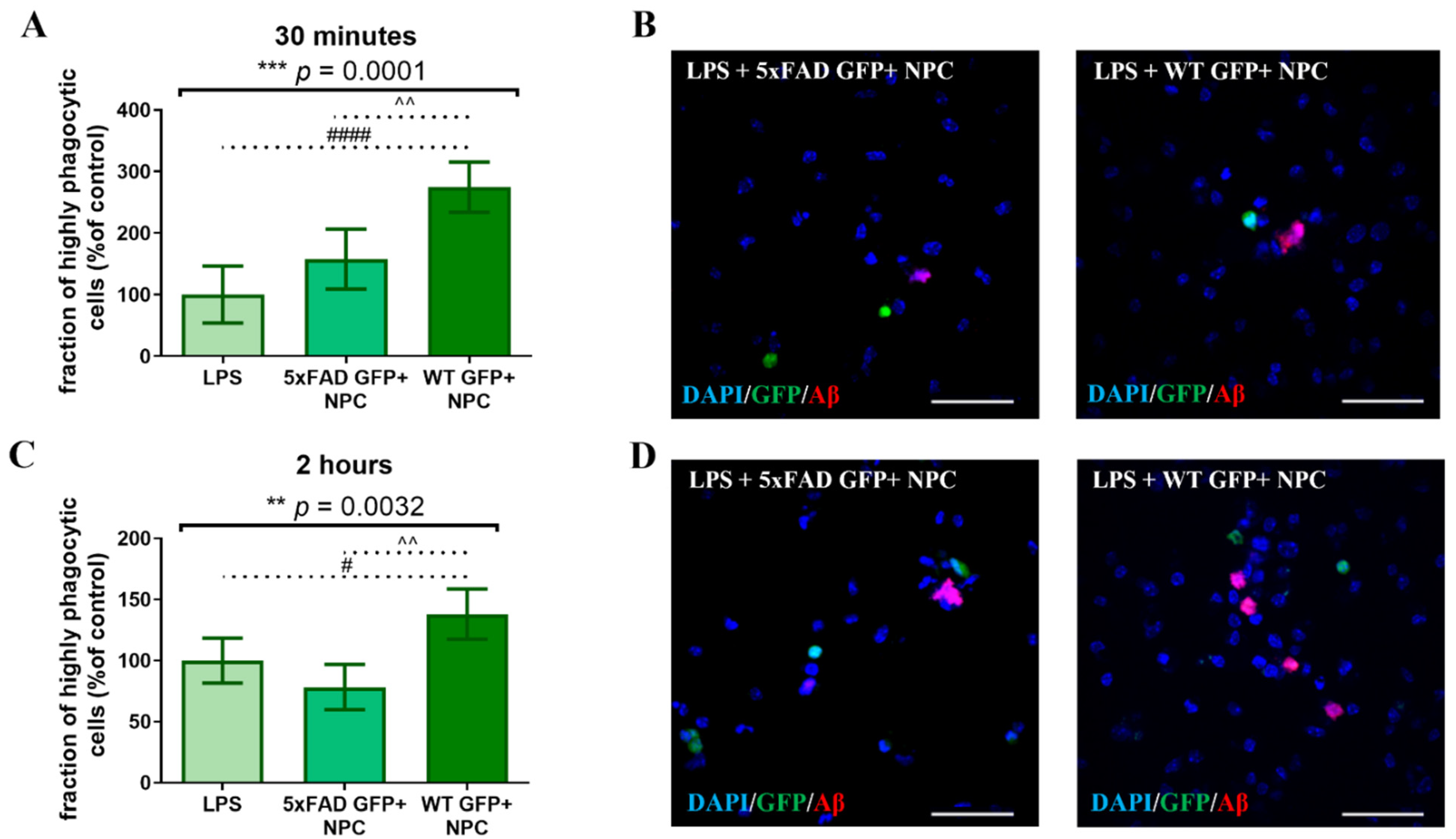
3.3. Neural Precursor Cells Induce an Increase in the Fraction of Microglia with High Aβ Phagocytic Activity
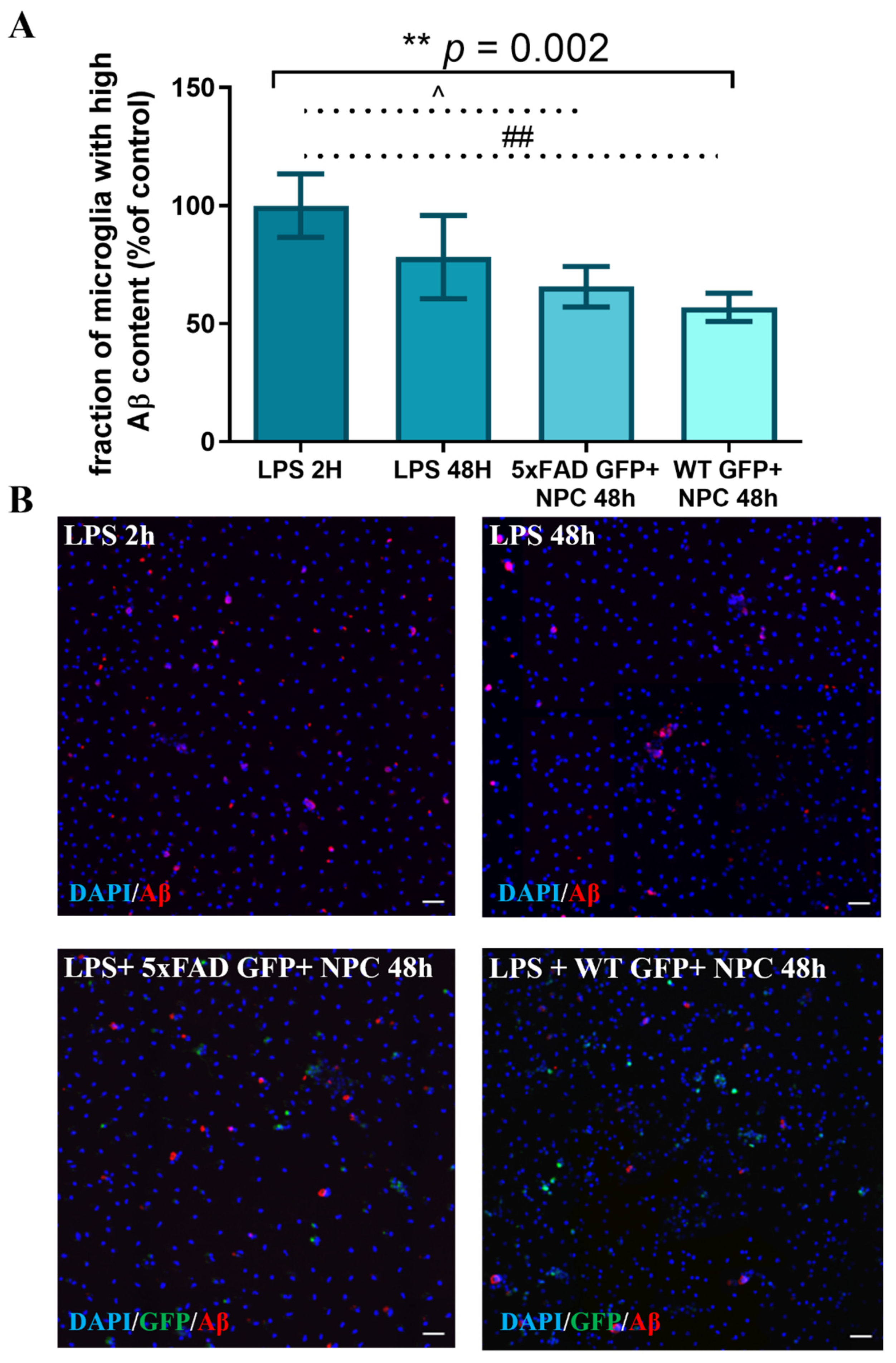
3.4. Wild Type NPCs Increase Aβ Phagocytosis by Microglia, but 5xFAD Subventricular Zone Extracts Enriched for NPCs Do Not
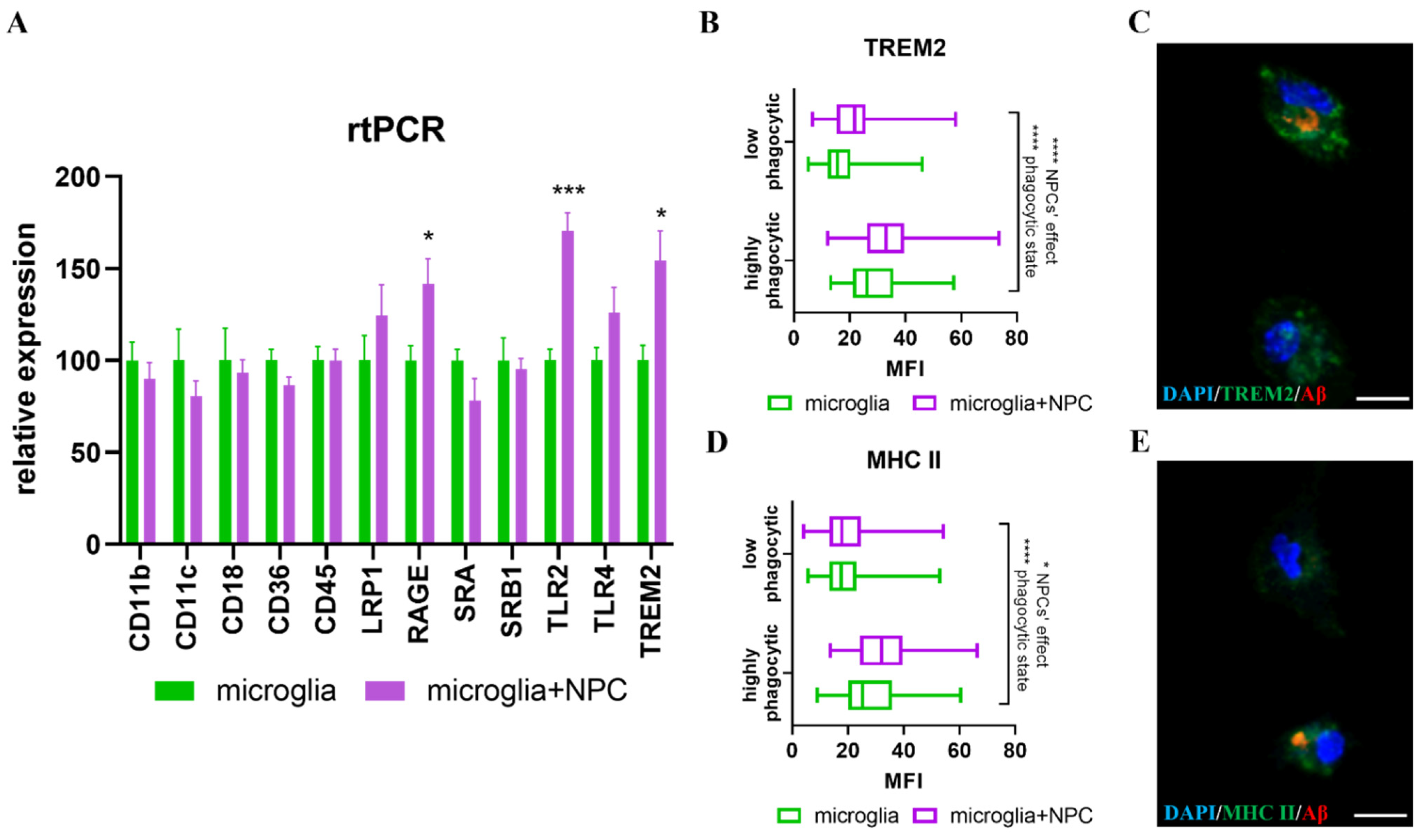
3.5. Isolated Nestin-GFP from Adult Wild Type NPCs, Induced an Increase in Microglial Aβ Phagocytosis, but Nestin-GFP 5xFAD NPCs Did Not
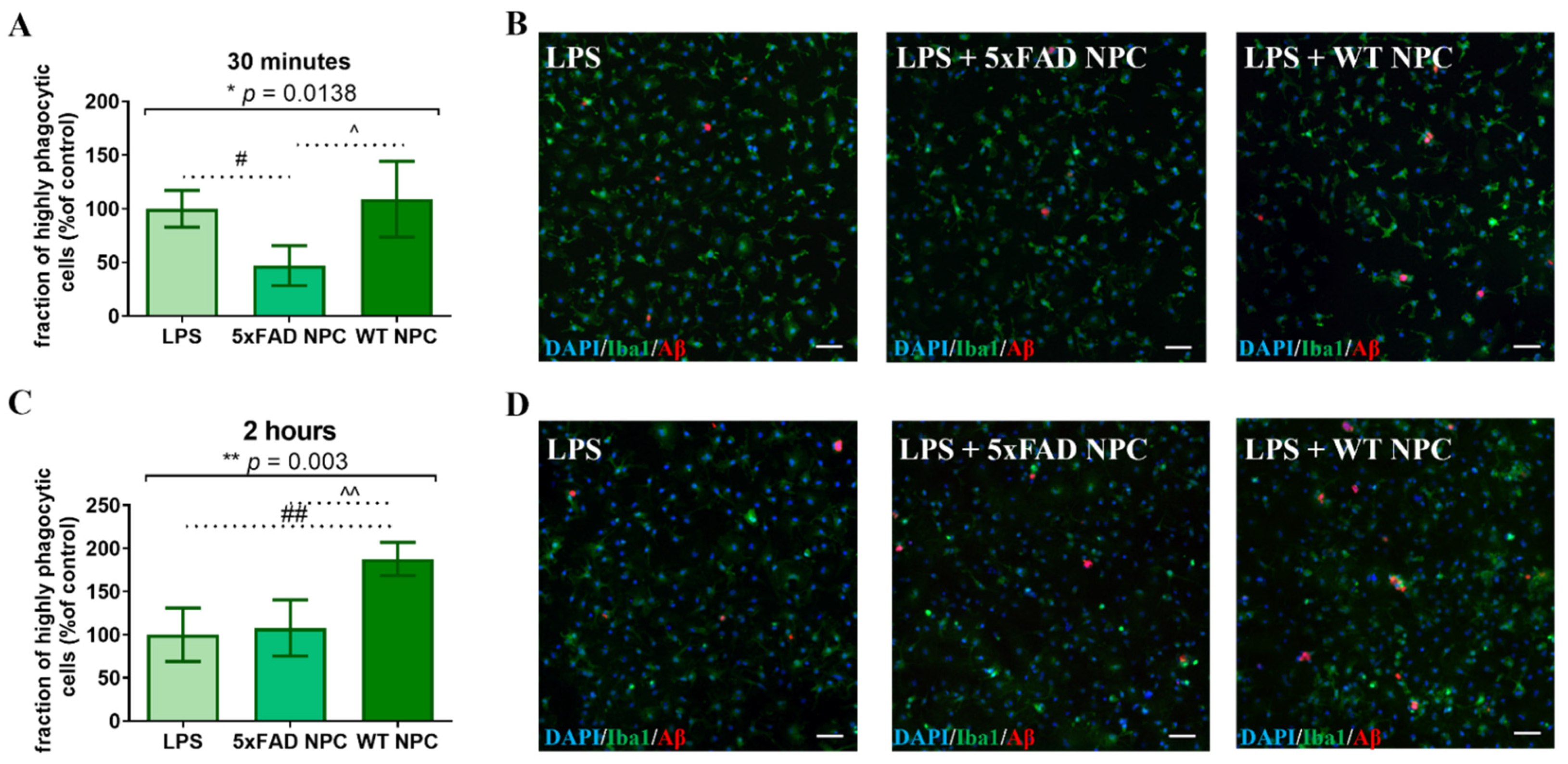
3.6. WT Newborn NPCs Increase Aβ Removal from 5xFAD Microglia
3.7. Nestin-GFP NPCs from Adult Mice Increases Aβ Removal from 5xFAD Microglia
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef]
- Fleisher, A.S.; Chen, K.; Quiroz, Y.T.; Jakimovich, L.J.; Gomez, M.G.; Langois, C.M.; Langbaum, J.B.; Ayutyanont, N.; Roontiva, A.; Thiyyagura, P.; et al. Florbetapir PET analysis of amyloid-beta deposition in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: A cross-sectional study. Lancet Neurol. 2012, 11, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Bartels, T.; De Schepper, S.; Hong, S. Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 2020, 370, 66–69. [Google Scholar] [CrossRef]
- Qin, Q.; Teng, Z.; Liu, C.; Li, Q.; Yin, Y.; Tang, Y. TREM2, microglia, and Alzheimer’s disease. Mech. Ageing Dev. 2021, 195, 111438. [Google Scholar] [CrossRef]
- Lee, C.Y.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef]
- Streit, W.J.; Khoshbouei, H.; Bechmann, I. Dystrophic microglia in late-onset Alzheimer’s disease. Glia 2020, 68, 845–854. [Google Scholar] [CrossRef]
- Einstein, O.; Ben-Hur, T. The changing face of neural stem cell therapy in neurologic diseases. Arch. Neurol. 2008, 65, 452–456. [Google Scholar] [CrossRef] [Green Version]
- Ottoboni, L.; De Feo, D.; Merlini, A.; Martino, G. Commonalities in immune modulation between mesenchymal stem cells (MSCs) and neural stem/precursor cells (NPCs). Immunol. Lett. 2015, 168, 228–239. [Google Scholar] [CrossRef]
- De Feo, D.; Merlini, A.; Laterza, C.; Martino, G. Neural stem cell transplantation in central nervous system disorders: From cell replacement to neuroprotection. Curr. Opin. Neurol. 2012, 25, 322–333. [Google Scholar] [CrossRef]
- Ben-Hur, T.; Fainstein, N.; Nishri, Y. Cell-based reparative therapies for multiple sclerosis. Curr. Neurol. Neurosci. Rep. 2013, 13, 397. [Google Scholar] [CrossRef]
- Ryu, J.K.; Cho, T.; Wang, Y.T.; McLarnon, J.G. Neural progenitor cells attenuate inflammatory reactivity and neuronal loss in an animal model of inflamed AD brain. J. Neuroinflamm. 2009, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wu, H.H.; Wang, Y.; Gu, G.J.; Zhang, W.; Xia, R. Neural stem cell transplantation decreases neuroinflammation in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2016, 136, 815–825. [Google Scholar] [CrossRef] [Green Version]
- McGinley, L.M.; Kashlan, O.N.; Bruno, E.S.; Chen, K.S.; Hayes, J.M.; Kashlan, S.R.; Raykin, J.; Johe, K.; Murphy, G.G.; Feldman, E.L. Human neural stem cell transplantation improves cognition in a murine model of Alzheimer’s disease. Sci. Rep. 2018, 8, 14776. [Google Scholar] [CrossRef]
- Zhang, W.; Gu, G.J.; Shen, X.; Zhang, Q.; Wang, G.M.; Wang, P.J. Neural stem cell transplantation enhances mitochondrial biogenesis in a transgenic mouse model of Alzheimer’s disease-like pathology. Neurobiol. Aging 2015, 36, 1282–1292. [Google Scholar] [CrossRef]
- Kim, J.A.; Ha, S.; Shin, K.Y.; Kim, S.; Lee, K.J.; Chong, Y.H.; Chang, K.A.; Suh, Y.H. Neural stem cell transplantation at critical period improves learning and memory through restoring synaptic impairment in Alzheimer’s disease mouse model. Cell Death Dis. 2015, 6, e1789. [Google Scholar] [CrossRef] [Green Version]
- Blurton-Jones, M.; Kitazawa, M.; Martinez-Coria, H.; Castello, N.A.; Muller, F.J.; Loring, J.F.; Yamasaki, T.R.; Poon, W.W.; Green, K.N.; LaFerla, F.M. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc. Natl. Acad. Sci. USA 2009, 106, 13594–13599. [Google Scholar] [CrossRef] [Green Version]
- Fainstein, N.; Dan-Goor, N.; Ben-Hur, T. Resident brain neural precursor cells develop age-dependent loss of therapeutic functions in Alzheimer’s mice. Neurobiol. Aging 2018, 72, 40–52. [Google Scholar] [CrossRef]
- Mosher, K.I.; Andres, R.H.; Fukuhara, T.; Bieri, G.; Hasegawa-Moriyama, M.; He, Y.; Guzman, R.; Wyss-Coray, T. Neural progenitor cells regulate microglia functions and activity. Nat. Neurosci. 2012, 15, 1485–1487. [Google Scholar] [CrossRef]
- De Feo, D.; Merlini, A.; Brambilla, E.; Ottoboni, L.; Laterza, C.; Menon, R.; Srinivasan, S.; Farina, C.; Garcia Manteiga, J.M.; Butti, E.; et al. Neural precursor cell-secreted TGF-beta2 redirects inflammatory monocyte-derived cells in CNS autoimmunity. J. Clin. Investig. 2017, 127, 3937–3953. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Vicario, N.; Costa, A.S.H.; Kwok, C.K.; Leonardi, T.; Booty, L.M.; Bicci, I.; Balzarotti, B.; Volpe, G.; et al. Macrophage-Derived Extracellular Succinate Licenses Neural Stem Cells to Suppress Chronic Neuroinflammation. Cell Stem Cell 2018, 22, 355–368.e313. [Google Scholar] [CrossRef] [Green Version]
- Bian, B.; Zhao, C.; He, X.; Gong, Y.; Ren, C.; Ge, L.; Zeng, Y.; Li, Q.; Chen, M.; Weng, C.; et al. Exosomes derived from neural progenitor cells preserve photoreceptors during retinal degeneration by inactivating microglia. J. Extracell. Vesicles 2020, 9, 1748931. [Google Scholar] [CrossRef] [Green Version]
- Talaveron, R.; Matarredona, E.R.; de la Cruz, R.R.; Macias, D.; Galvez, V.; Pastor, A.M. Implanted neural progenitor cells regulate glial reaction to brain injury and establish gap junctions with host glial cells. Glia 2014, 62, 623–638. [Google Scholar] [CrossRef]
- Einstein, O.; Ben-Menachem-Tzidon, O.; Mizrachi-Kol, R.; Reinhartz, E.; Grigoriadis, N.; Ben-Hur, T. Survival of neural precursor cells in growth factor-poor environment: Implications for transplantation in chronic disease. Glia 2006, 53, 449–455. [Google Scholar] [CrossRef]
- Goldfarb, S.; Fainstein, N.; Ben-Hur, T. Electroconvulsive stimulation attenuates chronic neuroinflammation. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Goshen, I.; Kreisel, T.; Ounallah-Saad, H.; Renbaum, P.; Zalzstein, Y.; Ben-Hur, T.; Levy-Lahad, E.; Yirmiya, R. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology 2007, 32, 1106–1115. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Eimer, W.A.; Vassar, R. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol. Neurodegener. 2013, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Stalder, M.; Phinney, A.; Probst, A.; Sommer, B.; Staufenbiel, M.; Jucker, M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am. J. Pathol. 1999, 154, 1673–1684. [Google Scholar] [CrossRef] [Green Version]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.C. The endotoxin hypothesis of neurodegeneration. J. Neuroinflamm. 2019, 16, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Lee, Y.K.; Yuk, D.Y.; Choi, D.Y.; Ban, S.B.; Oh, K.W.; Hong, J.T. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflamm. 2008, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Miller, R.G.; Gascon, R.; Champion, S.; Katz, J.; Lancero, M.; Narvaez, A.; Honrada, R.; Ruvalcaba, D.; McGrath, M.S. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 2009, 206, 121–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Jaber, V.; Lukiw, W.J. Secretory Products of the Human GI Tract Microbiome and Their Potential Impact on Alzheimer’s Disease (AD): Detection of Lipopolysaccharide (LPS) in AD Hippocampus. Front. Cell. Infect. Microbiol. 2017, 7, 318. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Stamova, B.; Jin, L.W.; De Carli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef] [Green Version]
- Pluchino, S.; Martino, G. The therapeutic plasticity of neural stem/precursor cells in multiple sclerosis. J. Neurol. Sci. 2008, 265, 105–110. [Google Scholar] [CrossRef]
- Liu, X.Y.; Yang, L.P.; Zhao, L. Stem cell therapy for Alzheimer’s disease. World J. Stem Cells 2020, 12, 787–802. [Google Scholar] [CrossRef]
- Liu, J.; Hjorth, E.; Zhu, M.; Calzarossa, C.; Samuelsson, E.B.; Schultzberg, M.; Akesson, E. Interplay between human microglia and neural stem/progenitor cells in an allogeneic co-culture model. J. Cell. Mol. Med. 2013, 17, 1434–1443. [Google Scholar] [CrossRef] [Green Version]
- Mittal, K.; Eremenko, E.; Berner, O.; Elyahu, Y.; Strominger, I.; Apelblat, D.; Nemirovsky, A.; Spiegel, I.; Monsonego, A. CD4 T Cells Induce A Subset of MHCII-Expressing Microglia that Attenuates Alzheimer Pathology. iScience 2019, 16, 298–311. [Google Scholar] [CrossRef] [Green Version]
- Mignone, J.L.; Kukekov, V.; Chiang, A.S.; Steindler, D.; Enikolopov, G. Neural stem and progenitor cells in nestin-GFP transgenic mice. J. Comp. Neurol. 2004, 469, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Carletti, B.; Piemonte, F.; Rossi, F. Neuroprotection: The emerging concept of restorative neural stem cell biology for the treatment of neurodegenerative diseases. Curr. Neuropharmacol. 2011, 9, 313–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemonnot, A.L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front. Aging Neurosci. 2019, 11, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sole-Domenech, S.; Cruz, D.L.; Capetillo-Zarate, E.; Maxfield, F.R. The endocytic pathway in microglia during health, aging and Alzheimer’s disease. Ageing Res. Rev. 2016, 32, 89–103. [Google Scholar] [CrossRef] [Green Version]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef]
- Olah, M.; Amor, S.; Brouwer, N.; Vinet, J.; Eggen, B.; Biber, K.; Boddeke, H.W. Identification of a microglia phenotype supportive of remyelination. Glia 2012, 60, 306–321. [Google Scholar] [CrossRef]
- Pannell, M.; Szulzewsky, F.; Matyash, V.; Wolf, S.A.; Kettenmann, H. The subpopulation of microglia sensitive to neurotransmitters/neurohormones is modulated by stimulation with LPS, interferon-gamma, and IL-4. Glia 2014, 62, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Paresce, D.M.; Ghosh, R.N.; Maxfield, F.R. Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron 1996, 17, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Daria, A.; Colombo, A.; Llovera, G.; Hampel, H.; Willem, M.; Liesz, A.; Haass, C.; Tahirovic, S. Young microglia restore amyloid plaque clearance of aged microglia. EMBO J. 2017, 36, 583–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribe-Querol, E.; Rosales, C. Phagocytosis: Our Current Understanding of a Universal Biological Process. Front. Immunol. 2020, 11, 1066. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lachish, M.; Fainstein, N.; Ganz, T.; Sofer, L.; Ben-Hur, T. Failure of Alzheimer’s Mice Brain Resident Neural Precursor Cells in Supporting Microglia-Mediated Amyloid β Clearance. Cells 2022, 11, 876. https://doi.org/10.3390/cells11050876
Lachish M, Fainstein N, Ganz T, Sofer L, Ben-Hur T. Failure of Alzheimer’s Mice Brain Resident Neural Precursor Cells in Supporting Microglia-Mediated Amyloid β Clearance. Cells. 2022; 11(5):876. https://doi.org/10.3390/cells11050876
Chicago/Turabian StyleLachish, Marva, Nina Fainstein, Tal Ganz, Lihi Sofer, and Tamir Ben-Hur. 2022. "Failure of Alzheimer’s Mice Brain Resident Neural Precursor Cells in Supporting Microglia-Mediated Amyloid β Clearance" Cells 11, no. 5: 876. https://doi.org/10.3390/cells11050876
APA StyleLachish, M., Fainstein, N., Ganz, T., Sofer, L., & Ben-Hur, T. (2022). Failure of Alzheimer’s Mice Brain Resident Neural Precursor Cells in Supporting Microglia-Mediated Amyloid β Clearance. Cells, 11(5), 876. https://doi.org/10.3390/cells11050876