
Lysosomal ATP Transporter SLC17A9 Controls Cell Viability via Regulating Cathepsin D



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Antibodies and Reagents
2.3. Immunocytochemistry
2.4. Confocal Microscopy
2.5. RNA Isolation and Plasmid Constructs
2.6. Western Blot
2.7. Cell Viability Assay
2.8. Data Analysis
3. Results
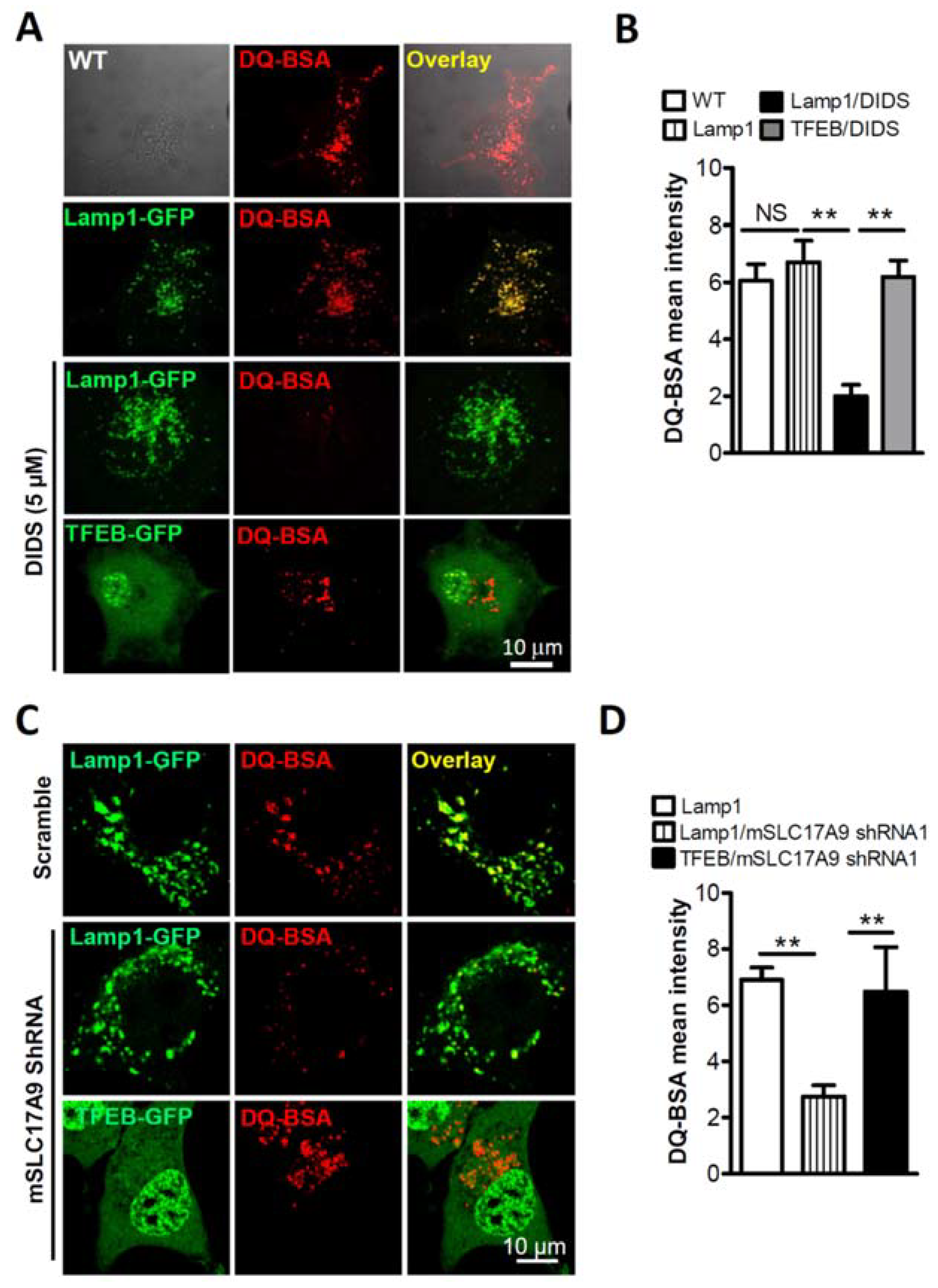
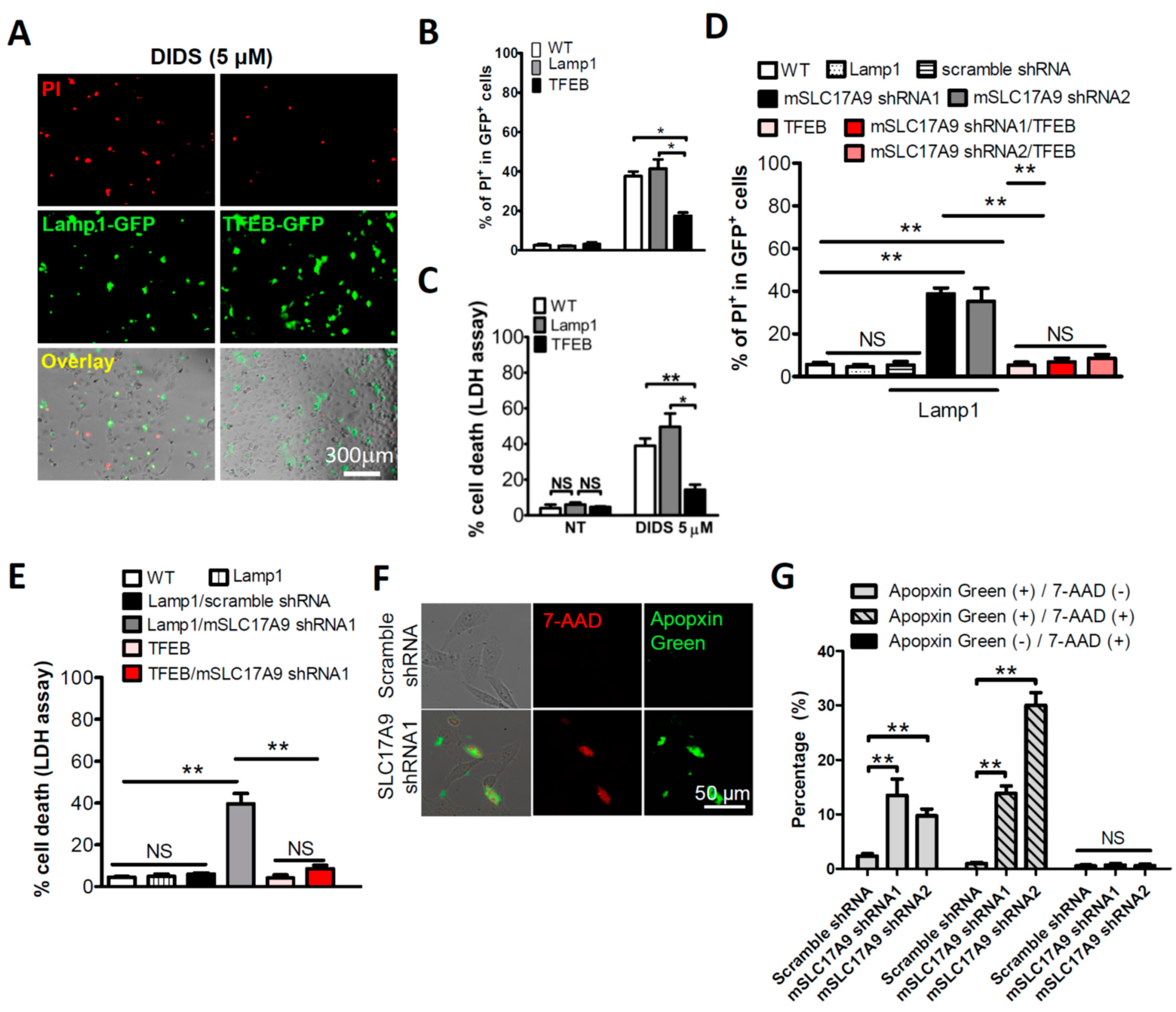
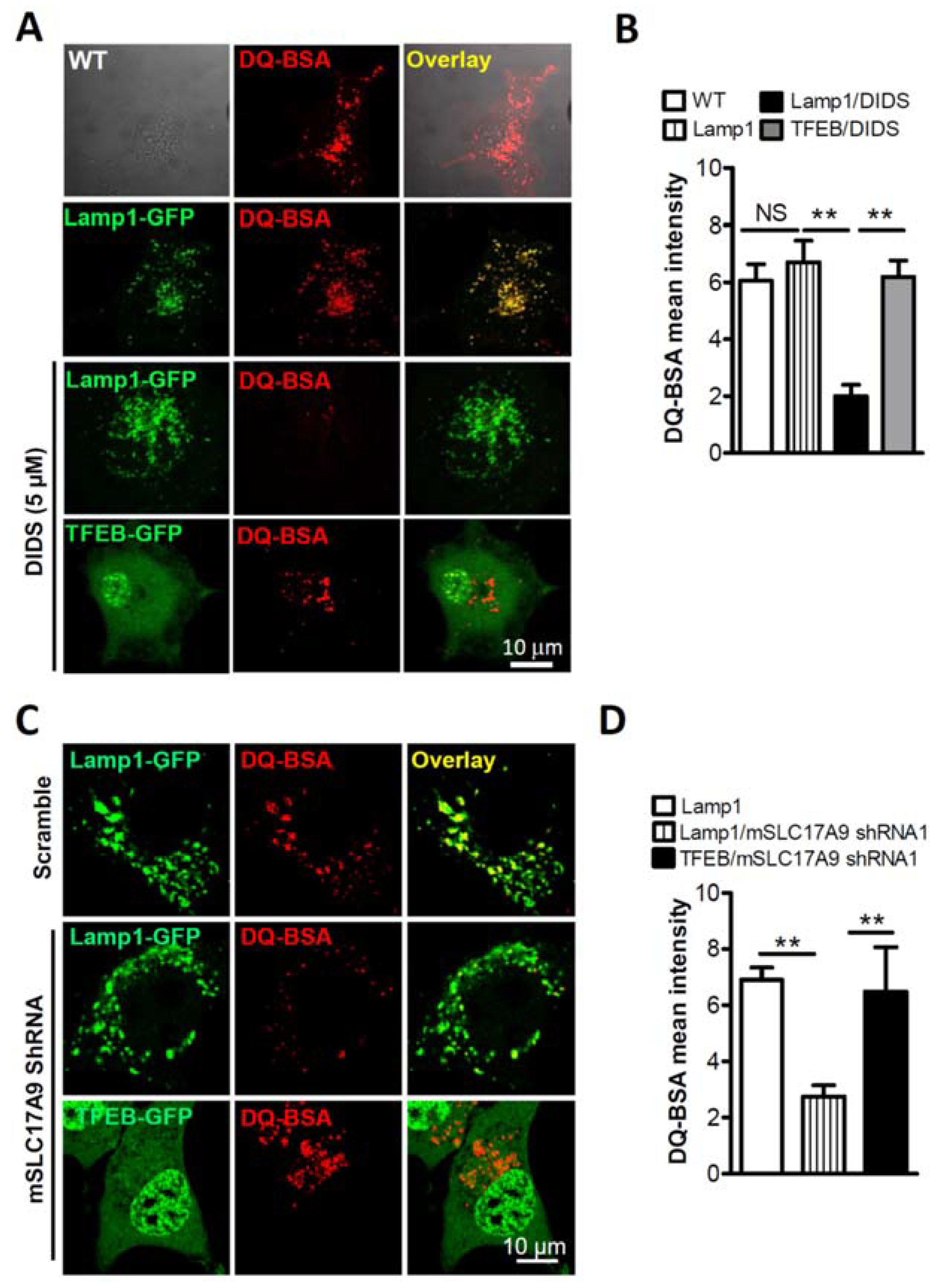
3.1. SLC17A9 Loss Associated Cell Death and Lysosome Dysfunction Are Rescued by TFEB Expression
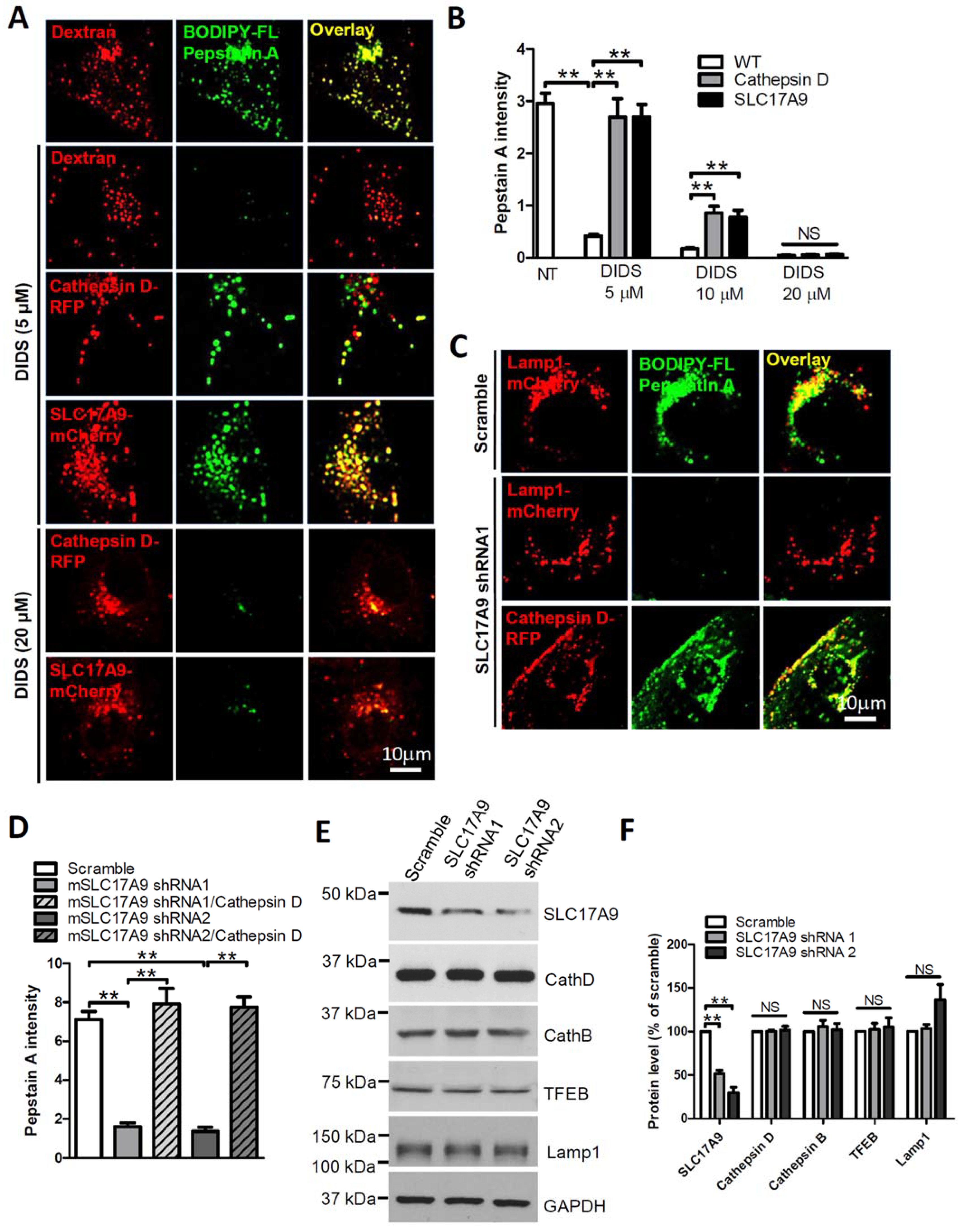
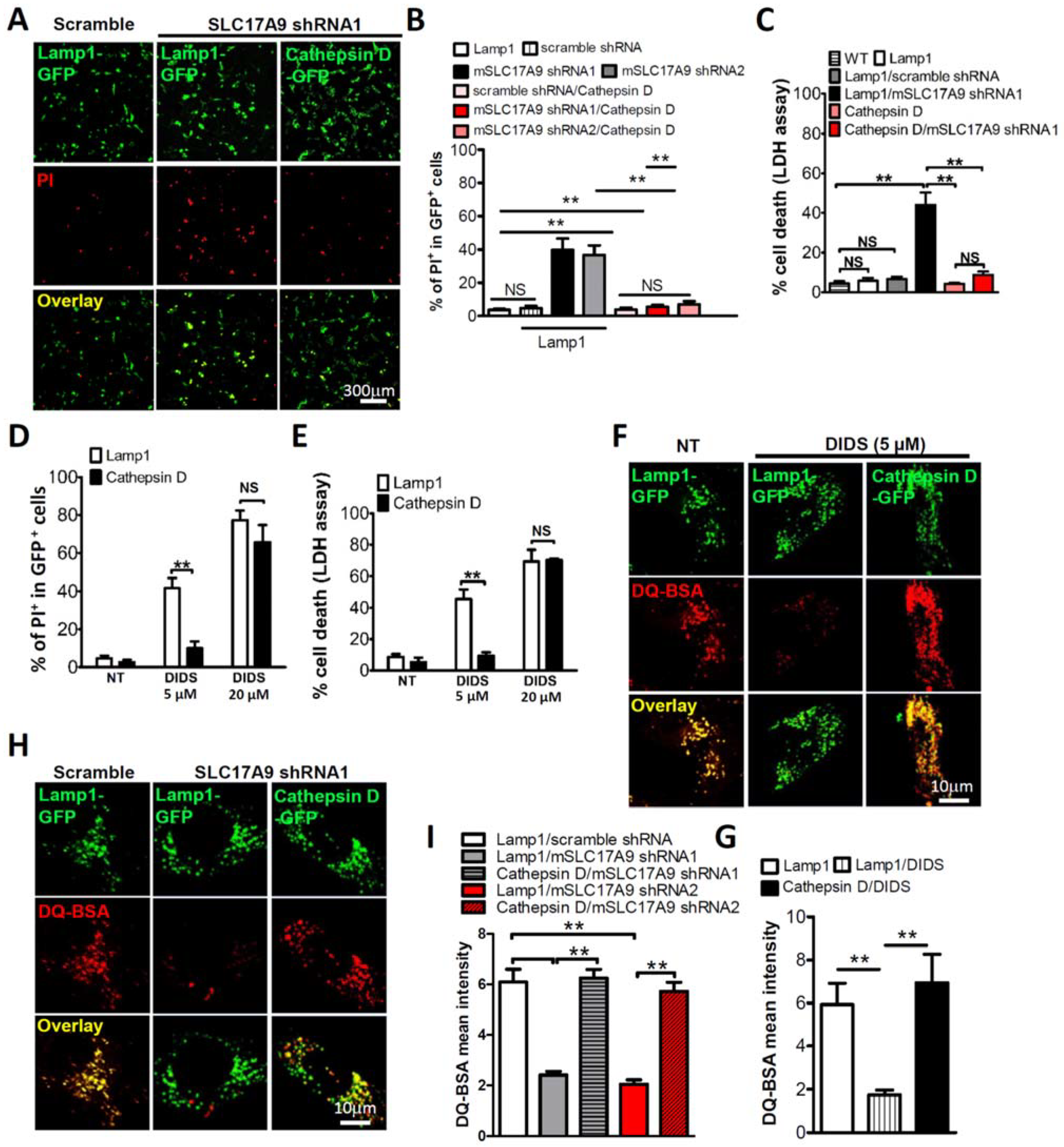
3.2. SLC17A9 Deficiency Results in Cathepsin D Dysfunction
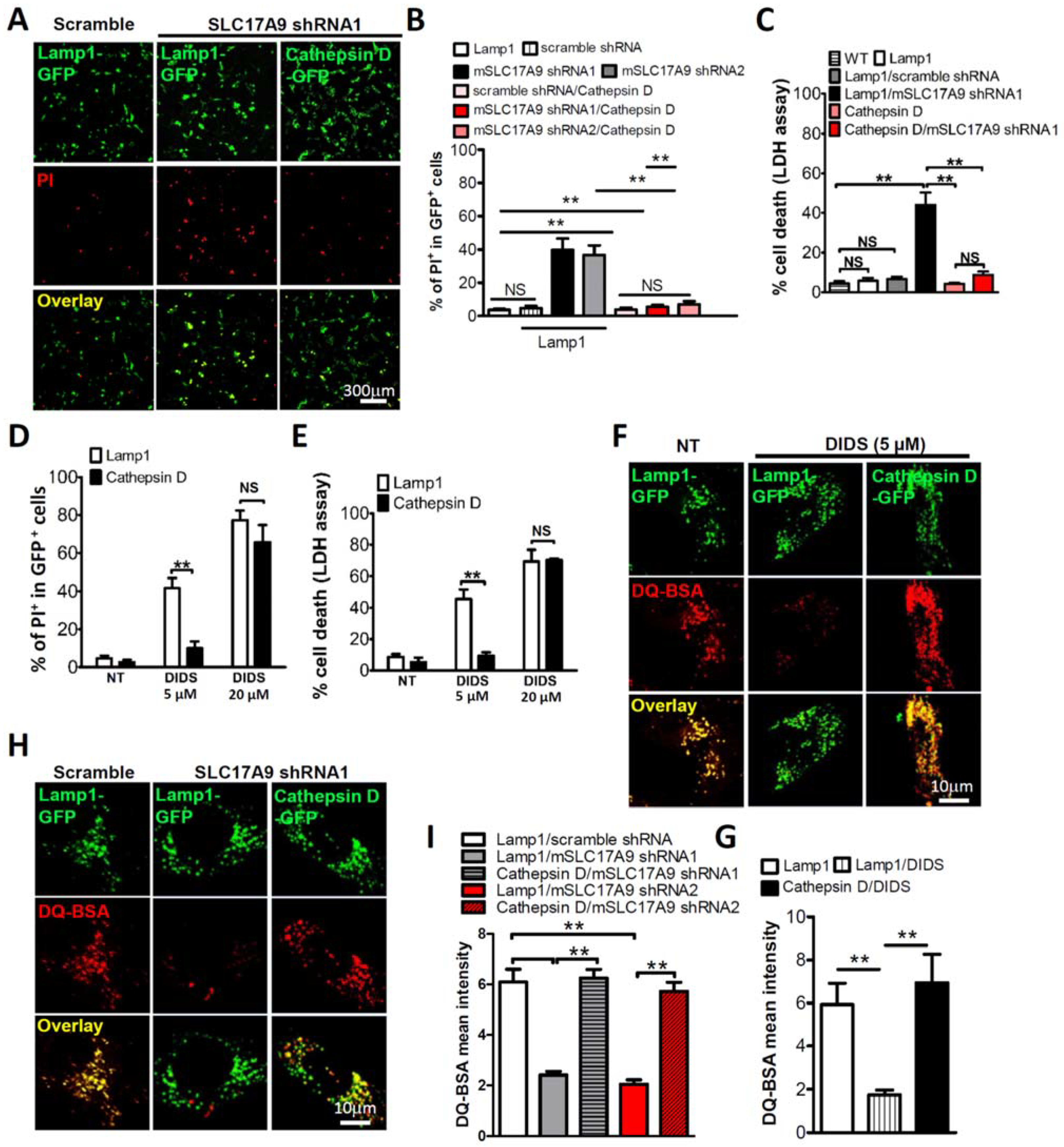
3.3. Cell Death and Lysosomal Dysfunction Induced by SLC17A9 Deficiency Are Rescued by Cathepsin D
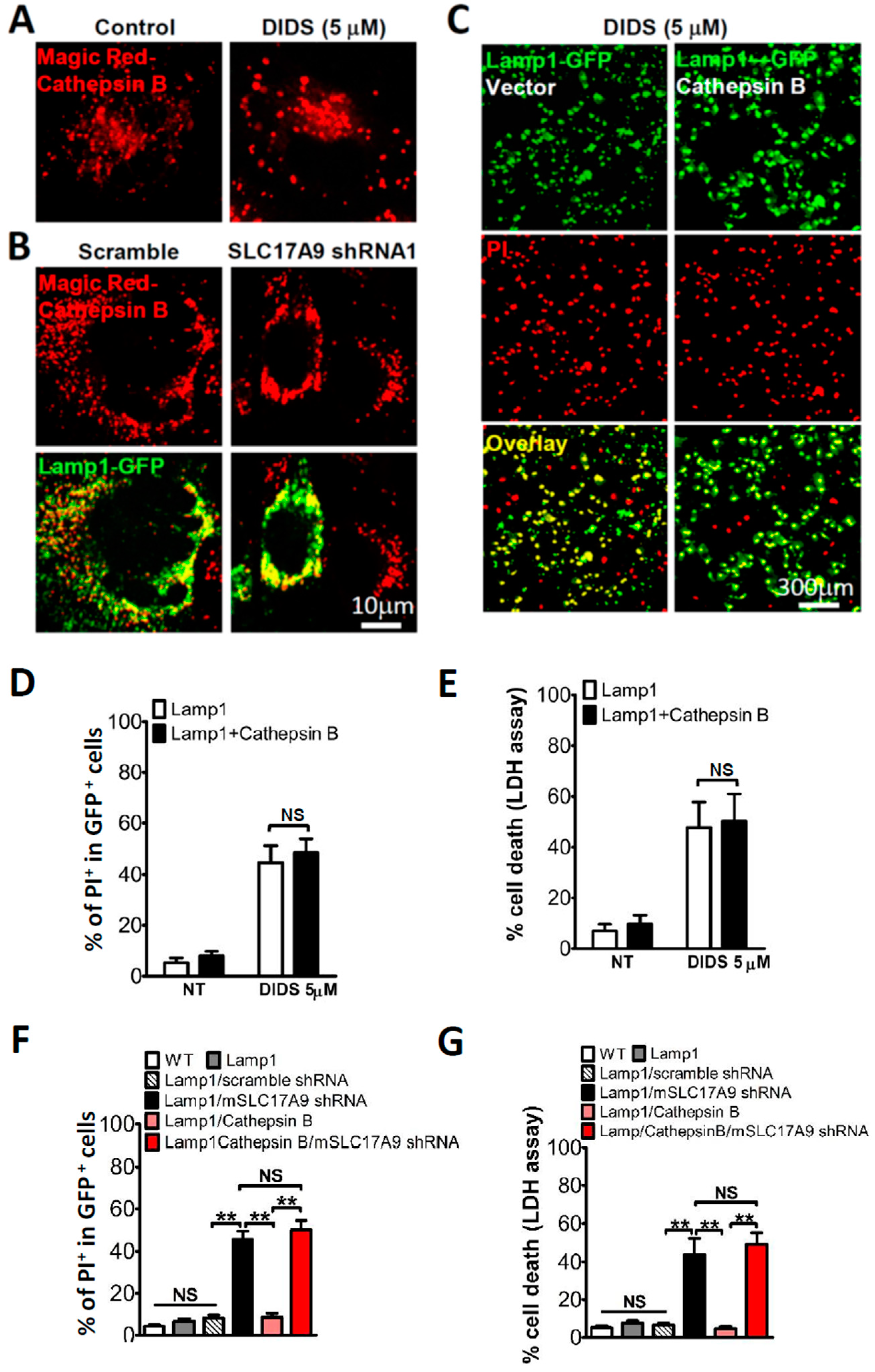
3.4. SLC17A9 Deficiency Associated Cell Death Is Not Rescued by Cathepsin B
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Fredriksson, R.; Nordstrom, K.J.; Stephansson, O.; Hagglund, M.G.; Schioth, H.B. The solute carrier (SLC) complement of the human genome: Phylogenetic classification reveals four major families. FEBS Lett. 2008, 582, 3811–3816. [Google Scholar] [CrossRef] [Green Version]
- Hediger, M.A.; Clemencon, B.; Burrier, R.E.; Bruford, E.A. The ABCs of membrane transporters in health and disease (SLC series): Introduction. Mol. Asp. Med. 2013, 34, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Hasuzawa, N.; Moriyama, S.; Moriyama, Y.; Nomura, M. Physiopathological roles of vesicular nucleotide transporter (VNUT), an essential component for vesicular ATP release. Biochim. Biophys Acta Biomembr. 2020, 1862, 183408. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, Y.; Hiasa, M.; Sakamoto, S.; Omote, H.; Nomura, M. Vesicular nucleotide transporter (VNUT): Appearance of an actress on the stage of purinergic signaling. Purinergic Signal. 2017, 13, 387–404. [Google Scholar] [CrossRef] [Green Version]
- Sawada, K.; Echigo, N.; Juge, N.; Miyaji, T.; Otsuka, M.; Omote, H.; Yamamoto, A.; Moriyama, Y. Identification of a vesicular nucleotide transporter. Proc. Natl. Acad. Sci. USA 2008, 105, 5683–5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Chen, G.; Zhou, W.; Song, A.; Xu, T.; Luo, Q.; Wang, W.; Gu, X.S.; Duan, S. Regulated ATP release from astrocytes through lysosome exocytosis. Nat. Cell Biol. 2007, 9, 945–953. [Google Scholar] [CrossRef]
- Dou, Y.; Wu, H.J.; Li, H.Q.; Qin, S.; Wang, Y.E.; Li, J.; Lou, H.F.; Chen, Z.; Li, X.M.; Luo, Q.M.; et al. Microglial migration mediated by ATP-induced ATP release from lysosomes. Cell Res. 2012, 22, 1022–1033. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Zou, Y.; Zhong, X.Z.; Cao, Q.; Zhao, K.; Zhu, M.X.; Murrell-Lagnado, R.; Dong, X.P. P2X4 forms functional ATP-activated cation channels on lysosomal membranes regulated by luminal pH. J. Biol. Chem. 2014, 289, 17658–17667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.Z.; Cao, Q.; Sun, X.; Dong, X.P. Activation of lysosomal P2X4 by ATP transported into lysosomes via VNUT/SLC17A9 using V-ATPase generated voltage gradient as the driving force. J. Physiol. 2016, 594, 4253–4266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Zhao, K.; Zhong, X.Z.; Zou, Y.; Yu, H.; Huang, P.; Xu, T.L.; Dong, X.P. SLC17A9 protein functions as a lysosomal ATP transporter and regulates cell viability. J. Biol. Chem. 2014, 289, 23189–23199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aits, S.; Jaattela, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, M.; Nakanishi, H.; Saftig, P.; Ezaki, J.; Isahara, K.; Ohsawa, Y.; Schulz-Schaeffer, W.; Watanabe, T.; Waguri, S.; Kametaka, S.; et al. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J. Neurosci. 2000, 20, 6898–6906. [Google Scholar] [CrossRef] [PubMed]
- Siintola, E.; Partanen, S.; Strömme, P.; Haapanen, A.; Haltia, M.; Maehlen, J.; Lehesjoki, A.E.; Tyynelä, J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain 2006, 129, 1438–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinfeld, R.; Reinhardt, K.; Schreiber, K.; Hillebrand, M.; Kraetzner, R.; Brück, W.; Saftig, P.; Gärtner, J. Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am. J. Hum. Genet. 2006, 78, 988–998. [Google Scholar] [CrossRef] [Green Version]
- Benes, P.; Vetvicka, V.; Fusek, M. Cathepsin D--many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 2008, 68, 12–28. [Google Scholar] [CrossRef] [Green Version]
- Pillai, S.; Zull, J.E. Effects of ATP, vanadate, and molybdate on cathepsin D-catalyzed proteolysis. J. Biol. Chem. 1985, 260, 8384–8389. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef]
- Song, W.; Wang, F.; Savini, M.; Ake, A.; di Ronza, A.; Sardiello, M.; Segatori, L. TFEB regulates lysosomal proteostasis. Hum. Mol. Genet. 2013, 22, 1994–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Kobayashi, H.; Higuchi, T.; Shimada, Y.; Ida, H.; Ohashi, T. TFEB overexpression promotes glycogen clearance of Pompe disease iPSC-derived skeletal muscle. Mol. Ther. Methods Clin. Dev. 2016, 3, 16054. [Google Scholar] [CrossRef] [PubMed]
- Awad, O.; Sarkar, C.; Panicker, L.M.; Miller, D.; Zeng, X.; Sgambato, J.A.; Lipinski, M.M.; Feldman, R.A. Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet. 2015, 24, 5775–5788. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef] [PubMed]
- Spampanato, C.; Feeney, E.; Li, L.; Cardone, M.; Lim, J.A.; Annunziata, F.; Zare, H.; Polishchuk, R.; Puertollano, R.; Parenti, G.; et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med. 2013, 5, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Gatto, F.; Rossi, B.; Tarallo, A.; Polishchuk, E.; Polishchuk, R.; Carrella, A.; Nusco, E.; Alvino, F.G.; Iacobellis, F.; De Leonibus, E.; et al. AAV-mediated transcription factor EB (TFEB) gene delivery ameliorates muscle pathology and function in the murine model of Pompe Disease. Sci. Rep. 2017, 7, 15089. [Google Scholar] [CrossRef] [PubMed]
- Rega, L.R.; Polishchuk, E.; Montefusco, S.; Napolitano, G.; Tozzi, G.; Zhang, J.; Bellomo, F.; Taranta, A.; Pastore, A.; Polishchuk, R.; et al. Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells. Kidney Int. 2016, 89, 862–873. [Google Scholar] [CrossRef] [Green Version]
- Sathe, M.N.; Woo, K.; Kresge, C.; Bugde, A.; Luby-Phelps, K.; Lewis, M.A.; Feranchak, A.P. Regulation of purinergic signaling in biliary epithelial cells by exocytosis of SLC17A9-dependent ATP-enriched vesicles. J. Biol. Chem. 2011, 286, 25363–25376. [Google Scholar] [CrossRef] [Green Version]
- Geisler, J.C.; Corbin, K.L.; Li, Q.; Feranchak, A.P.; Nunemaker, C.S.; Li, C. Vesicular nucleotide transporter-mediated ATP release regulates insulin secretion. Endocrinology 2013, 154, 675–684. [Google Scholar] [CrossRef]
- Tokunaga, A.; Tsukimoto, M.; Harada, H.; Moriyama, Y.; Kojima, S. Involvement of SLC17A9-dependent vesicular exocytosis in the mechanism of ATP release during T cell activation. J. Biol. Chem. 2010, 285, 17406–17416. [Google Scholar] [CrossRef] [Green Version]
- Katarkar, A.; Bottoni, G.; Clocchiatti, A.; Goruppi, S.; Bordignon, P.; Lazzaroni, F.; Gregnanin, I.; Ostano, P.; Neel, V.; Dotto, G.P. NOTCH1 gene amplification promotes expansion of cancer associated fibroblast populations in human skin. Nat. Commun. 2020, 11, 5126. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Hwang, E.S. Status of mTOR activity may phenotypically differentiate senescence and quiescence. Mol. Cells 2012, 33, 597–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.S.; Chen, W.N.; Zhou, M.; Arttamangkul, S.; Haugland, R.P. Probing the cathepsin D using a BODIPY FL–pepstatin A: Applications in fluorescence polarization and microscopy. J. Biochem. Biophys. Methods 2000, 42, 137–151. [Google Scholar] [CrossRef]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Farfel-Becker, T.; Roney, J.C.; Cheng, X.T.; Li, S.; Cuddy, S.; Sheng, Z.H. Neuronal soma-derived degradative lysosomes are continuously delivered to distal axons to maintain local degradation capacity. Cell Rep. 2019, 28, 51–64.e4. [Google Scholar] [CrossRef] [Green Version]
- Yuseff, M.I.; Reversat, A.; Lankar, D.; Diaz, J.; Fanget, I.; Pierobon, P.; Randrian, V.; Larochette, N.; Vascotto, F.; Desdouets, C.; et al. Polarized secretion of lysosomes at the B cell synapse couples antigen extraction to processing and presentation. Immunity 2011, 35, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Graves, A.R.; Curran, P.K.; Smith, C.L.; Mindell, J.A. The Cl-/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature 2008, 453, 788–792. [Google Scholar] [CrossRef]
- Steinberg, B.E.; Huynh, K.K.; Brodovitch, A.; Jabs, S.; Stauber, T.; Jentsch, T.J.; Grinstein, S. A cation counterflux supports lysosomal acidification. J. Cell Biol. 2010, 189, 1171–1186. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Tan, S.H.; Nicolas, V.; Bauvy, C.; Yang, N.D.; Zhang, J.; Xue, Y.; Codogno, P.; Shen, H.M. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013, 23, 508–523. [Google Scholar] [CrossRef] [Green Version]
- Chwieralski, C.E.; Welte, T.; Bühling, F. Cathepsin-regulated apoptosis. Apoptosis 2006, 11, 143–149. [Google Scholar] [CrossRef]
- Turk, B.; Turk, D.; Turk, V. Lysosomal cysteine proteases: More than scavengers. Biochim. Biophys. Acta 2000, 1477, 98–111. [Google Scholar] [CrossRef]
- Creasy, B.M.; Hartmann, C.B.; White, F.K.; McCoy, K.L. New assay using fluorogenic substrates and immunofluorescence staining to measure cysteine cathepsin activity in live cell subpopulations. Cytom. Part A 2007, 71, 114–123. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, P.; Cao, Q.; Xu, M.; Dong, X.-P. Lysosomal ATP Transporter SLC17A9 Controls Cell Viability via Regulating Cathepsin D. Cells 2022, 11, 887. https://doi.org/10.3390/cells11050887
Huang P, Cao Q, Xu M, Dong X-P. Lysosomal ATP Transporter SLC17A9 Controls Cell Viability via Regulating Cathepsin D. Cells. 2022; 11(5):887. https://doi.org/10.3390/cells11050887
Chicago/Turabian StyleHuang, Peng, Qi Cao, Mengnan Xu, and Xian-Ping Dong. 2022. "Lysosomal ATP Transporter SLC17A9 Controls Cell Viability via Regulating Cathepsin D" Cells 11, no. 5: 887. https://doi.org/10.3390/cells11050887
APA StyleHuang, P., Cao, Q., Xu, M., & Dong, X.-P. (2022). Lysosomal ATP Transporter SLC17A9 Controls Cell Viability via Regulating Cathepsin D. Cells, 11(5), 887. https://doi.org/10.3390/cells11050887