
Transcriptomics Reveals Discordant Lipid Metabolism Effects between In Vitro Models Exposed to Elafibranor and Liver Samples of NAFLD Patients after Bariatric Surgery

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. hSKP-HPC Cell Cultures
2.2. HepaRG Cell Cultures
2.3. Primary Human Hepatocyte Cell Cultures
2.4. Compound Preparation
2.5. RNA Extraction
2.6. Microarray
2.7. Publicly Available Datasets
2.8. Statistical Analysis
3. Results
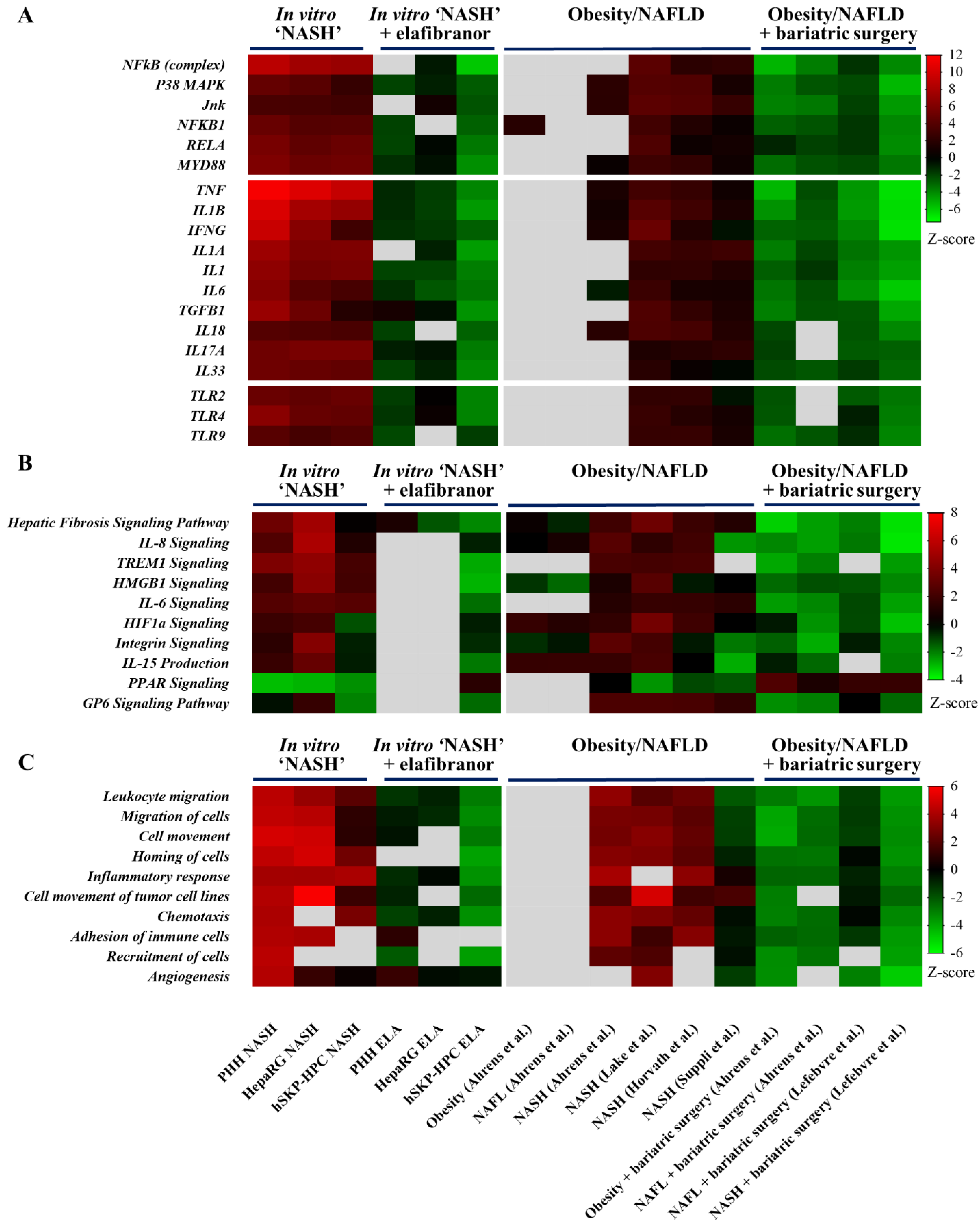
3.1. Transcriptomes of Human Hepatic In Vitro NASH Models Exhibit Similarities with Those of Human NASH Liver
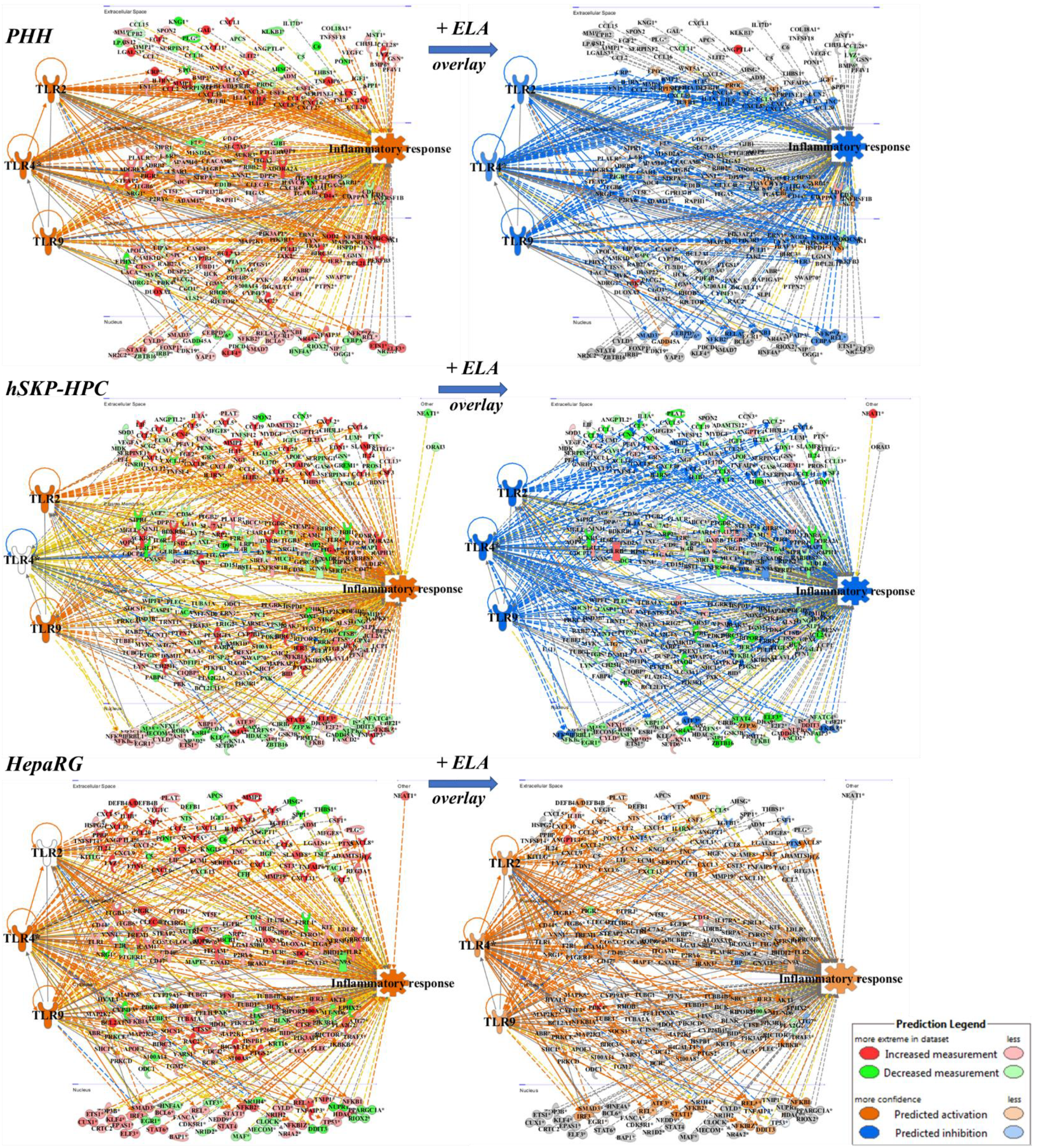
3.2. Elafibranor Reduces Toll-like Receptor-Dependent Inflammation in PHH and hSKP-HPC In Vitro NASH Models
3.3. Similarities in NASH-Related Upstream Regulators and Canonical Pathways in Hepatic In Vitro NASH Models Exposed to Elafibranor and Liver Samples of Patients after Bariatric Surgery
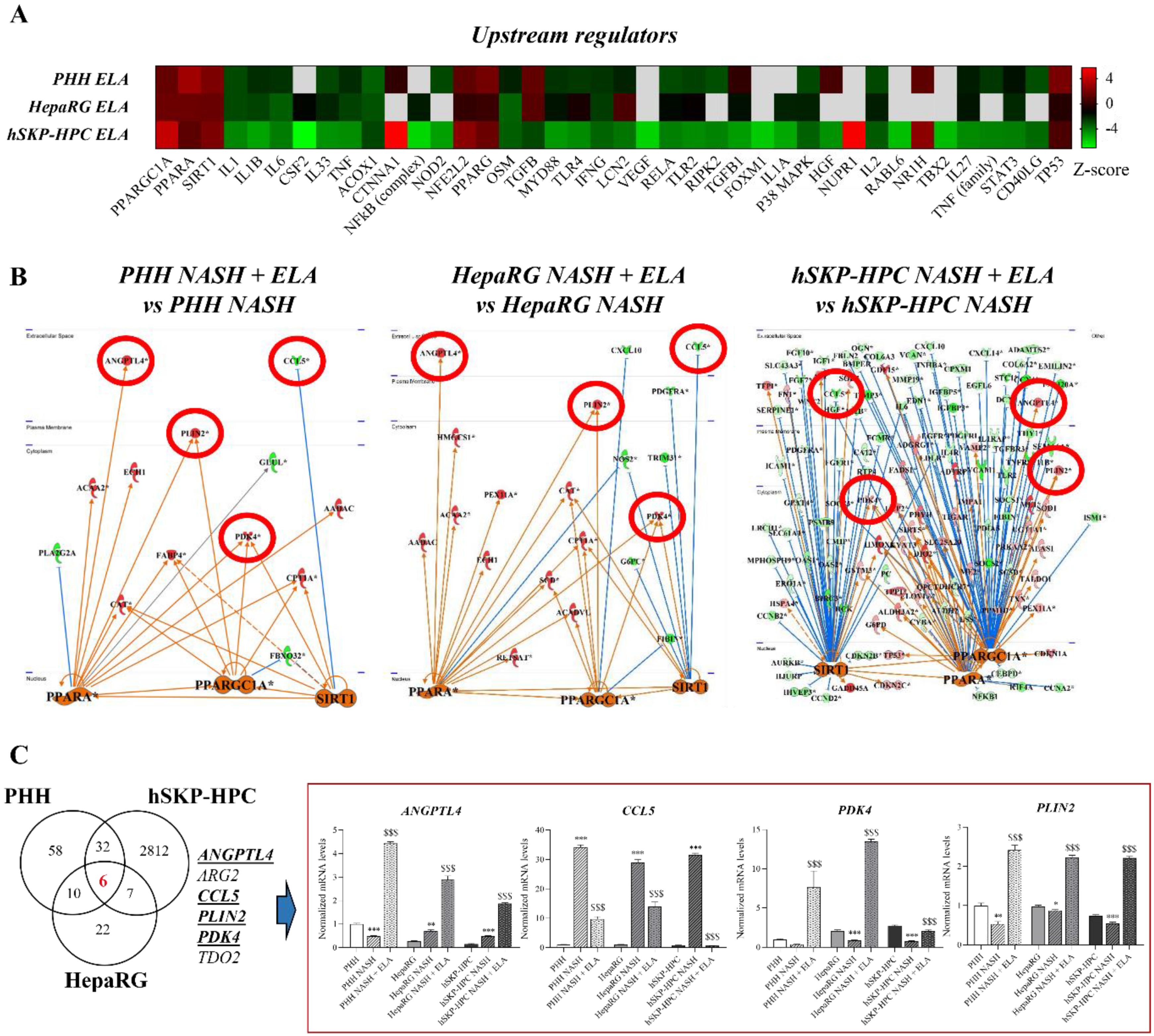
3.4. PPARGC1A, PPARA, and SIRT1 Regulate Shared Elafibranor-Induced Genes in NASH-Triggered PHH, HepaRG, and hSKP-HPC
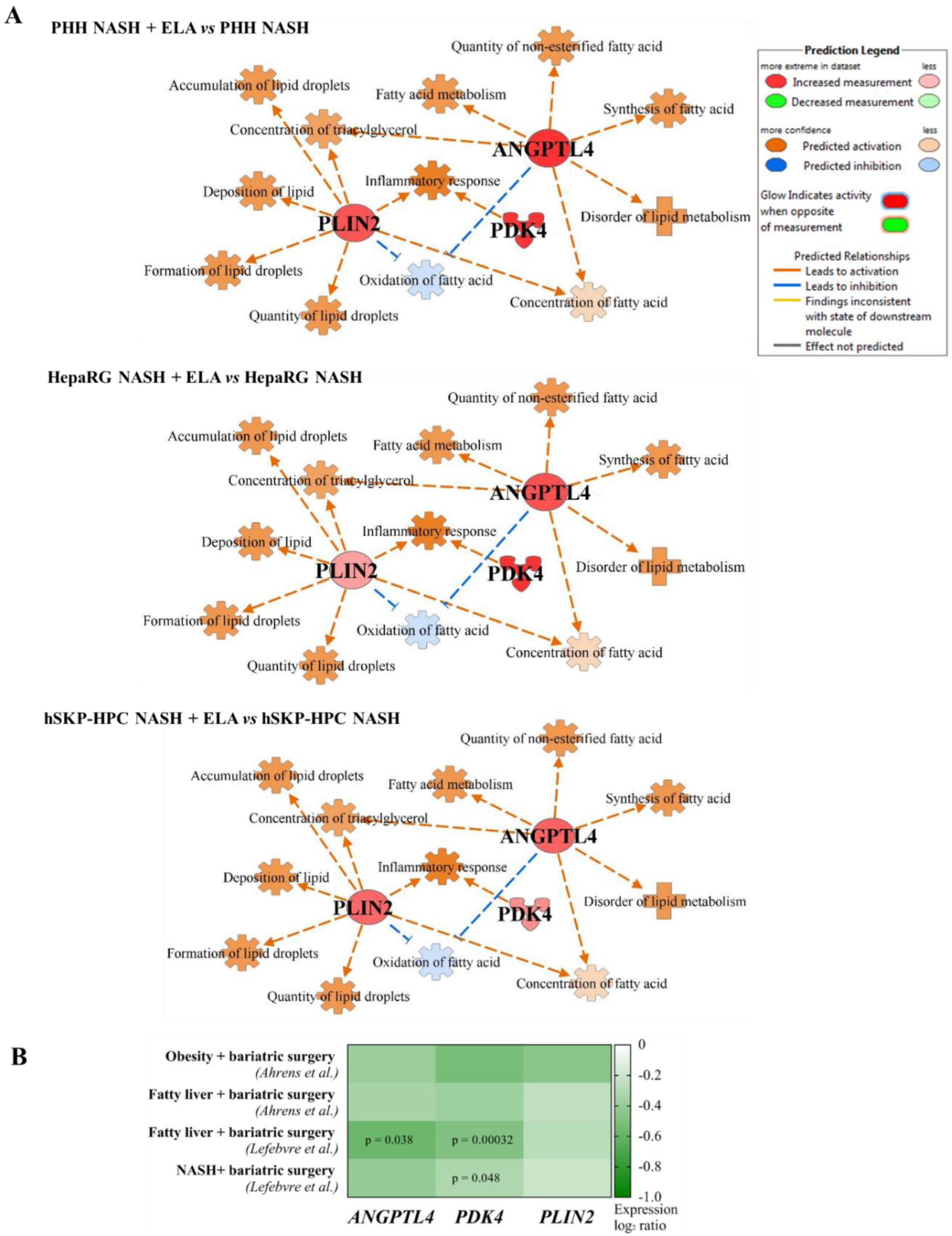
3.5. Elafibranor-Mediated Induction of ANGPTL4, PDK4, and PLIN2 In Vitro Predicts Induction of Steatosis, Contrarily to Anti-NASH Bariatric Surgery
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2020, 9, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A. Weight Loss via Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378. [Google Scholar] [CrossRef]
- Klebanoff, M.J.; Corey, K.E.; Chhatwal, J.; Kaplan, L.M.; Chung, R.T.; Hur, C. Bariatric Surgery for Nonalcoholic Steatohepatitis: A Clinical and Cost-Effectiveness Analysis. Hepatology 2017, 65, 1156–1164. [Google Scholar] [CrossRef]
- Boeckmans, J.; Buyl, K.; Natale, A.; Vandenbempt, V.; Branson, S.; De Boe, V.; Rogiers, V.; De Kock, J.; Rodrigues, R.M.; Vanhaecke, T. Elafibranor Restricts Lipogenic and Inflammatory Responses in a Human Skin Stem Cell-Derived Model of NASH. Pharmacol. Res. 2019, 144, 377–389. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Cami, B.; De Boe, V.; Heymans, A.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; et al. Human Hepatic in Vitro Models Reveal Distinct Anti-NASH Potencies of PPAR Agonists. Cell Biol. Toxicol. 2020, 37, 293–311. [Google Scholar] [CrossRef]
- GENFIT Announces Results from Interim Analysis of RESOLVE-IT Phase 3 Trial of Elafibranor in Adults with NASH and Fibrosis; Genfit: Loos, France, 2020; pp. 1–5.
- Harrison, S.A.; Ratziu, V.; Bedossa, P.; Dufour, J.-F.; Kruger, F.; Schattenberg, J.M.; Francque, S.M.; Arrese, M.; George, J.; Bugianesi, E.; et al. RESOLVE-IT Phase 3 Trial of Elafibranor in NASH: Final Results of the Week 72 Interim Surrogate Efficacy Analysis. Proceedings of AASLD 2020, Boston, MA, USA, 13–16 November 2020. [Google Scholar]
- Lefere, S.; Onghena, L.; Vanlander, A.; van Nieuwenhove, Y.; Devisscher, L.; Geerts, A. Bariatric Surgery and the Liver-Mechanisms, Benefits, and Risks. Obes. Rev. 2021, 22. [Google Scholar] [CrossRef]
- Chambers, A.P.; Jessen, L.; Ryan, K.K.; Sisley, S.; Wilsonpérez, H.E.; Stefater, M.A.; Gaitonde, S.G.; Sorrell, J.E.; Toure, M.; Berger, J.; et al. Weight-Independent Changes in Blood Glucose Homeostasis after Gastric Bypass or Vertical Sleeve Gastrectomy in Rats. Gastroenterology 2011, 141, 950–958. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, R.M.; De Kock, J.; Branson, S.; Vinken, M.; Meganathan, K.; Chaudhari, U.; Sachinidis, A.; Govaere, O.; Roskams, T.; De Boe, V.; et al. Human Skin-Derived Stem Cells as a Novel Cell Source for In Vitro Hepatotoxicity Screening of Pharmaceuticals. Stem Cells Dev. 2014, 23, 44–55. [Google Scholar] [CrossRef]
- Rodrigues, R.M.; Heymans, A.; De Boe, V.; Sachinidis, A.; Chaudhari, U.; Govaere, O.; Roskams, T.; Vanhaecke, T.; Rogiers, V.; De Kock, J. Toxicogenomics-Based Prediction of Acetaminophen-Induced Liver Injury Using Human Hepatic Cell Systems. Toxicol. Lett. 2016, 240, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, M.; Ammerpohl, O.; von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Ahrens, M.; Ammerpohl, O.; von Scho, W.; Teufel, A.; et al. DNA Methylation Analysis in Nonalcoholic Fatty Liver Disease Suggests Distinct Disease-Specific and Remodeling Signatures after Bariatric Surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, A.D.; Novak, P.; Fisher, C.D.; Jackson, J.P.; Hardwick, R.N.; Billheimer, D.D.; Klimecki, W.T.; Cherrington, N.J. Analysis of Global and Absorption, Distribution, Metabolism, and Elimination Gene Expression in the Progressive Stages of Human Nonalcoholic Fatty Liver Disease. Drug Metab. Dispos. 2011, 39, 1954–1960. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Erhart, W.; Brosch, M.; Ammerpohl, O.; Von Schönfels, W.; Ahrens, M.; Heits, N.; Bell, J.T.; Tsai, P.C.; Spector, T.D.; et al. Obesity Accelerates Epigenetic Aging of Human Liver. Proc. Natl. Acad. Sci. USA 2014, 111, 15538–15543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suppli, M.P.; Rigbolt, K.T.G.; Veidal, S.S.; Heebøll, S.; Eriksen, P.L.; Demant, M.; Bagger, J.I.; Nielsen, J.C.; Oró, D.; Thrane, S.W.; et al. Hepatic Transcriptome Signatures in Patients with Varying Degrees of Nonalcoholic Fatty Liver Disease Compared with Healthy Normal-Weight Individuals. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G462–G472. [Google Scholar] [CrossRef]
- Lefebvre, P.; Lalloyer, F.; Baugé, E.; Pawlak, M.; Gheeraert, C.; Dehondt, H.; Vanhoutte, J.; Woitrain, E.; Hennuyer, N.; Mazuy, C.; et al. Interspecies NASH Disease Activity Whole-Genome Profiling Identifies a Fibrogenic Role of PPARα-Regulated Dermatopontin. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Seki, E.; Ohnishi, H.; Brenner, D.A. Role of Toll-like Receptors and Their Downstream Molecules in the Development of Nonalcoholic Fatty Liver Disease. Gastroenterol. Res. Pract. 2010, 2010, 362847. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, J.; van Grunsven, L.A.; Tacke, F. Current and Emerging Pharmacotherapeutic Interventions for the Treatment of Liver Fibrosis. Expert Opin. Pharmacother. 2020, 21, 1637–1650. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Therapeutic Landscape for NAFLD in 2020. Gastroenterol 2020, 158, 1984–1998.e3. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Robim, M. Human-Based Systems: Mechanistic NASH Modelling Just around the Corner? Pharmacol. Res. 2018, 134, 257–267. [Google Scholar] [CrossRef]
- Frades, I.; Andreasson, E.; Mato, J.M.; Alexandersson, E.; Matthiesen, R.; Martínez-Chantar, M.L. Integrative Genomic Signatures Of Hepatocellular Carcinoma Derived from Nonalcoholic Fatty Liver Disease. PLoS ONE 2015, 10, e0124544. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and Resolution of Inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Gaskell, H.; Ge, X.; Nieto, N. High-Mobility Group Box-1 and Liver Disease. Hepatol. Commun. 2018, 2, 1005–1020. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Huang, J.; Shen, Z.; Xiang, C.; Zhang, M.; Lu, X. Inhibition of TREM-1 Attenuates Inflammation and Lipid Accumulation in Diet-Induced Nonalcoholic Fatty Liver Disease. J. Cell. Biochem. 2019, 120, 11867–11877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [Green Version]
- Scarpulla, R.C. Metabolic Control of Mitochondrial Biogenesis through the PGC-1 Family Regulatory Network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPAR-α Gene Expression Correlates with Severity and Histological Treatment Response in Patients with Non-Alcoholic Steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-Specific Deletion of SIRT1 Alters Fatty Acid Metabolism and Results in Hepatic Steatosis and Inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome Proliferator-Activated Receptor Alpha Target Genes. PPAR Res. 2010, 2010, 393–416. [Google Scholar] [CrossRef] [Green Version]
- Najt, C.P.; Lwande, J.S.; McIntosh, A.L.; Senthivinayagam, S.; Gupta, S.; Kuhn, L.A.; Atshaves, B.P. Structural and Functional Assessment of Perilipin 2 Lipid Binding Domain(s). Biochemistry 2014, 53, 7051–7066. [Google Scholar] [CrossRef]
- Orlicky, D.J.; Libby, A.E.; Bales, E.S.; McMahan, R.H.; Monks, J.; La Rosa, F.G.; McManaman, J.L. Perilipin-2 Promotes Obesity and Progressive Fatty Liver Disease in Mice through Mechanistically Distinct Hepatocyte and Extra-Hepatocyte Actions. J. Physiol. 2019, 597, 1565–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najt, C.P.; Senthivinayagam, S.; Aljazi, M.B.; Fader, K.A.; Olenic, S.D.; Brock, J.R.L.; Lydic, T.A.; Jones, A.D.; Atshaves, B.P. Liver-Specific Loss of Perilipin 2 Alleviates Diet-Induced Hepatic Steatosis, Inflammation, and Fibrosis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2016, 310, G726–G738. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Hulver, M.W.; McMillan, R.P.; Cline, M.A.; Gilbert, E.R. The Pivotal Role of Pyruvate Dehydrogenase Kinases in Metabolic Flexibility. Nutr. Metab. 2014, 11, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Zhao, Y.; Li, Z.; Wang, C. Pyruvate Dehydrogenase Kinase 4 Mediates Lipogenesis and Contributes to the Pathogenesis of Nonalcoholic Steatohepatitis. Biochem. Biophys. Res. Commun. 2018, 495, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Kotani, K.; Tanaka, K.; Egucih, Y.; Anzai, K. Therapeutic Approaches to Nonalcoholic Fatty Liver Disease: Exercise Intervention and Related Mechanisms. Front. Endocrinol. 2018, 9, 588. [Google Scholar] [CrossRef] [PubMed]
- Mandard, S.; Zandbergen, F.; Van Straten, E.; Wahli, W.; Kuipers, F.; Müller, M.; Kersten, S. The Fasting-Induced Adipose Factor/Angiopoietin-like Protein 4 Is Physically Associated with Lipoproteins and Governs Plasma Lipid Levels and Adiposity. J. Biol. Chem. 2006, 281, 934–944. [Google Scholar] [CrossRef] [Green Version]
- Xu, A.; Lam, M.C.; Chan, K.W.; Wang, Y.; Zhang, J.; Boo, R.L.C.; Xu, J.Y.; Chen, B.; Chow, W.S.; Tso, A.W.K.; et al. Angiopoietin-like Protein 4 Decreases Blood Glucose and Improves Glucose Tolerance but Induces Hyperlipidemia and Hepatic Steatosis in Mice. Proc. Natl. Acad. Sci. United States Am. 2005, 102, 6086–6091. [Google Scholar] [CrossRef] [Green Version]
- Sahini, N.; Selvaraj, S.; Borlak, J. Whole Genome Transcript Profiling of Drug Induced Steatosis in Rats Reveals a Gene Signature Predictive of Outcome. PLoS ONE 2014, 9, e114085. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boeckmans, J.; Gatzios, A.; Heymans, A.; Rombaut, M.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Transcriptomics Reveals Discordant Lipid Metabolism Effects between In Vitro Models Exposed to Elafibranor and Liver Samples of NAFLD Patients after Bariatric Surgery. Cells 2022, 11, 893. https://doi.org/10.3390/cells11050893
Boeckmans J, Gatzios A, Heymans A, Rombaut M, Rogiers V, De Kock J, Vanhaecke T, Rodrigues RM. Transcriptomics Reveals Discordant Lipid Metabolism Effects between In Vitro Models Exposed to Elafibranor and Liver Samples of NAFLD Patients after Bariatric Surgery. Cells. 2022; 11(5):893. https://doi.org/10.3390/cells11050893
Chicago/Turabian StyleBoeckmans, Joost, Alexandra Gatzios, Anja Heymans, Matthias Rombaut, Vera Rogiers, Joery De Kock, Tamara Vanhaecke, and Robim M. Rodrigues. 2022. "Transcriptomics Reveals Discordant Lipid Metabolism Effects between In Vitro Models Exposed to Elafibranor and Liver Samples of NAFLD Patients after Bariatric Surgery" Cells 11, no. 5: 893. https://doi.org/10.3390/cells11050893
APA StyleBoeckmans, J., Gatzios, A., Heymans, A., Rombaut, M., Rogiers, V., De Kock, J., Vanhaecke, T., & Rodrigues, R. M. (2022). Transcriptomics Reveals Discordant Lipid Metabolism Effects between In Vitro Models Exposed to Elafibranor and Liver Samples of NAFLD Patients after Bariatric Surgery. Cells, 11(5), 893. https://doi.org/10.3390/cells11050893