
Isolation and Crystallization of the D156C Form of Optogenetic ChR2

 , , ,
, , ,
Abstract
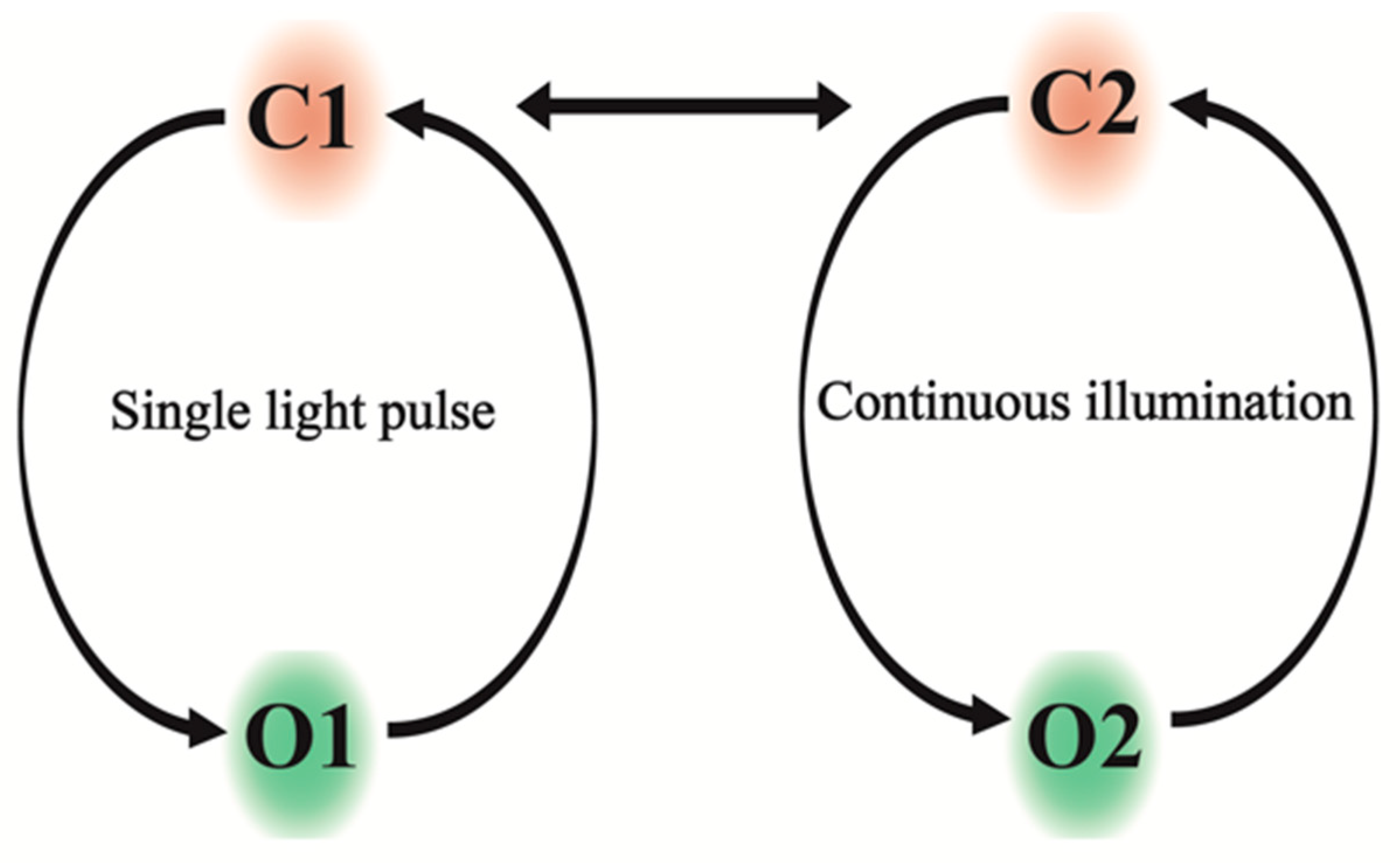
:1. Introduction
2. Materials and Methods
2.1. Construction and Overexpression of Recombinant Plasmids
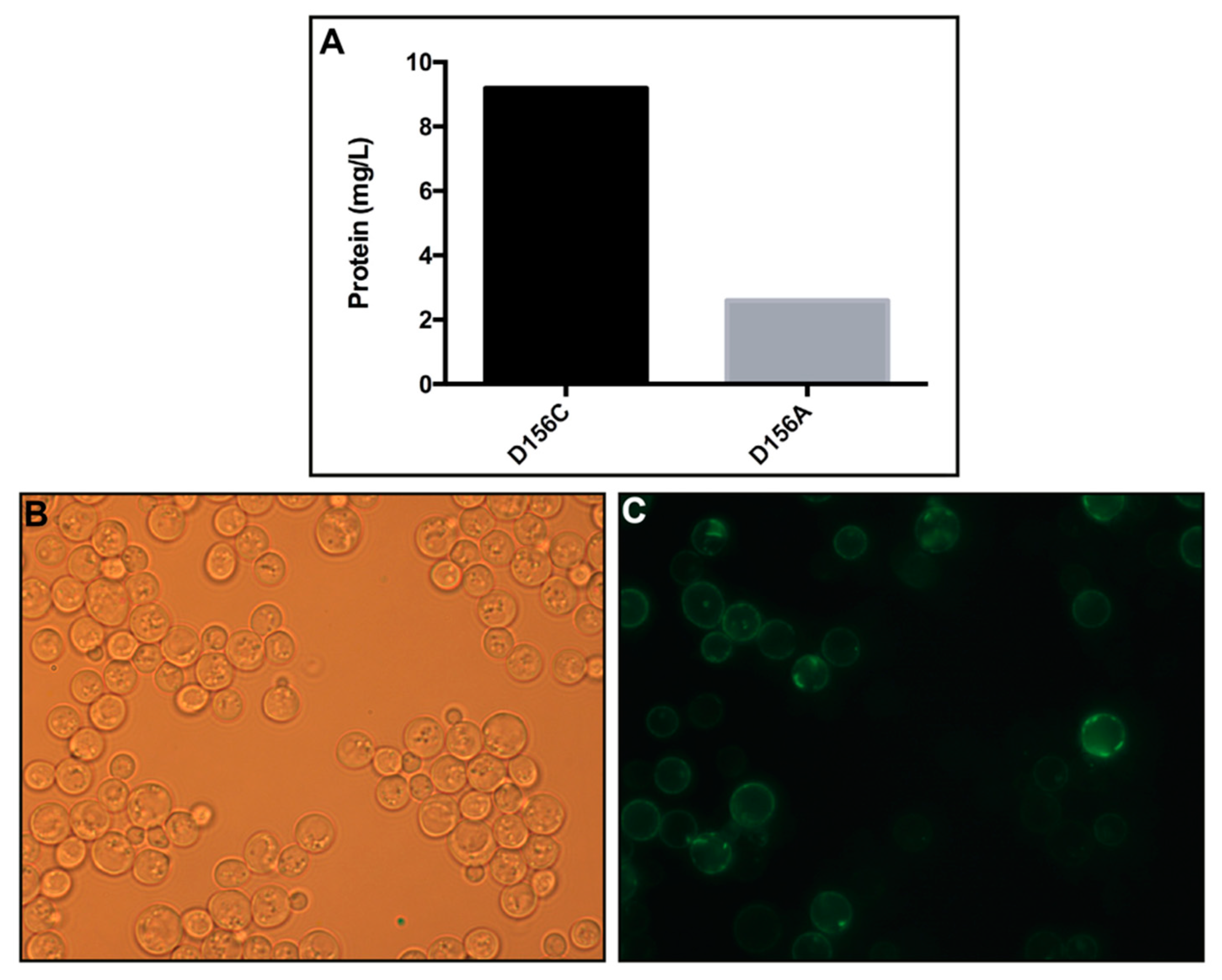
2.2. Protein Quantification Estimation by Whole-Cell Fluorescence
2.3. Bioimaging of Live Yeast Cells
2.4. Membrane Preparation
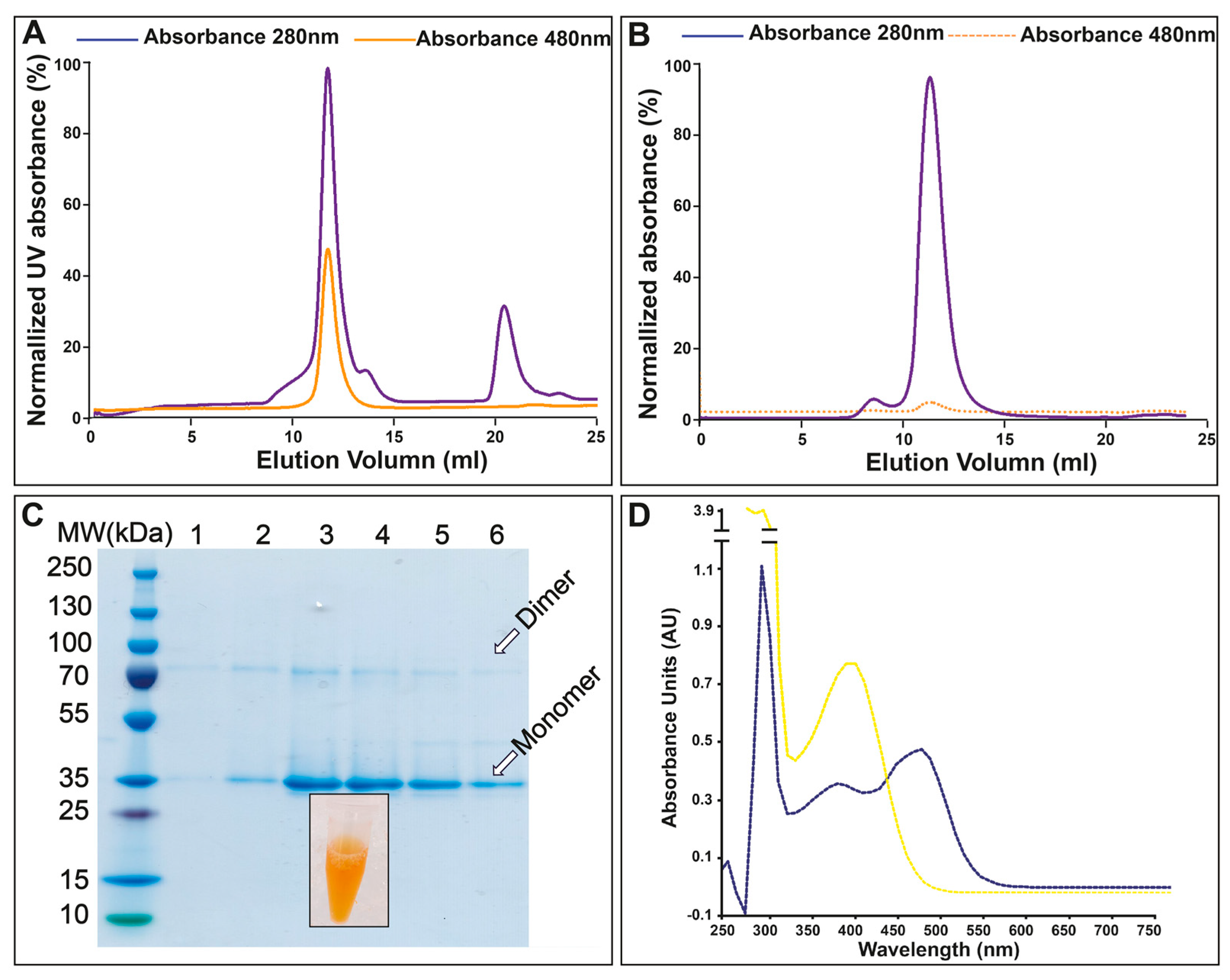
2.5. Protein Purification
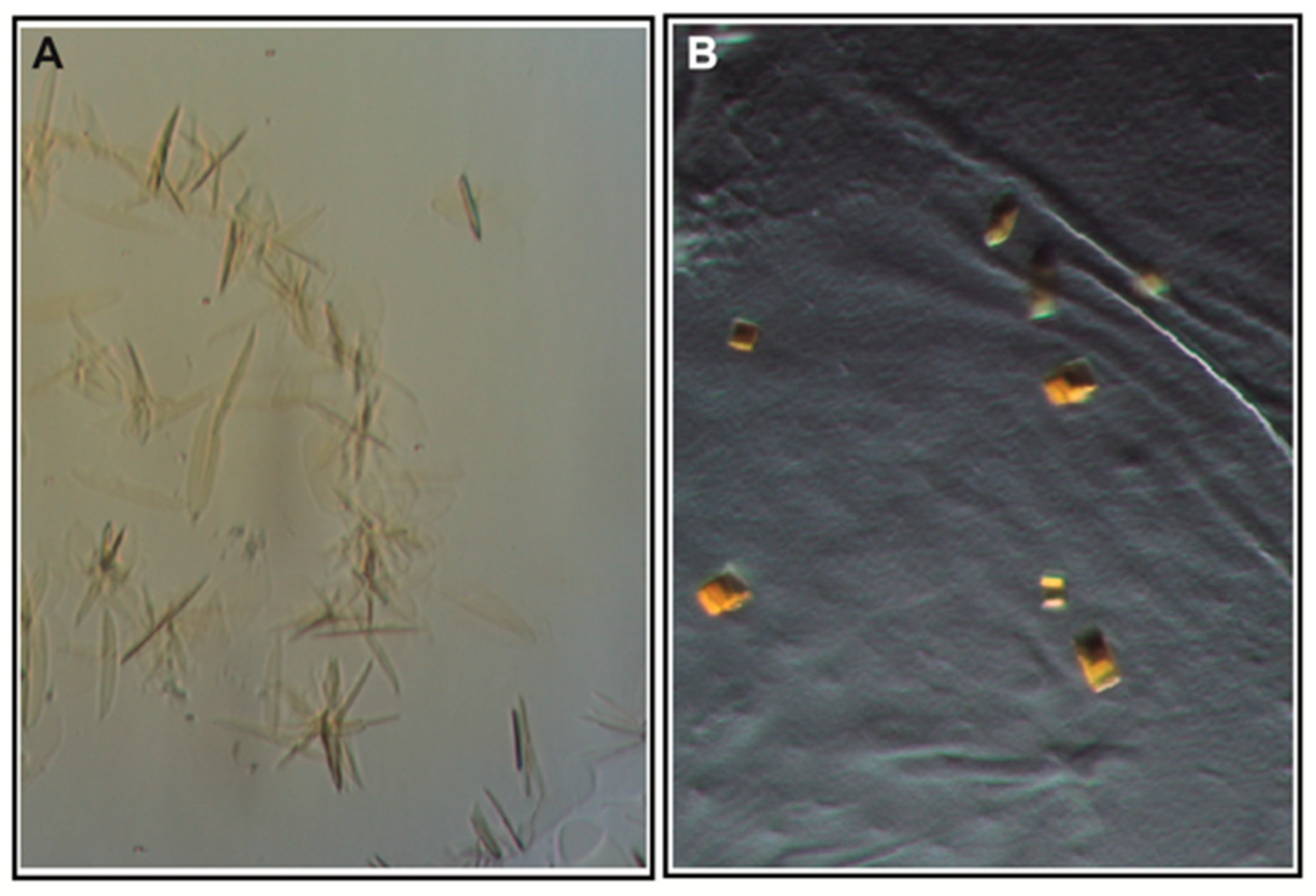
2.6. Crystallization and Optimization
2.7. Data Collection and Analysis
3. Results and Discussion
3.1. Production and Localization in S. cerevisiae
3.2. Purification and Absorption Spectra
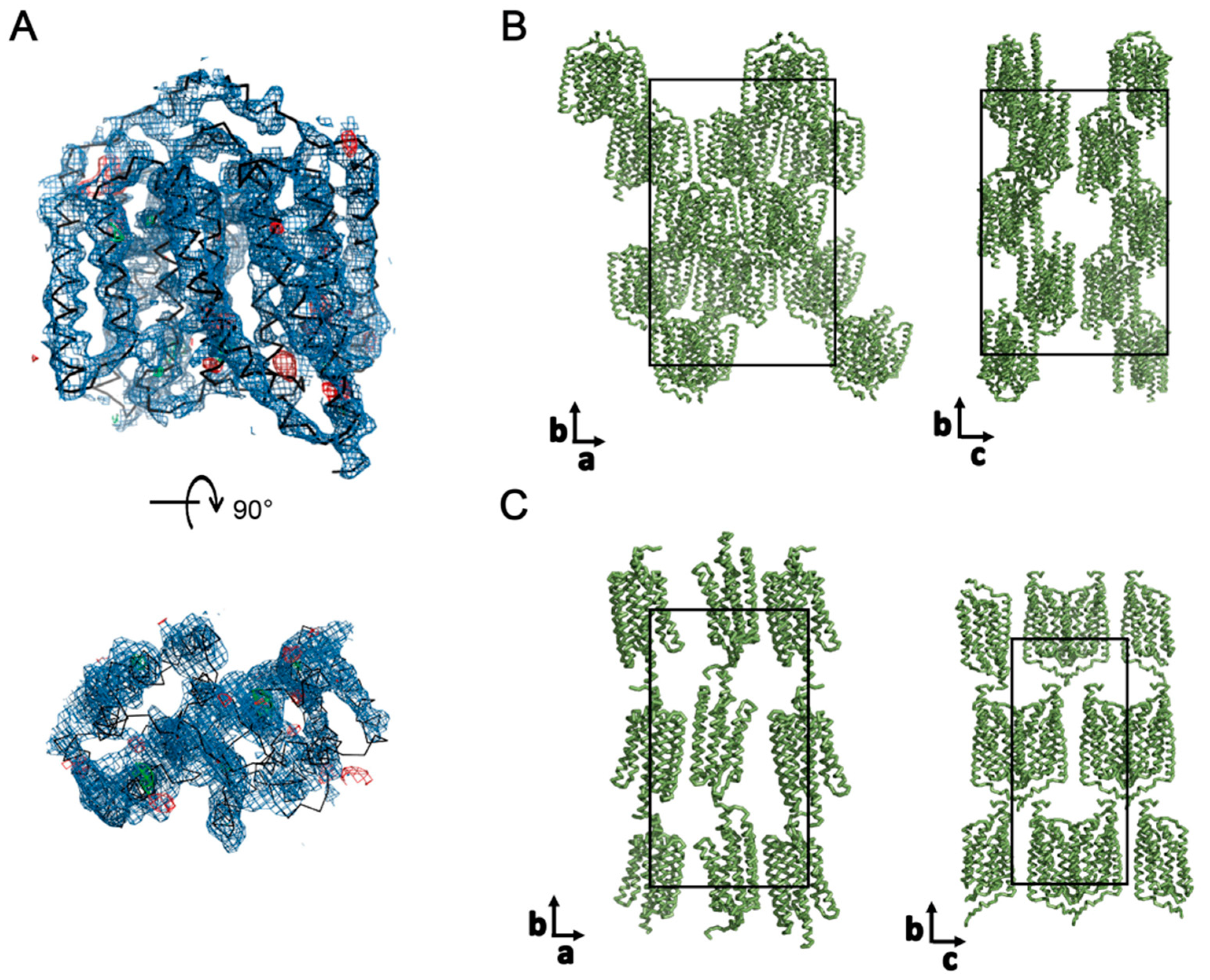
3.3. Crystallization
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arenkiel, B.R.; Peca, J.; Davison, I.G.; Feliciano, C.; Augustine, G.J.; Ehlers, M.D.; Feng, G. In Vivo Light-Induced Activation of Neural Circuitry in Transgenic Mice Expressing Channelrhodopsin-2. Neuron 2007, 54, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Peca, J.; Matsuzaki, M.; Matsuzaki, K.; Noguchi, J.; Qiu, L.; Wang, D.; Zhang, F.; Boyden, E.; Deisseroth, K.; et al. High-speed mapping of synaptic connectivity using photostimulation in Channelrhodopsin-2 transgenic mice. Proc. Natl. Acad. Sci. USA 2007, 104, 8143–8148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petreanu, L.; Huber, D.; Sobczyk, A.; Svoboda, K. Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat. Neurosci. 2007, 10, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Kateriya, S.; Berthold, P.; Bamberg, E.; Huhn, W.; Nagel, G.; Hegemann, P.; Adeishvili, N.; Szellas, T.; Ollig, D. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. USA 2003, 100, 13940–13945. [Google Scholar] [CrossRef] [Green Version]
- Ernst, O.P.; Hegemann, P.; Tsunoda, S.P.; Gradmann, D.; Mages, W.; Berthold, P. Channelrhodopsin-1 Initiates Phototaxis and Photophobic Responses in Chlamydomonas by Immediate Light-Induced Depolarization. Plant Cell Online 2008, 20, 1665–1677. [Google Scholar] [CrossRef] [Green Version]
- Josselyn, S.A. The past, present and future of light-gated ion channels and optogenetics. Elife 2018, 7, 1–5. [Google Scholar] [CrossRef]
- Steinbeck, J.A.; Choi, S.J.; Mrejeru, A.; Ganat, Y.; Deisseroth, K.; Sulzer, D.; Mosharov, E.V.; Studer, L. Optogenetics enables functional analysis of human embryonic stem cell-derived grafts in a Parkinson’s disease model. Nat. Biotechnol. 2015, 176, 139–148. [Google Scholar] [CrossRef]
- Scholl, H.P.N.; Strauss, R.W.; Singh, M.S.; Dalkara, D.; Roska, B.; Picaud, S.; Sahel, J.-A. Emerging therapies for inherited retinal degeneration. Sci. Transl. Med. 2016, 8, 368rv6. [Google Scholar] [CrossRef]
- Nagel, G.; Ollig, D.; Fuhrmann, M.; Kateriya, S.; Musti, A.M.; Bamberg, E.; Hegemann, P. Channelrhodopsin-1: A light-gated proton channel in green algae. Science 2002, 296, 2395–2398. [Google Scholar] [CrossRef]
- Harz, H.; Hegemann, P. Rhodopsin-regulated calcium currents in Chlamydomonas. Nature 1991, 351, 489–491. [Google Scholar] [CrossRef]
- Zhang, F.; Prigge, M.; Beyrière, F.; Tsunoda, S.P.; Mattis, J.; Yizhar, O.; Hegemann, P.; Deisseroth, K. Red-shifted optogenetic excitation: A tool for fast neural control derived from Volvox carteri. Nat. Neurosci. 2008, 11, 631–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klapoetke, N.C.; Murata, Y.; Kim, S.S.; Pulver, S.R.; Birdsey-Benson, A.; Cho, Y.K.; Morimoto, T.K.; Chuong, A.S.; Carpenter, E.J.; Tian, Z.; et al. Independent optical excitation of distinct neural populations. Nat. Methods 2014, 11, 338–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.Y. A user’s guide to channelrhodopsin variants: Features, limitations and future developments. Exp. Physiol. 2010, 96, 19–25. [Google Scholar] [CrossRef]
- Hegemann, P.; Gärtner, W.; Uhl, R. All-trans retinal constitutes the functional chromophore in Chlamydomonas rhodopsin. Biophys. J. 1991, 60, 1477–1489. [Google Scholar] [CrossRef] [Green Version]
- Ernst, O.P.; Sánchez Murcia, P.A.; Daldrop, P.; Tsunoda, S.P.; Kateriya, S.; Hegemann, P. Photoactivation of channelrhodopsin. J. Biol. Chem. 2008, 283, 1637–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, M.A.; Zacks, D.N.; Derguini, F.; Nakanishi, K.; Spudich, J.L. Retinal analog restoration of photophobic responses in a blind Chiamydomonas reinhardtii mutant. Biophys. J. 1991, 60, 1490–1498. [Google Scholar] [CrossRef] [Green Version]
- Bamann, C.; Kirsch, T.; Nagel, G.; Bamberg, E. Spectral Characteristics of the Photocycle of Channelrhodopsin-2 and Its Implication for Channel Function. J. Mol. Biol. 2008, 375, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Kandori, H.; Yamazaki, Y.; Sasaki, J.; Maeda, A.; Needleman, R.; Lanyi, J.K. Water-Mediated Proton Transfer in Proteins: An FTIR Study of Bacteriorhodopsin. J. Am. Chem. Soc. 1995, 117, 2118–2119. [Google Scholar] [CrossRef]
- Volkov, O.; Kovalev, K.; Polovinkin, V.; Borshchevskiy, V.; Bamann, C.; Astashkin, R.; Marin, E.; Popov, A.; Balandin, T.; Willbold, D.; et al. Structural insights into ion conduction by channelrhodopsin 2. Science 2017, 358. [Google Scholar] [CrossRef]
- Lórenz-Fonfría, V.A.; Heberle, J. Proton Transfer and Protein Conformation Dynamics in Photosensitive Proteins by Time-resolved Step-scan Fourier-transform Infrared Spectroscopy. J. Vis. Exp. 2014, 88, e51622. [Google Scholar] [CrossRef] [Green Version]
- Muders, V.; Kerruth, S.; Lórenz-Fonfría, V.A.; Bamann, C.; Heberle, J.; Schlesinger, R. Resonance Raman and FTIR spectroscopic characterization of the closed and open states of channelrhodopsin-1. FEBS Lett. 2014, 588, 2301–2306. [Google Scholar] [CrossRef] [PubMed]
- Ritter, E.; Puskar, L.; Bartl, F.J.; Aziz, E.F.; Hegemann, P.; Schade, U. Time-resolved infrared spectroscopic techniques as applied to channelrhodopsin. Front. Mol. Biosci. 2015, 2, 00038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Kato, H.E.; Taniguchi, R.; Iwata, T.; Nureki, O.; Kandori, H. Water-containing hydrogen-bonding network in the active center of channelrhodopsin. J. Am. Chem. Soc. 2014, 136, 3475–3482. [Google Scholar] [CrossRef] [PubMed]
- Hegemann, P.; Ehlenbeck, S.; Gradmann, D. Multiple photocycles of channelrhodopsin. Biophys. J. 2005, 89, 3911–3918. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, K.; Grossman, N.; Grubb, M.S.; Burrone, J.; Toumazou, C.; Degenaar, P. Photocycles of channelrhodopsin-2. Photochem. Photobiol. 2009, 85, 400–411. [Google Scholar] [CrossRef]
- Saita, M.; Pranga-Sellnau, F.; Resler, T.; Schlesinger, R.; Heberle, J.; Lorenz-Fonfria, V.A. Photoexcitation of the P4480State Induces a Secondary Photocycle That Potentially Desensitizes Channelrhodopsin-2. J. Am. Chem. Soc. 2018, 140, 9899–9903. [Google Scholar] [CrossRef] [Green Version]
- Kuhne, J.; Vierock, J.; Alexander, S.; Dreier, M.; Wietek, J. Title : A unifying photocycle model for light adaptation and temporal evolution of cation conductance in Channelrhodopsin-2. Proc. Natl. Acad. Sci. USA 2018, 116, 9380–9389. [Google Scholar] [CrossRef] [Green Version]
- Nack, M.; Radu, I.; Gossing, M.; Bamann, C.; Bamberg, E.; Von Mollard, G.F.; Heberle, J. The DC gate in channelrhodopsin-2: Crucial hydrogen bonding interaction between C128 and D156. Photochem. Photobiol. Sci. 2010, 9, 194–198. [Google Scholar] [CrossRef]
- Bamann, C.; Gueta, R.; Kleinlogel, S.; Nagel, G.; Bamberg, E. Structural guidance of the photocycle of channelrhodopsin-2 by an interhelical hydrogen bond. Biochemistry 2010, 49, 267–278. [Google Scholar] [CrossRef]
- Lorenz-Fonfria, V.A.; Resler, T.; Krause, N.; Nack, M.; Gossing, M.; Fischer von Mollard, G.; Bamann, C.; Bamberg, E.; Schlesinger, R.; Heberle, J. Transient protonation changes in channelrhodopsin-2 and their relevance to channel gating. Proc. Natl. Acad. Sci. USA 2013, 110, E1273–E1281. [Google Scholar] [CrossRef] [Green Version]
- Stehfest, K.; Ritter, E.; Berndt, A.; Bartl, F.; Hegemann, P. The branched photocycle of the slow-cycling channelrhodopsin-2 mutant C128T. J. Mol. Biol. 2010, 398, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Dawydow, A.; Gueta, R.; Ljaschenko, D.; Ullrich, S.; Hermann, M.; Ehmann, N.; Gao, S.; Fiala, A.; Langenhan, T.; Nagel, G.; et al. Channelrhodopsin-2-XXL, a powerful optogenetic tool for low-light applications. Proc. Natl. Acad. Sci. USA 2014, 111, 13972–13977. [Google Scholar] [CrossRef] [Green Version]
- Fenno, L.; Yizhar, O.; Deisseroth, K. The Development and Application of Optogenetics. Annu. Rev. Neurosci. 2011, 35, 2238–2256. [Google Scholar] [CrossRef] [PubMed]
- Felici, F.; Cesareni, G. Structure of the Saccharomyces cerevisiae gene encoding tRNAIle (IAU). Nucleic Acids Res. 1987, 15, 5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bomholt, J.; Helix-Nielsen, C.; Scharff-Poulsen, P.; Pedersen, P.A. Recombinant production of human Aquaporin-1 to an exceptional high membrane density in Saccharomyces cerevisiae. PLoS ONE 2013, 8, e56431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharff-Poulsen, P.; Pedersen, P.A. Saccharomyces cerevisiae-Based Platform for Rapid Production and Evaluation of Eukaryotic Nutrient Transporters and Transceptors for Biochemical Studies and Crystallography. PLoS ONE 2013, 8, e76851. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, J.R.; Pedersen, P.A. Role of phylogenetically conserved amino acids in folding of Na,K-ATPase. Biochemistry 2001, 40, 7301–7308. [Google Scholar] [CrossRef]
- Kabsch, W. Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.E.; Zhang, F.; Yizhar, O.; Ramakrishnan, C.; Nishizawa, T.; Hirata, K.; Ito, J.; Aita, Y.; Tsukazaki, T.; Hayashi, S.; et al. Crystal structure of the channelrhodopsin light-gated cation channel. Nature 2012, 482, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Mazza, F. Expression and Purification of Channelrhodopsin-2 in Pichia Pastoris: Generation of Blue-Shifted and Redshifted Optogenetic Variants. Ph.D. Thesis, University of Coimbra, Coimbra, Portugal, 2014. [Google Scholar]
- Ullrich, S.; Gueta, R.; Nagel, G. Degradation of channelopsin-2 in the absence of retinal and degradation resistance incertain mutants. Biol. Chem. 2013, 394, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Overproduction, P.; Hunter, C.N. Proteorhodopsin Overproduction Enhances the Long-Term Viability of Escherichia coli. Appl. Environ. Microbiol. 2020, 86, 1–14. [Google Scholar]
- Seki, T.; Fujishita, S.; Ito, M.; Matsuoka, N.; Tsukida, K. Retinoid composition in the compound eyes of insects. Exp. Biol. 1987, 47, 95–103. [Google Scholar]
- Waldo, G.S. Improving Protein Folding Efficiency by Directed Evolution Using the GFP Folding Reporter. In Directed Enzyme Evolution: Screening and Selection Methods; Arnold, F.H., Georgiou, G., Eds.; Humana Press: Totowa, NJ, USA, 2003; pp. 343–359. ISBN 978-1-59259-396-5. [Google Scholar]
- Gourdon, P.; Alfredsson, A.; Pedersen, A.; Malmerberg, E.; Nyblom, M.; Widell, M.; Berntsson, R.; Pinhassi, J.; Braiman, M.; Hansson, Ö.; et al. Optimized in vitro and in vivo expression of proteorhodopsin: A seven-transmembrane proton pump. Protein Expr. Purif. 2008, 58, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Krause, N. Structural Rearrangements upon Opening of Channelrhodopsin-2. Ph.D. Thesis, Freie Universität Berlin, Berlin, Germany, 2016. [Google Scholar]
- Bill, R.M.; Henderson, P.J.F.; Iwata, S.; Kunji, E.R.S.; Michel, H.; Neutze, R.; Newstead, S.; Poolman, B.; Tate, C.G.; Vogel, H. Overcoming barriers to membrane protein structure determination. Nat. Biotechnol. 2011, 29, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Gourdon, P.; Andersen, J.L.; Hein, K.L.; Bublitz, M.; Pedersen, B.P.; Liu, X.Y.; Yatime, L.; Nyblom, M.; Nielsen, T.T.; Olesen, C.; et al. HiLiDe-systematic approach to membrane protein crystallization in lipid and detergent. Cryst. Growth Des. 2011, 11, 2098–2106. [Google Scholar] [CrossRef]
- Caffrey, M. A comprehensive review of the lipid cubic phase or in meso method for crystallizing membrane and soluble proteins and complexes. Acta Crystallogr. Sect. FStructural Biol. Commun. 2015, 71, 3–18. [Google Scholar] [CrossRef] [Green Version]
- Royant, A.; Edman, K.; Ursby, T.; Pebay-Peyroula, E.; Landau, E.M.; Neutze, R. Helix deformation is coupled to vectorial proton transport in the photocycle of bacteriorhodopsin. Nature 2000, 406, 645–648. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Space Group | C222 |
|---|---|
| a, b, c, Å | 117.095 179.928 126.471 |
| α, β, γ, ° | 90.00 90.00 90.00 |
| Resolution, Å | 50–5.0 (5.3–5.0) |
| Rmerge % | 30.1 (337.5) |
| I/σ | 2.7 (0.5) |
| Completeness, % | 99.4 (98.4) |
| CC(1/2) | 100.0 (79.4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Wang, K.; Ning, S.; Pedersen, P.A.; Duelli, A.S.; Gourdon, P.E. Isolation and Crystallization of the D156C Form of Optogenetic ChR2. Cells 2022, 11, 895. https://doi.org/10.3390/cells11050895
Zhang L, Wang K, Ning S, Pedersen PA, Duelli AS, Gourdon PE. Isolation and Crystallization of the D156C Form of Optogenetic ChR2. Cells. 2022; 11(5):895. https://doi.org/10.3390/cells11050895
Chicago/Turabian StyleZhang, Liying, Kaituo Wang, Shuo Ning, Per Amstrup Pedersen, Annette Susanne Duelli, and Pontus Emanuel Gourdon. 2022. "Isolation and Crystallization of the D156C Form of Optogenetic ChR2" Cells 11, no. 5: 895. https://doi.org/10.3390/cells11050895