
Into the Tissues: Extracellular Matrix and Its Artificial Substitutes: Cell Signalling Mechanisms



Abstract
:
{kind=link}
{kind=link}
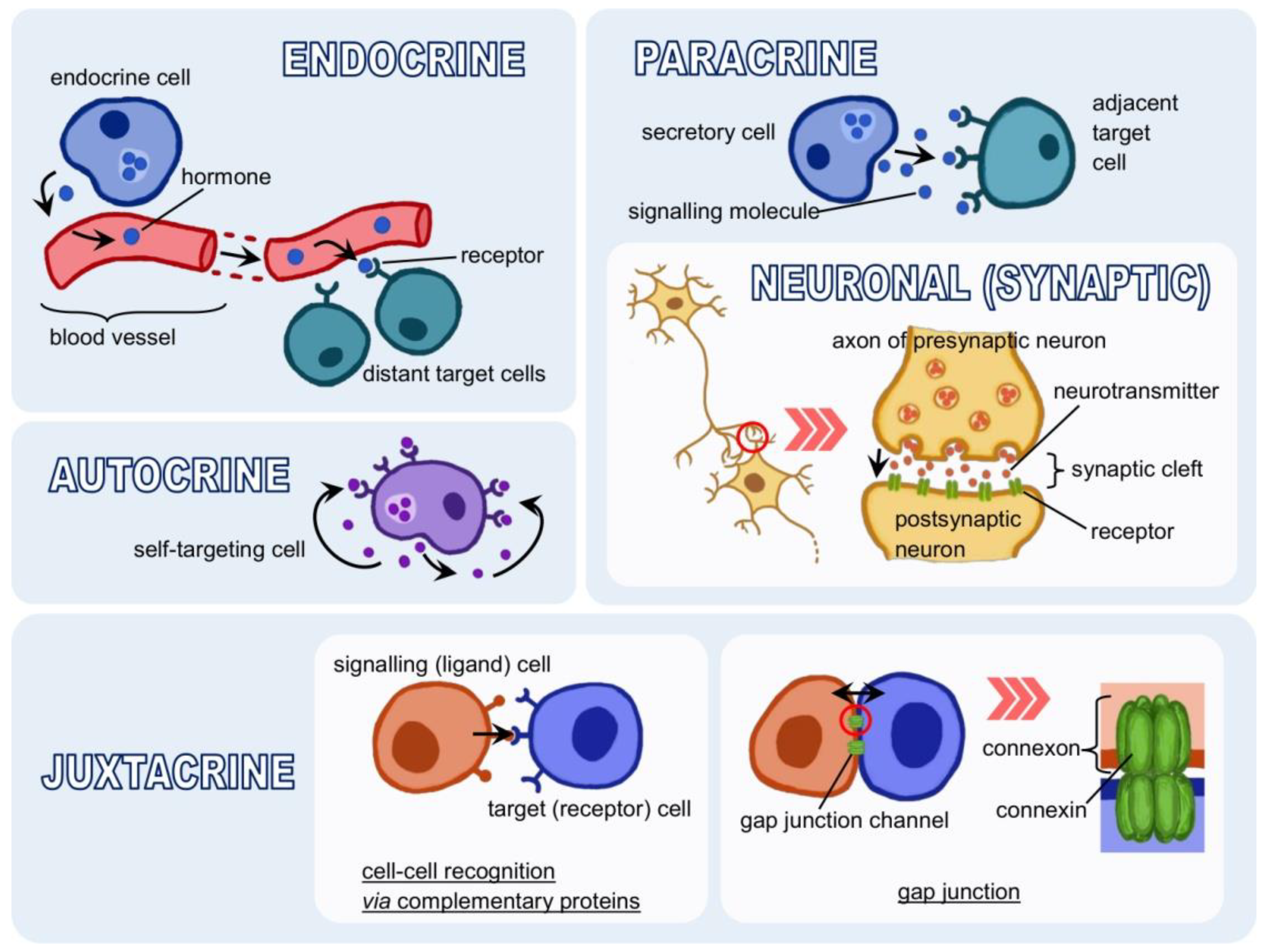{kind=link}
{kind=link}
{kind=link}
1. Introduction
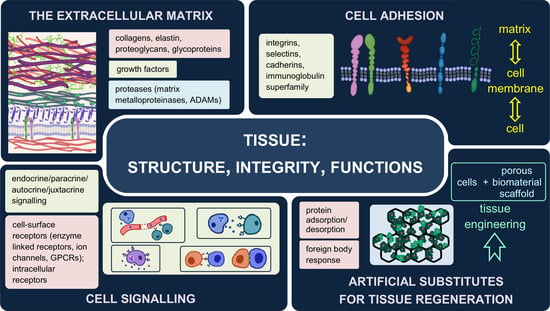
2. The Extracellular Matrix—Composition, Structure, Functions
2.1. Two Types of the Extracellular Matrix
2.2. Major Components of the Extracellular Matrix and Their Functions
2.2.1. Collagens
2.2.2. Elastin
2.2.3. Proteoglycans
2.2.4. Glycoproteins
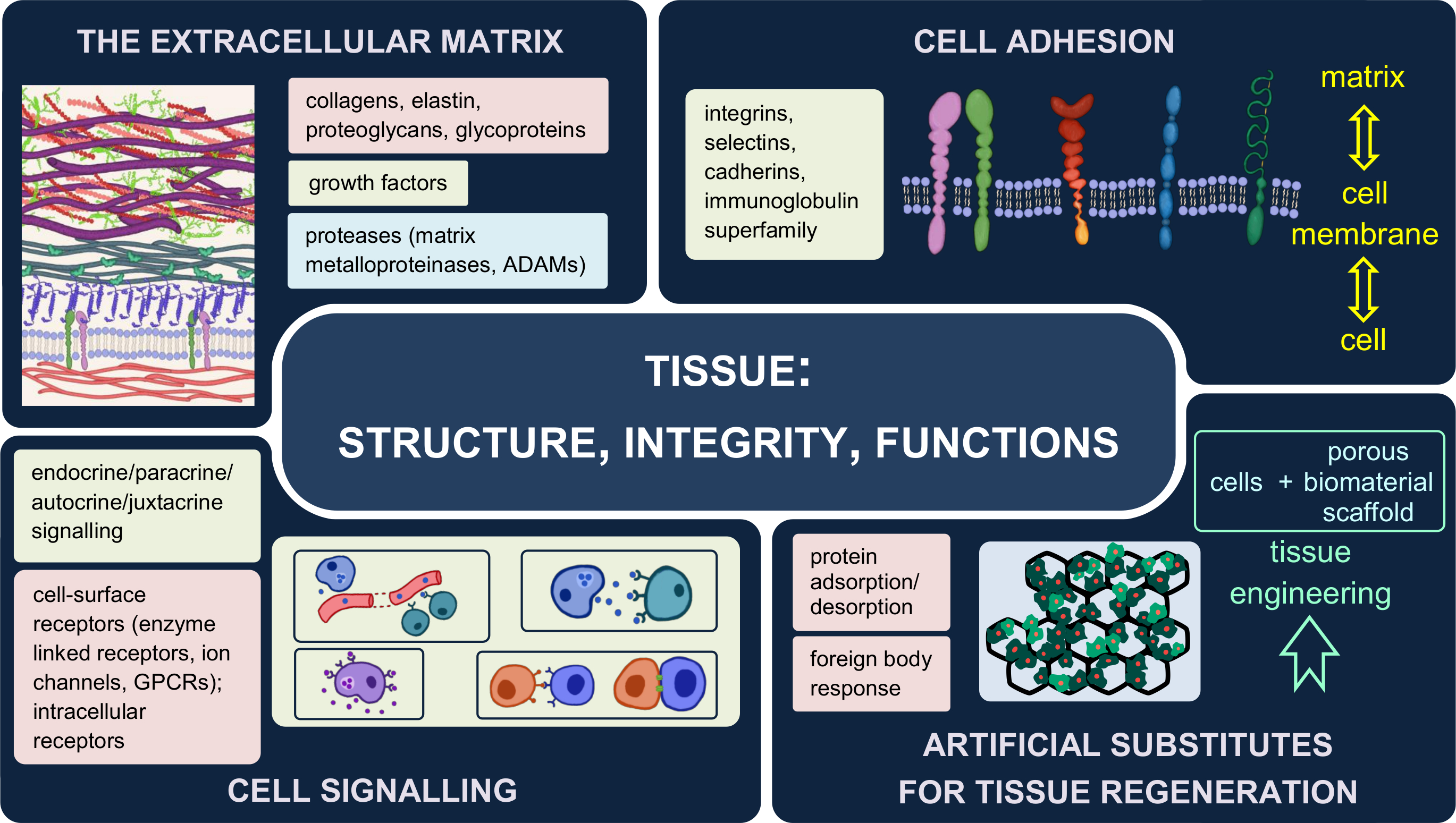
2.3. The Dynamic Structure of the Extracellular Matrix
2.4. The Extracellular Matrix as a Storehouse of Growth Factors
2.5. Anoikis—Programmed Death
2.6. Genetic Mutations of Matrix Components and Their Consequences
3. Interactions between Cells and Their Environment
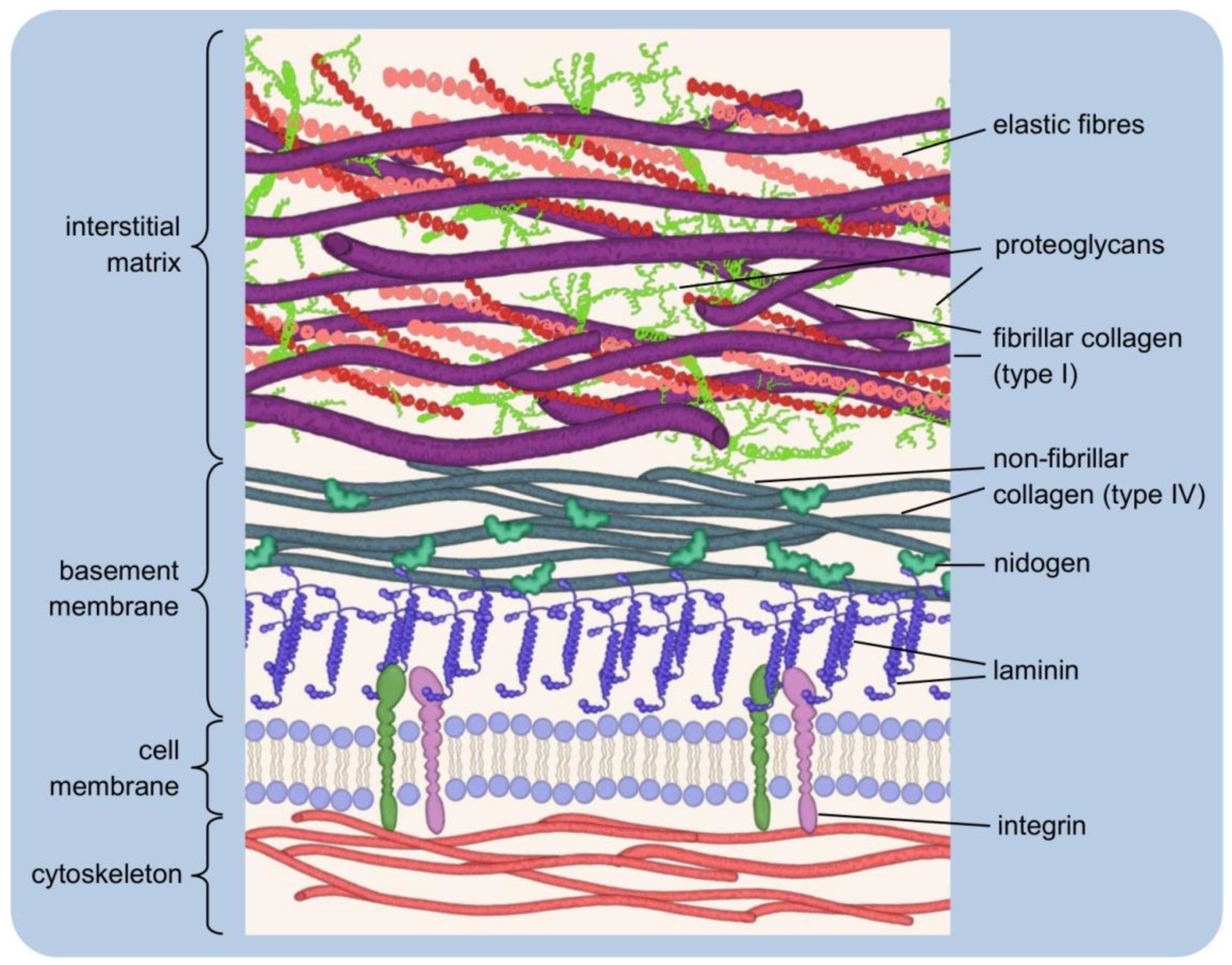
3.1. Forms of Signalling
3.2. Receptors
Cell Adhesion Molecules (CAMs)
4. Artificial Substitutes of the Extracellular Matrix
4.1. Host Response to Implantation
4.2. Influence of Material Properties on Cell Adhesion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Manou, D.; Caon, I.; Bouris, P.; Triantaphyllidou, I.E.; Giaroni, C.; Passi, A.; Karamanos, N.K.; Vigetti, D.; Theocharis, A.D. The complex interplay between extracellular matrix and cells in tissues. In Methods in Molecular Biology; Humana Press Inc.: Berlin/Heidelberg, Germany, 2019; Volume 1952, pp. 1–20. [Google Scholar]
- Muncie, J.M.; Weaver, V.M. The Physical and Biochemical Properties of the Extracellular Matrix Regulate Cell Fate. In Current Topics in Developmental Biology; Academic Press Inc.: Amsterdam, The Netherlands, 2018; Volume 130, pp. 1–37. [Google Scholar]
- Berrier, A.L.; Yamada, K.M. Cell-matrix adhesion. J. Cell. Physiol. 2007, 213, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Chauhan, P.; Saha, B.; Kubatzky, K.F. Conceptual evolution of cell signaling. Int. J. Mol. Sci. 2019, 20, 3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; King, M.W. Biodegradable Polymers as the Pivotal Player in the Design of Tissue Engineering Scaffolds. Adv. Healthc. Mater. 2020, 9, 1901358. [Google Scholar] [CrossRef] [PubMed]
- Bosman, F.T.; Stamenkovic, I. Functional structure and composition of the extracellular matrix. J. Pathol. 2003, 200, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- McKee, T.J.; Perlman, G.; Morris, M.; Komarova, S.V. Extracellular matrix composition of connective tissues: A systematic review and meta-analysis. Sci. Rep. 2019, 9, 10542. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, M.; Kaiser, E.; Milz, S. Structure-function relationships in tendons: A review. J. Anat. 2008, 212, 211–228. [Google Scholar] [CrossRef]
- Gentili/snm, C.; Cancedda, R. Cartilage and Bone Extracellular Matrix. Curr. Pharm. Des. 2009, 15, 1334–1348. [Google Scholar] [CrossRef]
- Chun, S.Y.; Lim, J.O.; Lee, E.H.; Han, M.-H.; Ha, Y.-S.; Lee, J.N.; Kim, B.S.; Park, M.J.; Yeo, M.; Jung, B.; et al. Preparation and Characterization of Human Adipose Tissue-Derived Extracellular Matrix, Growth Factors, and Stem Cells: A Concise Review. Tissue Eng. Regen. Med. 2019, 16, 385. [Google Scholar] [CrossRef]
- Phan, S.H. Biology of Fibroblasts and Myofibroblasts. Proc. Am. Thorac. Soc. 2008, 5, 334. [Google Scholar] [CrossRef]
- Shaw, T.J.; Rognoni, E. Dissecting Fibroblast Heterogeneity in Health and Fibrotic Disease. Curr. Rheumatol. Rep. 2020, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Manou, D.; Karamanos, N.K. The extracellular matrix as a multitasking player in disease. FEBS J. 2019, 286, 2830–2869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kular, J.K.; Basu, S.; Sharma, R.I. The extracellular matrix: Structure, composition, age-related differences, tools for analysis and applications for tissue engineering. J. Tissue Eng. 2014, 5, 112. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Halfter, W.; Candiello, J.; Hu, H.; Zhang, P.; Schreiber, E.; Balasubramani, M. Protein composition and biomechanical properties of in vivo-derived basement membranes. Cell Adhes. Migr. 2013, 7, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Pozzi, A.; Yurchenco, P.D.; Iozzo, R.V. The nature and biology of basement membranes. Matrix Biol. 2017, 57–58, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yurchenco, P.; Patton, B. Developmental and Pathogenic Mechanisms of Basement Membrane Assembly. Curr. Pharm. Des. 2009, 15, 1277–1294. [Google Scholar] [CrossRef] [Green Version]
- Bornstein, P.; Sage, E.H. Matricellular proteins: Extracellular modulators of cell function. Curr. Opin. Cell Biol. 2002, 14, 608–616. [Google Scholar] [CrossRef]
- Murphy-Ullrich, J.E.; Sage, E.H. Revisiting the matricellular concept. Matrix Biol. 2014, 37, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.E.; Kelley, M.J.; Acott, T.S. Extracellular matrix gene alternative splicing by trabecular meshwork cells in response to mechanical stretching. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, P. Matricellular proteins: An overview. J. Cell Commun. Signal. 2009, 3, 163–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosset, E.M.; Bradshaw, A.D. SPARC/osteonectin in mineralized tissue. Matrix Biol. 2016, 52–54, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornstein, P. Thrombospondins as matricellular modulators of cell function. J. Clin. Investig. 2001, 107, 929–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, S.E.; Torricelli, A.A.M.; Marino, G.K. Corneal epithelial basement membrane: Structure, function and regeneration. Exp. Eye Res. 2020, 194, 108002. [Google Scholar] [CrossRef]
- Kvist, A.J.; Nyström, A.; Hultenby, K.; Sasaki, T.; Talts, J.F.; Aspberg, A. The major basement membrane components localize to the chondrocyte pericellular matrix-A cartilage basement membrane equivalent? Matrix Biol. 2008, 27, 22–33. [Google Scholar] [CrossRef]
- Zhang, Z. Chondrons and the Pericellular Matrix of Chondrocytes. Tissue Eng.-Part B Rev. 2015, 21, 267–277. [Google Scholar] [CrossRef]
- Youn, I.; Choi, J.B.; Cao, L.; Setton, L.A.; Guilak, F. Zonal variations in the three-dimensional morphology of the chondron measured in situ using confocal microscopy. Osteoarthr. Cartil. 2006, 14, 889–897. [Google Scholar] [CrossRef] [Green Version]
- Poole, C.A. Review. Articular cartilage chondrons: Form, function and failure. J. Anat. 1997, 191, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fraser, S.A.; Crawford, A.; Frazer, A.; Dickinson, S.; Hollander, A.P.; Brook, I.M.; Hatton, P.V. Localization of type VI collagen in tissue-engineered cartilage on polymer scaffolds. Tissue Eng. 2006, 12, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome-An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, M. Update on Collagens: What You Need to Know and Consider. Plast. Surg. Nurs. 2019, 39, 112–115. [Google Scholar] [CrossRef]
- Exposito, J.Y.; Valcourt, U.; Cluzel, C.; Lethias, C. The fibrillar collagen family. Int. J. Mol. Sci. 2010, 11, 407–426. [Google Scholar] [CrossRef] [Green Version]
- Bella, J.; Hulmes, D.J.S. Fibrillar collagens. Subcell. Biochem. 2017, 82, 457–490. [Google Scholar] [CrossRef]
- Mienaltowski, M.J.; Birk, D.E. Structure, Physiology, and Biochemistry of Collagens. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2014; Volume 802, pp. 5–29. [Google Scholar]
- Shaw, L.M.; Olsen, B.R. FACIT collagens: Diverse molecular bridges in extracellular matrices. Trends Biochem. Sci. 1991, 16, 191–194. [Google Scholar] [CrossRef]
- Ricard-Blum, S.; Ruggiero, F. The collagen superfamily: From the extracellular matrix to the cell membrane. Pathol. Biol. 2005, 53, 430–442. [Google Scholar] [CrossRef]
- Ivanova, V.P.; Krivchenko, A.I. Current viewpoint on structure and on evolution of collagens. II. Fibril-associated collagens. J. Evol. Biochem. Physiol. 2014, 50, 273–285. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef] [Green Version]
- Harsha, L.; Brundha, M.P. Role of collagen in wound healing. Drug Invent. Today 2020, 13, 55–57. [Google Scholar]
- Heino, J. The collagen family members as cell adhesion proteins. BioEssays 2007, 29, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Luckman, S.P.; Rees, E.; Kwan, A.P.L. Partial characterization of cell-type X collagen interactions. Biochem. J. 2003, 372, 485–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smethurst, P.A.; Onley, D.J.; Jarvis, G.E.; O’Connor, M.N.; Graham Knight, C.; Herr, A.B.; Ouwehand, W.H.; Farndale, R.W. Structural basis for the platelet-collagen interaction: The smallest motif within collagen that recognizes and activates platelet Glycoprotein VI contains two glycine-proline-hydroxyproline triplets. J. Biol. Chem. 2007, 282, 1296–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paavola, K.J.; Sidik, H.; Zuchero, J.B.; Eckart, M.; Talbot, W.S. Type IV collagen is an activating ligand for the adhesion G protein-coupled receptor GPR126. Sci. Signal. 2014, 7, ra76. [Google Scholar] [CrossRef] [Green Version]
- Wolf, K.; Alexander, S.; Schacht, V.; Coussens, L.M.; von Andrian, U.H.; van Rheenen, J.; Deryugina, E.; Friedl, P. Collagen-based cell migration models in vitro and in vivo. Semin. Cell Dev. Biol. 2009, 20, 931–941. [Google Scholar] [CrossRef] [Green Version]
- Guido, S.; Tranquillo, R.T. A methodology for the systematic and quantitative study of cell contact guidance in oriented collagen gels. Correlation of fibroblast orientation and gel birefringence. J. Cell Sci. 1993, 105, 317–331. [Google Scholar] [CrossRef]
- Borgne-Rochet, M.L.; Angevin, L.; Bazellières, E.; Ordas, L.; Comunale, F.; Denisov, E.V.; Tashireva, L.A.; Perelmuter, V.M.; Bièche, I.; Vacher, S.; et al. P-cadherin-induced decorin secretion is required for collagen fiber alignment and directional collective cell migration. J. Cell Sci. 2019, 132, 3189. [Google Scholar] [CrossRef]
- Canty, E.G.; Kadler, K.E. Collagen fibril biosynthesis in tendon: A review and recent insights. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2002, 133, 979–985. [Google Scholar] [CrossRef]
- Zhang, G.; Young, B.B.; Birk, D.E. Differential expression of type XII collagen in developing chicken metatarsal tendons. J. Anat. 2003, 202, 411–420. [Google Scholar] [CrossRef]
- Ricard-Blum, S. The Collagen Family. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parenteau-Bareil, R.; Gauvin, R.; Berthod, F. Collagen-based biomaterials for tissue engineering applications. Materials 2010, 3, 1863–1887. [Google Scholar] [CrossRef] [Green Version]
- Cocciolone, A.J.; Hawes, J.Z.; Staiculescu, M.C.; Johnson, E.O.; Murshed, M.; Wagenseil, J.E. Elastin, arterial mechanics, and cardiovascular disease. Am. J. Physiol.-Hear Circ. Physiol. 2018, 315, H189–H205. [Google Scholar] [CrossRef] [PubMed]
- Mariani, T.J.; Dunsmore, S.E.; Li, Q.; Ye, X.; Pierce, R.A. Regulation of lung fibroblast tropoelastin expression by alveolar epithelial cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1998, 274, L47–L57. [Google Scholar] [CrossRef] [PubMed]
- Mecham, R.P.; Madaras, J.; McDonald, J.A.; Ryan, U. Elastin production by cultured calf pulmonary artery endothelial cells. J. Cell. Physiol. 1983, 116, 282–288. [Google Scholar] [CrossRef]
- Kajiya, H.; Tanaka, N.; Inazumi, T.; Seyama, Y.; Tajima, S.; Ishibashi, A. Cultured human keratinocytes express tropoelastin. J. Investig. Dermatol. 1997, 109, 641–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, A.S.; Sandberg, L.B.; Ross, R.; Layman, D.L. The smooth muscle cell: III. Elastin synthesis in arterial smooth muscle cell culture. J. Cell Biol. 1976, 68, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Nishizaki, T. PKCϵ Increases Extracellular Elastin and Fibulin-5/DANCE in Dermal Fibroblasts. Cell. Physiol. Biochem. 2018, 46, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Vrhovski, B.; Weiss, A.S. Biochemistry of tropoelastin. Eur. J. Biochem. 1998, 258, 1–18. [Google Scholar] [CrossRef]
- Ozsvar, J.; Yang, C.; Cain, S.A.; Baldock, C.; Tarakanova, A.; Weiss, A.S. Tropoelastin and Elastin Assembly. Front. Bioeng. Biotechnol. 2021, 9, 138. [Google Scholar] [CrossRef]
- Vindin, H.; Mithieux, S.M.; Weiss, A.S. Elastin architecture. Matrix Biol. 2019, 84, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Lucero, H.A.; Kagan, H.M. Lysyl oxidase: An oxidative enzyme and effector of cell function. Cell. Mol. Life Sci. 2006, 63, 2304–2316. [Google Scholar] [CrossRef] [PubMed]
- Wagenseil, J.E.; Mecham, R.P. New insights into elastic fiber assembly. Birth Defects Res. Part C-Embryo Today Rev. 2007, 81, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.K.; Simpson, A.; Steer, R.; Cain, S.A.; Kielty, C.M. Elastic fibres in health and disease. Expert Rev. Mol. Med. 2013, 15, 23. [Google Scholar] [CrossRef] [Green Version]
- Thomson, J.; Singh, M.; Eckersley, A.; Cain, S.A.; Sherratt, M.J.; Baldock, C. Fibrillin microfibrils and elastic fibre proteins: Functional interactions and extracellular regulation of growth factors. Semin. Cell Dev. Biol. 2019, 89, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Ritty, T.M.; Ditsios, K.; Starcher, B.C. Distribution of the elastic fiber and associated proteins in flexor tendon reflects function. Anat. Rec. 2002, 268, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Gabriela Espinosa, M.; Catalin Staiculescu, M.; Kim, J.; Marin, E.; Wagenseil, J.E. Elastic Fibers and Large Artery Mechanics in Animal Models of Development and Disease. J. Biomech. Eng. 2018, 140, 0208031. [Google Scholar] [CrossRef]
- Dubick, M.A.; Rucker, R.B.; Cross, C.E.; Last, J.A. Elastin metabolism in rodent lung. BBA-Gen. Subj. 1981, 672, 303–306. [Google Scholar] [CrossRef]
- Davidson, J.M.; Smith, K.; Shibahara, S.; Tolstoshev, P.; Crystal, R.G. Regulation of elastin synthesis in developing sheep nuchal ligament by elastin mRNA levels. J. Biol. Chem. 1982, 257, 747–754. [Google Scholar] [CrossRef]
- Burnett, W.; Finnigan-Bunick, A.; Yoon, K.; Rosenbloom, J. Analysis of elastin gene expression in the developing chick aorta using cloned elastin cDNA. J. Biol. Chem. 1982, 257, 1569–1572. [Google Scholar] [CrossRef]
- Kühl, T.; Mezger, M.; Hausser, I.; Guey, L.T.; Handgretinger, R.; Bruckner-Tuderman, L.; Nyström, A. Collagen VII Half-Life at the Dermal-Epidermal Junction Zone: Implications for Mechanisms and Therapy of Genodermatoses. J. Investig. Dermatol. 2016, 136, 1116–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, S.D.; Endicott, S.K.; Province, M.A.; Pierce, J.A.; Campbell, E.J. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J. Clin. Investig. 1991, 87, 1828–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagisawa, H.; Wagenseil, J. Elastic fibers and biomechanics of the aorta: Insights from mouse studies. Matrix Biol. 2020, 85–86, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, H.; Davis, E.C. Unraveling the mechanism of elastic fiber assembly: The roles of short fibulins. Int. J. Biochem. Cell Biol. 2010, 42, 1084–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef] [PubMed]
- Perrimon, N.; Bernfield, M. Cellular functions of proteoglycans—An overview. Semin. Cell Dev. Biol. 2001, 12, 65–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardingham, T.E.; Fosang, A.J. Proteoglycans: Many forms and many functions. FASEB J. 1992, 6, 861–870. [Google Scholar] [CrossRef]
- Rozario, T.; DeSimone, D.W. The Extracellular Matrix In Development and Morphogenesis: A Dynamic View. Dev. Biol. 2010, 341, 126. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, U. A personal voyage through the proteoglycan field. Matrix Biol. 2014, 35, 3–7. [Google Scholar] [CrossRef]
- Vynios, D.H.; Karamanos, N.K.; Tsiganos, C.P. Advances in analysis of glycosaminoglycans: Its application for the assessment of physiological and pathological states of connective tissues. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2002, 781, 21–38. [Google Scholar] [CrossRef]
- Kjellén, L.; Lindahl, U. Proteoglycans: Structures and interactions. Annu. Rev. Biochem. 1991, 60, 443–475. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Mancera, R.L. The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Cai, H.; Long, X.; Cheng, K.; Xu, X.; Zhang, D.; Li, J. Hyaluronic acid bioinspired polymers for the regulation of cell chondrogenic and osteogenic differentiation. Int. J. Biol. Macromol. 2020, 161, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Ghiselli, G. Drug-Mediated Regulation of Glycosaminoglycan Biosynthesis. Med. Res. Rev. 2017, 37, 1051–1094. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, T.E.; Ewins, R.J.F.; Muir, H. Cartilage proteoglycans. Structure and heterogeneity of the protein core and the effects of specific protein modifications on the binding to hyaluronate. Biochem. J. 1976, 157, 127–143. [Google Scholar] [CrossRef] [Green Version]
- Hascall, V.C. Interaction of cartilage proteoglycans with hyaluronic acid. J. Supramol. Cell. Biochem. 1977, 7, 101–120. [Google Scholar] [CrossRef]
- Rosenberg, L.; Hellmann, W.; Kleinschmidt, A.K. Electron microscopic studies of proteoglycan aggregates from bovine articular cartilage. J. Biol. Chem. 1975, 250, 1877–1883. [Google Scholar] [CrossRef]
- Kobayashi, T.; Chanmee, T.; Itano, N. Hyaluronan: Metabolism and function. Biomolecules 2020, 10, 1525. [Google Scholar] [CrossRef]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Esko, J.D. Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 2014, 83, 129–157. [Google Scholar] [CrossRef]
- Dudhia, J. Aggrecan, aging and assembly in articular cartilage. Cell. Mol. Life Sci. 2005, 62, 2241–2256. [Google Scholar] [CrossRef] [PubMed]
- Walimbe, T.; Panitch, A. Proteoglycans in biomedicine: Resurgence of an underexploited class of ECM molecules. Front. Pharmacol. 2020, 10, 1661. [Google Scholar] [CrossRef] [PubMed]
- Elfenbein, A.; Simons, M. Syndecan-4 signaling at a glance. J. Cell Sci. 2013, 126, 3799–3804. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chung, H.; Jung, H.; Couchman, J.R.; Oh, E.S. Syndecans as cell surface receptors: Unique structure equates with functional diversity. Matrix Biol. 2011, 30, 93–99. [Google Scholar] [CrossRef]
- Kolset, S.O.; Tveit, H. Serglycin-Structure and biology. Cell. Mol. Life Sci. 2008, 65, 1073–1085. [Google Scholar] [CrossRef]
- Henningsson, F.; Hergeth, S.; Cortelius, R.; Åbrink, M.; Pejler, G. A role for serglycin proteoglycan in granular retention and processing of mast cell secretory granule components. FEBS J. 2006, 273, 4901–4912. [Google Scholar] [CrossRef] [PubMed]
- Åbrink, M.; Grujic, M.; Pejler, G. Serglycin is essential for maturation of mast cell secretory granule. J. Biol. Chem. 2004, 279, 40897–40905. [Google Scholar] [CrossRef] [Green Version]
- Whitelock, J.M.; Graham, L.D.; Melrose, J.; Murdoch, A.D.; Iozzo, R.V.; Anne Underwood, P. Human perlecan immunopurified from different endothelial cell sources has different adhesive properties for vascular cells. Matrix Biol. 1999, 18, 163–178. [Google Scholar] [CrossRef]
- Sher, I.; Zisman-Rozen, S.; Eliahu, L.; Whitelock, J.M.; Maas-Szabowski, N.; Yamada, Y.; Breitkreutz, D.; Fusenig, N.E.; Arikawa-Hirasawa, E.; Iozzo, R.V.; et al. Targeting perlecan in human keratinocytes reveals novel roles for perlecan in epidermal formation. J. Biol. Chem. 2006, 281, 5178–5187. [Google Scholar] [CrossRef] [Green Version]
- Nugent, M.A.; Nugent, H.M.; Iozzo, R.V.; Sanchack, K.; Edelman, E.R. Perlecan is required to inhibit thrombosis after deep vascular injury and contributes to endothelial cell-mediated inhibition of intimal hyperplasia. Proc. Natl. Acad. Sci. USA 2000, 97, 6722–6727. [Google Scholar] [CrossRef] [Green Version]
- Iozzo, R.V. Basement membrane proteoglycans: From cellar to ceiling. Nat. Rev. Mol. Cell Biol. 2005, 6, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Bezakova, G.; Ruegg, M.A. New insights into the roles of agrin. Nat. Rev. Mol. Cell Biol. 2003, 4, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yang, L.; Li, G.; Jin, Y.; Wu, D.; Wang, Q.M.; Huang, P. Agrin Involvement in Synaptogenesis Induced by Exercise in a Rat Model of Experimental Stroke. Neurorehabil. Neural Repair 2020, 34, 1124–1137. [Google Scholar] [CrossRef] [PubMed]
- Jan, A.T.; Lee, E.J.; Choi, I. Fibromodulin: A regulatory molecule maintaining cellular architecture for normal cellular function. Int. J. Biochem. Cell Biol. 2016, 80, 66–70. [Google Scholar] [CrossRef]
- Ezura, Y.; Chakravarti, S.; Oldberg, A.; Chervoneva, I.; Birk, D.E. Differential expression of lumican and fibromodulin regulate collagen fibrillogenesis in developing mouse tendons. J. Cell Biol. 2000, 151, 779–787. [Google Scholar] [CrossRef]
- Davis, B.G. Synthesis of glycoproteins. Chem. Rev. 2002, 102, 579–601. [Google Scholar] [CrossRef]
- Kornfeld, R.; Kornfeld, S. Comparative aspects of glycoprotein structure. Annu. Rev. Biochem. 1976, 45, 217–237. [Google Scholar] [CrossRef]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Mecham, R.P. Overview of extracellular matrix. Curr. Protoc. Cell Biol. 2012, 57, 10. [Google Scholar] [CrossRef]
- Dwek, R.A. Glycobiology: Toward understanding the function of sugars. Chem. Rev. 1996, 96, 683–720. [Google Scholar] [CrossRef]
- Preissner, K.T.; Reuning, U. Vitronectin in vascular context: Facets of a multitalented matricellular protein. Semin. Thromb. Hemost. 2011, 37, 408–424. [Google Scholar] [CrossRef] [PubMed]
- Weeterings, C.; Adelmeijer, J.; Myles, T.; De Groot, P.G.; Lisman, T. Glycoprotein Ibα-mediated platelet adhesion and aggregation to immobilized thrombin under conditions of flow. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 670–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockhausen, I.; Kuhns, W. Role of Glycoproteins of the Immune and Blood Coagulation Systems; Springer: Berlin/Heidelberg, Germany, 1997; pp. 77–84. [Google Scholar]
- Rudd, P.M.; Elliott, T.; Cresswell, P.; Wilson, I.A.; Dwek, R.A. Glycosylation and the immune system. Science 2001, 291, 2370–2376. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, J.C.; Kaplan, H.A.; Woloski, B.M.R.N.J. Glycoprotein biosynthesis during the acute-phase response to inflammation. Can. J. Biochem. Cell Biol. 1983, 61, 1041–1048. [Google Scholar] [CrossRef]
- Gupta, S.K. Role of zona pellucida glycoproteins during fertilization in humans. J. Reprod. Immunol. 2015, 108, 90–97. [Google Scholar] [CrossRef]
- Timpl, R.; Sasaki, T.; Kostka, G.; Chu, M.L. Fibulins: A versatile family of extracellular matrix proteins. Nat. Rev. Mol. Cell Biol. 2003, 4, 479–489. [Google Scholar] [CrossRef]
- Chiquet-Ehrismann, R.; Tucker, R.P. Tenascins and the importance of adhesion modulation. Cold Spring Harb. Perspect. Biol. 2011, 3, a004960. [Google Scholar] [CrossRef] [Green Version]
- Weisel, J.W. Fibrinogen and fibrin. Adv. Protein Chem. 2005, 70, 247–299. [Google Scholar] [CrossRef]
- Leavesley, D.I.; Kashyap, A.S.; Croll, T.; Sivaramakrishnan, M.; Shokoohmand, A.; Hollier, B.G.; Upton, Z. Vitronectin-Master controller or micromanager? IUBMB Life 2013, 65, 807–818. [Google Scholar] [CrossRef] [Green Version]
- Ganss, B.; Kim, R.H.; Sodek, J. Bone sialoprotein. Crit. Rev. Oral Biol. Med. 1999, 10, 79–98. [Google Scholar] [CrossRef]
- Fatemi, S.H. Reelin glycoprotein: Structure, biology and roles in health and disease. Mol. Psychiatry 2005, 10, 251–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, M.; Norman, D.; Willis, A.; Campbell, I.D. Structure of the fibronectin type 1 module. Nature 1990, 345, 642–646. [Google Scholar] [CrossRef]
- Potts, J.R.; Campbell, I.D. Fibronectin structure and assembly. Curr. Opin. Cell Biol. 1994, 6, 648–655. [Google Scholar] [CrossRef]
- McDonald, J.A.; Kelley, D.G.; Broekelmann, T.J. Role of fibronectin in collagen deposition: Fab’ to the gelatin-binding domain of fibronectin inhibits both fibronectin and collagen organization in fibroblast extracellular matrix. J. Cell Biol. 1982, 92, 485–492. [Google Scholar] [CrossRef]
- Tamkun, J.W.; DeSimone, D.W.; Fonda, D.; Patel, R.S.; Buck, C.; Horwitz, A.F.; Hynes, R.O. Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell 1986, 46, 271–282. [Google Scholar] [CrossRef]
- Mao, Y.; Schwarzbauer, J.E. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005, 24, 389–399. [Google Scholar] [CrossRef]
- Sekiguchi, K.; Hakomori, S. Functional domain structure of fibronectin. Proc. Natl. Acad. Sci. USA 1980, 77, 2661–2665. [Google Scholar] [CrossRef] [Green Version]
- Hymes, J.P.; Klaenhammer, T.R. Stuck in the middle: Fibronectin-binding proteins in gram-positive bacteria. Front. Microbiol. 2016, 7, 1504. [Google Scholar] [CrossRef] [Green Version]
- To, W.S.; Midwood, K.S. Plasma and cellular fibronectin: Distinct and independent functions during tissue repair. Fibrogenes. Tissue Repair 2011, 4, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sottile, J.; Hocking, D.C. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol. Biol. Cell 2002, 13, 3546–3559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, T.; Johnson, K.J.; Murozono, M.; Sakai, K.; Magnuson, M.A.; Wieloch, T.; Cronberg, T.; Isshiki, A.; Erickson, H.P.; Fässler, R. Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat. Med. 2001, 7, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Maurer, L.M.; Ma, W.; Mosher, D.F. Dynamic structure of plasma fibronectin. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzbauer, J.E. Identification of the fibronectin sequences required for assembly of a fibrillar matrix. J. Cell Biol. 1991, 113, 1463–1473. [Google Scholar] [CrossRef] [Green Version]
- Schwarzbauer, J.E.; Tamkun, J.W.; Lemischka, I.R.; Hynes, R.O. Three different fibronectin mRNAs arise by alternative splicing within the coding region. Cell 1983, 35, 421–431. [Google Scholar] [CrossRef]
- Engel, J.; Odermatt, E.; Engel, A.; Madri, J.A.; Furthmayr, H.; Rohde, H.; Timpl, R. Shapes, domain organizations and flexibility of laminin and fibronectin, two multifunctional proteins of the extracellular matrix. J. Mol. Biol. 1981, 150, 97–120. [Google Scholar] [CrossRef]
- Senyürek, I.; Kempf, W.E.; Klein, G.; Maurer, A.; Kalbacher, H.; Schäfer, L.; Wanke, I.; Christ, C.; Stevanovic, S.; Schaller, M.; et al. Processing of laminin α chains generates peptides involved in wound healing and host defense. J. Innate Immun. 2014, 6, 467–484. [Google Scholar] [CrossRef]
- Aumailley, M.; Bruckner-Tuderman, L.; Carter, W.G.; Deutzmann, R.; Edgar, D.; Ekblom, P.; Engel, J.; Engvall, E.; Hohenester, E.; Jones, J.C.R.; et al. A simplified laminin nomenclature. Matrix Biol. 2005, 24, 326–332. [Google Scholar] [CrossRef]
- Hohenester, E. Structural biology of laminins. Essays Biochem. 2019, 63, 285–295. [Google Scholar]
- Odenthal, U.; Haehn, S.; Tunggal, P.; Merkl, B.; Schomburg, D.; Frie, C.; Paulsson, M.; Smyth, N. Molecular analysis of laminin N-terminal domains mediating self-interactions. J. Biol. Chem. 2004, 279, 44504–44512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schittny, J.C.; Yurchenco, P.D. Terminal short arm domains of basement membrane laminin are critical for its self-assembly. J. Cell Biol. 1990, 110, 825–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, S.A.; Carafoli, F.; Hohenester, E. Determinants of laminin polymerization revealed by the structure of the α5 chain amino-terminal region. EMBO Rep. 2011, 12, 276–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohenester, E.; Yurchenco, P.D. Laminins in basement membrane assembly. Cell Adhes. Migr. 2013, 7, 56–63. [Google Scholar] [CrossRef] [Green Version]
- McKee, K.K.; Hohenester, E.; Aleksandrova, M.; Yurchenco, P.D. Organization of the laminin polymer node. Matrix Biol. 2021, 98, 49–63. [Google Scholar] [CrossRef]
- Sasaki, T.; Fässler, R.; Hohenester, E. Laminin: The crux of basement membrane assembly. J. Cell Biol. 2004, 164, 959–963. [Google Scholar] [CrossRef] [Green Version]
- Colognato, H.; Yurchenco, P.D. Form and function: The laminin family of heterotrimers. Dev. Dyn. 2000, 218, 213–234. [Google Scholar] [CrossRef]
- Colognato, H.; Winkelmann, D.A.; Yurchenco, P.D. Laminin polymerization induces a receptor-cytoskeleton network. J. Cell Biol. 1999, 145, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Pöschl, E.; Schlötzer-Schrehardt, U.; Brachvogel, B.; Saito, K.; Ninomiya, Y.; Mayer, U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 2004, 131, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Costell, M.; Gustafsson, E.; Aszódi, A.; Mörgelin, M.; Bloch, W.; Hunziker, E.; Addicks, K.; Timpl, R.; Fässler, R. Perlecan maintains the integrity of cartilage and some basement membranes. J. Cell Biol. 1999, 147, 1109–1122. [Google Scholar] [CrossRef]
- McKee, K.K.; Aleksandrova, M.; Yurchenco, P.D. Chimeric protein identification of dystrophic, Pierson and other laminin polymerization residues. Matrix Biol. 2018, 67, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Böse, K.; Nischt, R.; Page, A.; Bader, B.L.; Paulsson, M.; Smyth, N. Loss of nidogen-1 and -2 results in syndactyly and changes in limb development. J. Biol. Chem. 2006, 281, 39620–39629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Liquari, P.; McKee, K.K.; Harrison, D.; Patel, R.; Lee, S.; Yurchenco, P.D. Laminin-sulfatide binding initiates basement membrane assembly and enables receptor signaling in Schwann cells and fibroblasts. J. Cell Biol. 2005, 169, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.M.; Mei, R. Basement Membrane Type IV Collagen and Laminin: An Overview of Their Biology and Value as Fibrosis Biomarkers of Liver Disease. Anat. Rec. 2017, 300, 1371–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambi, A.; Chavrier, P. Tissue remodeling by invadosomes. Fac. Rev. 2021, 10, 39. [Google Scholar] [CrossRef]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44–46, 247–254. [Google Scholar] [CrossRef]
- Cornfine, S.; Himmel, M.; Kopp, P.; el Azzouzi, K.; Wiesner, C.; Krüger, M.; Rudel, T.; Linder, S. The kinesin KIF9 and reggie/flotillin proteins regulate matrix degradation by macrophage podosomes. Mol. Biol. Cell 2011, 22, 202. [Google Scholar] [CrossRef]
- Cawston, T.E.; Young, D.A. Proteinases involved in matrix turnover during cartilage and bone breakdown. Cell Tissue Res. 2010, 339, 221–235. [Google Scholar] [CrossRef]
- Oda, K. New families of carboxyl peptidases: Serine-carboxyl peptidases and glutamic peptidases. J. Biochem. 2012, 151, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1. [Google Scholar] [CrossRef] [Green Version]
- Kapoor; Vaidya, S.; Wadhwan, V.; Hitesh; Kaur, G.; Pathak, A. Seesaw of matrix metalloproteinases (MMPs). J. Cancer Res. Ther. 2016, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Jin, M.; Yang, F.; Zhu, J.; Xiao, Q.; Zhang, L. Matrix metalloproteinases: Inflammatory regulators of cell behaviors in vascular formation and remodeling. Mediators Inflamm. 2013, 2013, 8315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 17, 463–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, K.; Hiramatsu, A.; Fukushima, D.; Pierschbacher, M.D.; Okada, Y. Degradation of decorin by matrix metalloproteinases: Identification of the cleavage sites, kinetic analyses and transforming growth factor-β1 release. Biochem. J. 1997, 322, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Falk-Marzillier, J.; Schiraldi, O.; Stetler-Stevenson, W.G.; Quaranta, V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science 1997, 277, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Stetler-Stevenson, W.G. Matrix metalloproteinases in angiogenesis: A moving target for therapeutic intervention. J. Clin. Investig. 1999, 103, 1237–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anacker, J.; Segerer, S.E.; Hagemann, C.; Feix, S.; Kapp, M.; Bausch, R.; Kämmerer, U. Human decidua and invasive trophoblasts are rich sources of nearly all human matrix metalloproteinases. Mol. Hum. Reprod. 2011, 17, 637–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yushchenko, M.; Weber, F.; Mäder, M.; Schöll, U.; Maliszewska, M.; Tumani, H.; Felgenhauer, K.; Beuche, W. Matrix metalloproteinase-9 (MMP-9) in human cerebrospinal fluid (CSF): Elevated levels are primarily related to CSF cell count. J. Neuroimmunol. 2000, 110, 244–251. [Google Scholar] [CrossRef]
- Hartung, H.-P.; Kieseier, B.C. The role of matrix metalloproteinases in autoimmune damage to the central and peripheral nervous system. J. Neuroimmunol. 2000, 107, 140–147. [Google Scholar] [CrossRef]
- Gonzalez-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. Hematol. 2019, 137, 57–83. [Google Scholar] [CrossRef] [PubMed]
- Blobel, C.P. Metalloprotease-Disintegrins: Links to Cell Adhesion and Cleavage of TNFα and Notch. Cell 1997, 90, 589–592. [Google Scholar] [CrossRef] [Green Version]
- Werb, Z. ECM and Cell Surface Proteolysis: Regulating Cellular Ecology. Cell 1997, 91, 439–442. [Google Scholar] [CrossRef] [Green Version]
- Blobel, C.P. ADAMs: Key components in egfr signalling and development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef]
- Murphy, G. The ADAMs: Signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 2008, 8, 929–941. [Google Scholar] [CrossRef]
- Rose-John, S. ADAM17, shedding, TACE as therapeutic targets. Pharmacol. Res. 2013, 71, 19–22. [Google Scholar] [CrossRef]
- Marczynska, J.; Ozga, A.; Wlodarczyk, A.; Majchrzak-Gorecka, M.; Kulig, P.; Banas, M.; Michalczyk-Wetula, D.; Majewski, P.; Hutloff, A.; Schwarz, J.; et al. The Role of Metalloproteinase ADAM17 in Regulating ICOS Ligand–Mediated Humoral Immune Responses. J. Immunol. 2014, 193, 2753–2763. [Google Scholar] [CrossRef] [Green Version]
- Hsia, H.E.; Tüshaus, J.; Brummer, T.; Zheng, Y.; Scilabra, S.D.; Lichtenthaler, S.F. Functions of ‘A disintegrin and metalloproteases (ADAMs)’ in the mammalian nervous system. Cell. Mol. Life Sci. 2019, 76, 3055–3081. [Google Scholar] [CrossRef]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta-Mol. Cell Res. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef]
- Van Goor, H.; Melenhorst, W.B.W.H.; Turner, A.J.; Holgate, S.T. Adamalysins in biology and disease. J. Pathol. 2009, 219, 277–286. [Google Scholar] [CrossRef]
- Boudreau, N.J.; Jones, P.L. Extracellular matrix and integrin signalling: The shape of things to come. Biochem. J. 1999, 339, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Creemers, E.E.; Kassiri, Z. Matrix as an interstitial transport system. Circ. Res. 2014, 114, 889–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, G.S.; Wysocki, A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009, 17, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Rifkin, D.B. Extracellular matrix regulation of growth factor and protease activity. Curr. Opin. Cell Biol. 1991, 3, 817–823. [Google Scholar] [CrossRef]
- O’Callaghan, P.; Zhang, X.; Li, J.P. Heparan Sulfate Proteoglycans as Relays of Neuroinflammation. J. Histochem. Cytochem. 2018, 66, 305–319. [Google Scholar] [CrossRef] [Green Version]
- Li, J.P.; Kusche-Gullberg, M. Heparan Sulfate: Biosynthesis, Structure, and Function. In International Review of Cell and Molecular Biology; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 325, pp. 215–273. ISBN 9780128048061. [Google Scholar]
- Billings, P.C.; Pacifici, M. Interactions of signaling proteins, growth factors and other proteins with heparan sulfate: Mechanisms and mysteries. Connect. Tissue Res. 2015, 56, 272–280. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, I.; Kimura-Yoshida, C. Extracellular distribution of diffusible growth factors controlled by heparan sulfate proteoglycans during mammalian embryogenesis. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130545. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.H.; Zhu, Y.; Zhu, Y.Y.; Wei, H.; Zhang, N.N.; Qin, J.S.; Zhu, X.L.; Yu, M.; Li, Y.F. Abnormalities in FGF family members and their roles in modulating depression-related molecules. Eur. J. Neurosci. 2021, 53, 140–150. [Google Scholar] [CrossRef]
- Malkowski, A.; Sobolewski, K.; Jaworski, S.; Bankowski, E. FGF binding by extracellular matrix components of Wharton’s jelly. Acta Biochim. Pol. 2007, 54, 357–363. [Google Scholar] [CrossRef]
- Loo, B.M.; Kreuger, J.; Jalkanen, M.; Lindahl, U.; Salmivirta, M. Binding of Heparin/Heparan Sulfate to Fibroblast Growth Factor Receptor 4. J. Biol. Chem. 2001, 276, 16868–16876. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rifkin, D.B.; Rifkin, W.J.; Zilberberg, L. LTBPs in biology and medicine: LTBP diseases. Matrix Biol. 2018, 71–72, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Todorovic, V.; Rifkin, D.B. LTBPs, more than just an escort service. J. Cell. Biochem. 2012, 113, 410–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, R.M.; Gentry, L.E.; Purchio, A.F.; Moses, H.L. Mechanism of activation of latent recombinant transforming growth factor beta 1 by plasmin. J. Cell Biol. 1990, 110, 1361–1367. [Google Scholar] [CrossRef]
- Jenkins, G. The role of proteases in transforming growth factor-β activation. Int. J. Biochem. Cell Biol. 2008, 40, 1068–1078. [Google Scholar] [CrossRef]
- Munger, J.S.; Harpel, J.G.; Giancotti, F.G.; Rifkin, D.B. Interactions between growth factors and integrins: Latent forms of transforming growth factor-β are ligands for the integrin αvβ1. Mol. Biol. Cell 1998, 9, 2627–2638. [Google Scholar] [CrossRef]
- Tatler, A.L.; John, A.E.; Jolly, L.; Habgood, A.; Porte, J.; Brightling, C.; Knox, A.J.; Pang, L.; Sheppard, D.; Huang, X.; et al. Integrin αvβ5-Mediated TGF-β Activation by Airway Smooth Muscle Cells in Asthma. J. Immunol. 2011, 187, 6094–6107. [Google Scholar] [CrossRef] [Green Version]
- Sarrazy, V.; Koehler, A.; Chow, M.L.; Zimina, E.; Li, C.X.; Kato, H.; Caldarone, C.A.; Hinz, B. Integrins αvβ5 and αvβ3 promote latent TGF-β1 activation by human cardiac fibroblast contraction. Cardiovasc. Res. 2014, 102, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Murphy-Ullrich, J.E.; Poczatek, M. Activation of latent TGF-beta by thrombospondin-1: Mechanisms and physiology. Cytokine Growth Factor Rev. 2000, 11, 59–69. [Google Scholar] [CrossRef]
- Ribeiro, S.M.; Poczatek, M.; Schultz-Cherry, S.; Villain, M.; Murphy-Ullrich, J.E. The activation sequence of thrombospondin-1 interacts with the latency-associated peptide to regulate activation of latent transforming growth factor-beta. J. Biol. Chem. 1999, 274, 13586–13593. [Google Scholar] [CrossRef] [Green Version]
- Barcellos-Hoff, M.H.; Derynck, R.; Tsang, M.L.S.; Weatherbee, J.A. Transforming growth factor-β activation in irradiated murine mammary gland. J. Clin. Investig. 1994, 93, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, E.J.; Segarini, P.; Tsang, M.L.-S.; Carroll, A.G.; Barcellos-Hoff, M.H. Latent transforming growth factor β1 activation in situ: Quantitative and functional evidence after low-dose γ-irradiation 1. FASEB J. 1997, 11, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Kottmann, R.M.; Kulkarni, A.A.; Smolnycki, K.A.; Lyda, E.; Dahanayake, T.; Salibi, R.; Honnons, S.; Jones, C.; Isern, N.G.; Hu, J.Z.; et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am. J. Respir. Crit. Care Med. 2012, 186, 740–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jullien, P.; Berg, T.M.; Lawrence, D.A. Acidic cellular environments: Activation of latent tgf-β and sensitization of cellular responses to tgf-β and egf. Int. J. Cancer 1989, 43, 886–891. [Google Scholar] [CrossRef]
- Brownh, P.D.; Wakefiel, L.M.; Levinson, A.D.; Sporn, M.B. Physicochemical activation of recombinant latent transforming growth factor-beta’s 1, 2, and 3. Growth Factors 1990, 3, 35–43. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Dix, T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996, 10, 1077–1083. [Google Scholar] [CrossRef] [Green Version]
- Reddig, P.J.; Juliano, R.L. Clinging to life: Cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 2005, 24, 425–439. [Google Scholar] [CrossRef]
- Merten, O.W. Advances in cell culture: Anchorage dependence. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140040. [Google Scholar] [CrossRef] [Green Version]
- Chiarugi, P.; Giannoni, E. Anoikis: A necessary death program for anchorage-dependent cells. Biochem. Pharmacol. 2008, 76, 1352–1364. [Google Scholar] [CrossRef]
- Frisch, S.M.; Ruoslahti, E. Integrins and anoikis. Curr. Opin. Cell Biol. 1997, 9, 701–706. [Google Scholar] [CrossRef]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, S.M.; Screaton, R.A. Anoikis mechanisms. Curr. Opin. Cell Biol. 2001, 13, 555–562. [Google Scholar] [CrossRef]
- Stoker, M.; O’Neill, C.; Berryman, S.; Waxman, V. Anchorage and growth regulation in normal and virus-transformed cells. Int. J. Cancer 1968, 3, 683–693. [Google Scholar] [CrossRef] [PubMed]
- McGill, G.; Shimamura, A.; Bates, R.C.; Savage, R.E.; Fisher, D.E. Loss of matrix adhesion triggers rapid transformation-selective apoptosis in fibroblasts. J. Cell Biol. 1997, 138, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Hungerford, J.E.; Compton, M.T.; Matter, M.L.; Hoffstrom, B.G.; Otey, C.A. Inhibition of pp125FAK in cultured fibroblasts results in apoptosis. J. Cell Biol. 1996, 135, 1383–1390. [Google Scholar] [CrossRef] [Green Version]
- Fukai, F.; Mashimo, M.; Akiyama, K.; Goto, T.; Tanuma, S.I.; Katayama, T. Modulation of apoptotic cell death by extracellular matrix proteins and a fibronectin-derived antiadhesive peptide. Exp. Cell Res. 1998, 242, 92–99. [Google Scholar] [CrossRef]
- Aoudjit, F.; Vuori, K. Matrix attachment regulates Fas-induced apoptosis in endothelial cells: A role for c-Flip and implications for anoikis. J. Cell Biol. 2001, 153, 633–643. [Google Scholar] [CrossRef]
- Marconi, A.; Atzei, P.; Panza, C.; Fila, C.; Tiberio, R.; Truzzi, F.; Wachter, T.; Leverkus, M.; Pincelli, C. FLICE/caspace-8 activation triggers anoikis induced by β 1-integrin blockade in human keratinocytes. J. Cell Sci. 2004, 117, 5815–5823. [Google Scholar] [CrossRef] [Green Version]
- Tiberio, R.; Marconi, A.; Fila, C.; Fumelli, C.; Pignatti, M.; Krajewski, S.; Giannetti, A.; Reed, J.C.; Pincelli, C. Keratinocytes enriched for stem cells are protected from anoikis via an integrin signaling pathway in a Bcl-2 dependent manner. FEBS Lett. 2002, 524, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Abe, Y.; Yamamoto, T.; Sugiyama, Y.; Watanabe, T.; Saito, N.; Kayama, H.; Kumagai, T. “Anoikis” of Oligodendrocytes Induced by Wallerian Degeneration: Ultrastructural Observations. J. Neurotrauma 2004, 21, 119–124. [Google Scholar] [CrossRef]
- Marchionini, D.M.; Collier, T.J.; Camargo, M.; McGuire, S.; Pitzer, M.; Sortwell, C.E. Interference with anoikis-induced cell death of dopamine neurons: Implications for augmenting embryonic graft survival in a rat model of Parkinson’s disease. J. Comp. Neurol. 2003, 464, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Gray, S.; Pham, T.; Delgardo, M.; Nguyen, A.; Do, S.; Ireland, S.K.; Chen, R.; Abdel-Mageed, A.B.; Biliran, H. Downregulation of Bit1 expression promotes growth, anoikis resistance, and transformation of immortalized human bronchial epithelial cells via Erk activation-dependent suppression of E-cadherin. Biochem. Biophys. Res. Commun. 2018, 495, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Aoshiba, K.; Rennard, S.I.; Spurzem, J.R. Cell-matrix and cell-cell interactions modulate apoptosis of bronchial epithelial cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1997, 272, L28–L37. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, H.; Derouet, M.; Berezkin, A.; Sasazuki, T.; Shirasawa, S.; Rosen, K. Oncogenic ras inhibits anoikis of intestinal epithelial cells by preventing the release of a mitochondrial pro-apoptotic protein Omi/HtrA2 into the cytoplasm. J. Biol. Chem. 2006, 281, 14738–14747. [Google Scholar] [CrossRef] [Green Version]
- Beauséjour, M.; Thibodeau, S.; Demers, M.J.; Bouchard, V.; Gauthier, R.; Beaulieu, J.F.; Vachon, P.H. Suppression of anoikis in human intestinal epithelial cells: Differentiation state-selective roles of α2β1, α3β1, α5β1, and α6β4 integrins. BMC Cell Biol. 2013, 14, 53. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, J.; Mohr, S.; Lapetina, E.G.; Fiocchi, C.; Levine, A.D. Sequential and rapid activation of select caspases during apoptosis of normal intestinal epithelial cells. Am. J. Physiol.-Gastrointest. Liver Physiol. 1998, 274, G1117–G1124. [Google Scholar] [CrossRef]
- Farrelly, N.; Lee, Y.J.; Oliver, J.; Dive, C.; Streuli, C.H. Extracellular matrix regulates apoptosis in mammary epithelium through a control on insulin signaling. J. Cell Biol. 1999, 144, 1337–1347. [Google Scholar] [CrossRef]
- Kruidering, M.; Evan, G. Caspase-8 in Apoptosis: The Beginning of “The End”? IUBMB Life 2000, 50, 85–90. [Google Scholar] [CrossRef]
- Bozzo, C.; Sabbatini, M.; Tiberio, R.; Piffanelli, V.; Santoro, C.; Cannas, M. Activation of caspase-8 triggers anoikis in human neuroblastoma cells. Neurosci. Res. 2006, 56, 145–153. [Google Scholar] [CrossRef]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta-Mol. Cell Res. 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [Green Version]
- Aleksandrushkina, N.I.; Vanyushin, B.F. Endonucleases and apoptosis in animals. Biochemistry 2012, 77, 1436–1451. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Hu, C.; Yang, Z.; Zhang, X.; Zhao, L.; Xiong, J.; Ma, J.; Chen, L.; Qian, H.; Luo, X.; et al. Midkine promotes hepatocellular carcinoma metastasis by elevating anoikis resistance of circulating tumor cells. Oncotarget 2017, 8, 32523–32535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Chun, J.; Pan, C.; Kumar, A.; Zhang, G.; Ha, Y.; Li, D.; Alesi, G.N.; Kang, Y.; Zhou, L.; et al. The PLAG1-GDH1 Axis Promotes Anoikis Resistance and Tumor Metastasis through CamKK2-AMPK Signaling in LKB1-Deficient Lung Cancer. Mol. Cell 2018, 69, 87–99.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.N.; Zeng, Z.L.; Lu, J.; Wang, Y.; Liu, Z.X.; He, M.M.; Zhao, Q.; Wang, Z.X.; Li, T.; Lu, Y.X.; et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene 2018, 37, 6025–6040. [Google Scholar] [CrossRef]
- Guha, D.; Saha, T.; Bose, S.; Chakraborty, S.; Dhar, S.; Khan, P.; Adhikary, A.; Das, T.; Sa, G. Integrin-EGFR interaction regulates anoikis resistance in colon cancer cells. Apoptosis 2019, 24, 958–971. [Google Scholar] [CrossRef]
- Lamandé, S.R.; Bateman, J.F. Genetic Disorders of the Extracellular Matrix. Anat. Rec. 2020, 303, 1527–1542. [Google Scholar] [CrossRef]
- Bateman, J.F.; Boot-Handford, R.P.; Lamandé, S.R. Genetic diseases of connective tissues: Cellular and extracellular effects of ECM mutations. Nat. Rev. Genet. 2009, 10, 173–183. [Google Scholar] [CrossRef]
- Marini, J.C.; Forlino, A.; Bächinger, H.P.; Bishop, N.J.; Byers, P.H.; De Paepe, A.; Fassier, F.; Fratzl-Zelman, N.; Kozloff, K.M.; Krakow, D.; et al. Osteogenesis imperfecta. Nat. Rev. Dis. Prim. 2017, 3, 1377–1385. [Google Scholar] [CrossRef]
- Morello, R. Osteogenesis imperfecta and therapeutics. Matrix Biol. 2018, 71–72, 294–312. [Google Scholar] [CrossRef]
- Sillence, D.O.; Senn, A.; Danks, D.M. Genetic heterogeneity in osteogenesis imperfecta. J. Med. Genet. 1979, 16, 101–116. [Google Scholar] [CrossRef] [Green Version]
- Marini, J.C.; Reich, A.; Smith, S.M. Osteogenesis imperfecta due to mutations in non-collagenous genes: Lessons in the biology of bone formation. Curr. Opin. Pediatr. 2014, 26, 500–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boote, C.; Sigal, I.A.; Grytz, R.; Hua, Y.; Nguyen, T.D.; Girard, M.J.A. Scleral structure and biomechanics. Prog. Retin. Eye Res. 2020, 74, 100773. [Google Scholar] [CrossRef] [PubMed]
- Evereklioglu, C.; Madenci, E.; Bayazit, Y.A.; Yilmaz, K.; Balat, A.; Bekir, N.A. Central corneal thickness is lower in osteogenesis imperfecta and negatively correlates with the presence of blue sclera. Ophthalmic Physiol. Opt. 2002, 22, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Sobey, G. Ehlers-Danlos syndrome-A commonly misunderstood group of conditions. Clin. Med. J. R. Coll. Physicians Lond. 2014, 14, 432–436. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, A.; Malfait, F. The Ehlers-Danlos syndrome, a disorder with many faces. Clin. Genet. 2012, 82, 1–11. [Google Scholar] [CrossRef]
- Miller, E.; Grosel, J.M. A review of Ehlers-Danlos syndrome. J. Am. Acad. Physician Assist. 2020, 33, 23–28. [Google Scholar] [CrossRef]
- Piérard, G.E.; Trinh, L.; Piérard-Franchimont, C.; Lapiére, C.M. Morphometric Study of Cauliflower Collagen Fibrils in Ehlers-Danlos Syndrome Type I. Top. Catal. 1988, 8, 453–457. [Google Scholar] [CrossRef]
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and Genetic Features of Ehlers–Danlos Syndrome Type IV, the Vascular Type. N. Engl. J. Med. 2000, 342, 673–680. [Google Scholar] [CrossRef]
- Joseph, A.W.; Joseph, S.S.; Francomano, C.A.; Kontis, T.C. Characteristics, diagnosis, and management of Ehlers-Danlos syndromes: A review. In JAMA Facial Plastic Surgery; American Medical Association: Chicago, IL, USA, 2018; Volume 20, pp. 70–75. [Google Scholar]
- Berglund, B.; Nordström, G.; Lützén, K. Living a restricted life with Ehlers-Danlos Syndrome (EDS). Int. J. Nurs. Stud. 2000, 37, 111–118. [Google Scholar] [CrossRef]
- Biery, N.J.; Eldadah, Z.A.; Moore, C.S.; Stetten, G.; Spencer, F.; Dietz, H.C. Revised genomic organization of FBN1 and significance for regulated gene expression. Genomics 1999, 56, 70–77. [Google Scholar] [CrossRef]
- Ferruzzi, J.; Collins, M.J.; Yeh, A.T.; Humphrey, J.D. Mechanical assessment of elastin integrity in fibrillin-1-deficient carotid arteries: Implications for Marfan syndrome. Cardiovasc. Res. 2011, 92, 287–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyeritz, R.E. The Marfan syndrome. Annu. Rev. Med. 2000, 51, 481–510. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A.H.; Zaradzki, M.; Arif, R.; Remes, A.; Müller, O.J.; Kallenbach, K. Marfan syndrome: A therapeutic challenge for long-term care. Biochem. Pharmacol. 2019, 164, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Pyeritz, R.E. Marfan syndrome: Improved clinical history results in expanded natural history. Genet. Med. 2019, 21, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Mckusick, V.A. The cardiovascular aspects of Marfan’s syndrome: A heritable disorder of connective tissue. Circulation 1955, 11, 321–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarevic, A.M.; Nakatani, S.; Okita, Y.; Marinkovic, J.; Takeda, Y.; Hirooka, K.; Matsuo, H.; Kitamura, S.; Yamagishi, M.; Miyatake, K. Determinants of rapid progression of aortic root dilatation and complications in Marfan syndrome. Int. J. Cardiol. 2006, 106, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Meester, J.A.N.; Verstraeten, A.; Schepers, D.; Alaerts, M.; van Laer, L.; Loeys, B.L. Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann. Cardiothorac. Surg. 2017, 6, 582–594. [Google Scholar] [CrossRef] [Green Version]
- Bitterman, A.D.; Sponseller, P.D. Marfan Syndrome: A Clinical Update. J. Am. Acad. Orthop. Surg. 2017, 25, 603–609. [Google Scholar] [CrossRef]
- Gray, J.R.; Bridges, A.B.; West, R.R.; McLeish, L.; Stuart, A.G.; Dean, J.C.S.; Porteous, M.E.M.; Boxer, M.; Davies, S.J. Life expectancy in British Marfan syndrome populations. Clin. Genet. 1998, 54, 124–128. [Google Scholar] [CrossRef]
- Snead, M.P.; Yates, J.R.W. Clinical and molecular genetics of Stickler syndrome. J. Med. Genet. 1999, 36, 353–359. [Google Scholar]
- Boothe, M.; Morris, R.; Robin, N. Stickler syndrome: A review of clinical manifestations and the genetics evaluation. J. Pers. Med. 2020, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Caria, F.; Cescon, M.; Gualandi, F.; Pichiecchio, A.; Rossi, R.; Rimessi, P.; Cotti Piccinelli, S.; Gallo Cassarino, S.; Gregorio, I.; Galvagni, A.; et al. Autosomal recessive Bethlem myopathy: A clinical, genetic and functional study. Neuromuscul. Disord. 2019, 29, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Lamandé, S.R.; Bateman, J.F.; Hutchison, W.; Gardner, R.J.M.K.; Bower, S.P.; Byrne, E.; Dahl, H.H.M. Reduced collagen VI causes Bethlem myopathy: A heterozygous COL6A1 nonsense mutation results in mRNA decay and functional haploinsufficiency. Hum. Mol. Genet. 1998, 7, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Peat, R.A.; Baker, N.L.; Jones, K.J.; North, K.N.; Lamandé, S.R. Variable penetrance of COL6A1 null mutations: Implications for prenatal diagnosis and genetic counselling in Ullrich congenital muscular dystrophy families. Neuromuscul. Disord. 2007, 17, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.L.; Mörgelin, M.; Peat, R.; Goemans, N.; North, K.N.; Bateman, J.F.; Lamandé, S.R. Dominant collagen VI mutations are a common cause of Ulrich congenital muscular dystrophy. Hum. Mol. Genet. 2005, 14, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Hovnanian, A.; Rochat, A.; Bodemer, C.; Petit, E.; Rivers, C.A.; Prost, C.; Fraitag, S.; Christiano, A.M.; Uitto, J.; Lathrop, M.; et al. Characterization of 18 new mutations in COL7A1 in recessive dystrophic epidermolysis bullosa provides evidence for distinct molecular mechanisms underlying defective anchoring fibril formation. Am. J. Hum. Genet. 1997, 61, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Christiano, A.M.; Amano, S.; Eichenfield, L.F.; Burgeson, R.E.; Uitto, J. Premature termination codon mutations in the type VII collagen gene in recessive dystrophic epidermolysis bullosa result in nonsense-mediated mRNA decay and absence of functional protein. J. Investig. Dermatol. 1997, 109, 390–394. [Google Scholar] [CrossRef] [Green Version]
- Stum, M.; Davoine, C.S.; Vicart, S.; Guillot-Noël, L.; Topaloglu, H.; Carod-Artal, F.J.; Kayserili, H.; Hentati, F.; Merlini, L.; Urtizberea, J.A.; et al. Spectrum of HSPG2 (perlecan) mutations in patients with Schwartz-Jampel syndrome. Hum. Mutat. 2006, 27, 1082–1091. [Google Scholar] [CrossRef]
- Arikawa-Hirasawa, E.; Le, A.H.; Nishino, I.; Nonaka, I.; Ho, N.C.; Francomano, C.A.; Govindraj, P.; Hassell, J.R.; Devaney, J.M.; Spranger, J.; et al. Structural and functional mutations of the perlecan gene cause Schwartz-Jampel syndrome, with myotonic myopathy and chondrodysplasia. Am. J. Hum. Genet. 2002, 70, 1368–1375. [Google Scholar] [CrossRef] [Green Version]
- Matejas, V.; Hinkes, B.; Alkandari, F.; Al-Gazali, L.; Annexstad, E.; Aytac, M.B.; Barrow, M.; Bláhová, K.; Bockenhauer, D.; Cheong, H.I.; et al. Mutations in the human laminin β2 (LAMB2) gene and the associated phenotypic spectrum. Hum. Mutat. 2010, 31, 992–1002. [Google Scholar] [CrossRef] [Green Version]
- Funk, S.D.; Lin, M.H.; Miner, J.H. Alport syndrome and Pierson syndrome: Diseases of the glomerular basement membrane. Matrix Biol. 2018, 71–72, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Lotery, A.J.; Baas, D.; Ridley, C.; Jones, R.P.O.; Klaver, C.C.W.; Stone, E.; Nakamura, T.; Luff, A.; Griffiths, H.; Wang, T.; et al. Reduced secretion of fibulin 5 in age-related macular degeneration and cutis laxa. Hum. Mutat. 2006, 27, 568–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullins, R.F.; Olvera, M.A.; Clark, A.F.; Stone, E.M. Fibulin-5 distribution in human eyes: Relevance to age-related macular degeneration. Exp. Eye Res. 2007, 84, 378–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toda, S.; Frankel, N.W.; Lim, W.A. Engineering cell–cell communication networks: Programming multicellular behaviors. Curr. Opin. Chem. Biol. 2019, 52, 31–38. [Google Scholar] [CrossRef]
- Xu, F.; Zhao, Z.J. Cell density regulates tyrosine phosphorylation and localization of focal adhesion kinase. Exp. Cell Res. 2001, 262, 49–58. [Google Scholar] [CrossRef]
- Shah, N.H.; Kuriyan, J. Understanding molecular mechanisms in cell signaling through natural and artificial sequence variation. Nat. Struct. Mol. Biol. 2019, 26, 25–34. [Google Scholar] [CrossRef]
- Müller, P.; Schier, A.F. Extracellular Movement of Signaling Molecules. Dev. Cell 2011, 21, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Pasiakos, S.M. Exercise and amino acid anabolic cell signaling and the regulation of skeletal muscle mass. Nutrients 2012, 4, 740–758. [Google Scholar] [CrossRef]
- Zingg, J.M. Vitamin E: Regulatory Role on Signal Transduction. IUBMB Life 2019, 71, 456–478. [Google Scholar] [CrossRef]
- Carballo, G.B.; Honorato, J.R.; De Lopes, G.P.F.; de Spohr, T.C.L. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 1–15. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Pessin, J.E. Insulin signaling pathways in time and space. Trends Cell Biol. 2002, 12, 65–71. [Google Scholar] [CrossRef]
- Watowich, S.S. The erythropoietin receptor: Molecular structure and hematopoietic signaling pathways. J. Investig. Med. 2011, 59, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Mosaad, E.; Peiris, H.N.; Holland, O.; Morean Garcia, I.; Mitchell, M.D. The Role(s) of Eicosanoids and Exosomes in Human Parturition. Front. Physiol. 2020, 11, 1540. [Google Scholar] [CrossRef] [PubMed]
- Psarra, A.M.G.; Sekeris, C.E. Steroid and thyroid hormone receptors in mitochondria. IUBMB Life 2008, 60, 210–223. [Google Scholar] [CrossRef]
- Naomi Handly, L.; Pilko, A.; Wollman, R. Paracrine communication maximizes cellular response fidelity in wound signaling. eLife 2015, 4, e09652. [Google Scholar] [CrossRef]
- Tse, L.H.; Wong, Y.H. GPCRs in autocrine and paracrine regulations. Front. Endocrinol. 2019, 10, 428. [Google Scholar] [CrossRef]
- Kennedy, M.B. Synaptic signaling in learning and memory. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.W.; Poo, M. Retrograde signaling at central synapses. Proc. Natl. Acad. Sci. USA 2001, 98, 11009–11015. [Google Scholar] [CrossRef] [Green Version]
- González, M.I.; Robinson, M.B. Neurotransmitter transporters: Why dance with so many partners? Curr. Opin. Pharmacol. 2004, 4, 30–35. [Google Scholar] [CrossRef]
- Sporn, M.B.; Todaro, G.J. Autocrine Secretion and Malignant Transformation of Cells. N. Engl. J. Med. 1980, 303, 878–880. [Google Scholar] [CrossRef]
- Segers, V.F.M.; De Keulenaer, G.W. Autocrine signaling in cardiac remodeling: A rich source of therapeutic targets. J. Am. Heart Assoc. 2021, 10, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Piepkorn, M.; Pittelkow, M.R.; Cook, P.W. Autocrine regulation of keratinocytes: The emerging role of heparin- binding, epidermal growth factor-related growth factors. J. Investig. Dermatol. 1998, 111, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, G.S.; Valiunas, V.; Brink, P.R. Selective permeability of gap junction channels. Biochim. Biophys. Acta-Biomembr. 2004, 1662, 96–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, W.H.; De Vuyst, E.; Leybaert, L. The gap junction cellular internet: Connexin hemichannels enter the signalling limelight. Biochem. J. 2006, 397, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Prado, N.; Bukauskas, F.F. Heterotypic gap junction channels as voltage-sensitive valves for intercellular signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 14855–14860. [Google Scholar] [CrossRef] [Green Version]
- Dejana, E. Endothelial cell-cell junctions: Happy together. Nat. Rev. Mol. Cell Biol. 2004, 5, 261–270. [Google Scholar] [CrossRef]
- Zimmerman, G.A.; Lorant, D.E.; McIntyre, T.M.; Prescott, S.M. Juxtacrine intercellular signaling: Another way to do it. Am. J. Respir. Cell Mol. Biol. 1993, 9, 573–577. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Precision medicine for human cancers with Notch signaling dysregulation (Review). Int. J. Mol. Med. 2020, 45, 279–297. [Google Scholar] [CrossRef] [Green Version]
- Hruby, V.J. Designing peptide receptor agonists and antagonists. Nat. Rev. Drug Discov. 2002, 1, 847–858. [Google Scholar] [CrossRef]
- Pleuvry, B.J. Receptors, agonists and antagonists. Anaesth. Intensive Care Med. 2004, 5, 350–352. [Google Scholar] [CrossRef]
- Tompa, P. The principle of conformational signaling. Chem. Soc. Rev. 2016, 45, 4252–4284. [Google Scholar] [CrossRef] [PubMed]
- Ferrell, J.E. Tripping the switch fantastic: How a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem. Sci. 1996, 21, 460–466. [Google Scholar] [CrossRef]
- Govern, C.C.; Chakraborty, A.K. Signaling Cascades Modulate the Speed of Signal Propagation through Space. PLoS ONE 2009, 4, e4639. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.L. The hippo pathway: Biology and pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef] [Green Version]
- Bali, A.; Jaggi, A.S. Angiotensin II-triggered kinase signaling cascade in the central nervous system. Rev. Neurosci. 2016, 27, 301–315. [Google Scholar] [CrossRef]
- Catozzi, S.; Di-Bella, J.P.; Ventura, A.C.; Sepulchre, J.A. Signaling cascades transmit information downstream and upstream but unlikely simultaneously. BMC Syst. Biol. 2016, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Gorski, J.; Gannon, F. Current models of steroid hormone action: A critique. Annu. Rev. Physiol. 1976, 38, 425–450. [Google Scholar] [CrossRef]
- Plagemann, P.G.W.; Erbe, J. Glucocorticoids-uptake by simple diffusion by cultured reuber and novikoff rat hepatoma cells. Biochem. Pharmacol. 1976, 25, 1489–1494. [Google Scholar] [CrossRef]
- Oren, I.; Fleishman, S.J.; Kessel, A.; Ben-Tal, N. Free diffusion of steroid hormones across biomembranes: A simplex search with implicit solvent model calculations. Biophys. J. 2004, 87, 768–779. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, N.; Viswanatha, R.; Bittar, R.; Li, Z.; Haga-Yamanaka, S.; Perrimon, N.; Yamanaka, N. A Membrane Transporter Is Required for Steroid Hormone Uptake in Drosophila. Dev. Cell 2018, 47, 294–305.e7. [Google Scholar] [CrossRef] [Green Version]
- Neuman, S.D.; Bashirullah, A. Reconsidering the Passive Diffusion Model of Steroid Hormone Cellular Entry. Dev. Cell 2018, 47, 261–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, R.J.; Bailey, J.C. Effects of acetylcholine on action potential characteristics of atrial and ventricular myocardium after bilateral cervical vagotomy in the cat. Circ. Res. 1985, 56, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, E.J. Three types of acetylcholine response in bivalve heart muscle cells. J. Physiol. 1980, 300, 283–302. [Google Scholar] [CrossRef] [PubMed]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a Neuromodulator: Cholinergic Signaling Shapes Nervous System Function and Behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Saradhi, M.; Chaturvedi, N.K.; Tyagi, R.K. Intracellular localization and nucleocytoplasmic trafficking of steroid receptors: An overview. Mol. Cell. Endocrinol. 2006, 246, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Aranda, A.; Pascual, A. Nuclear hormone receptors and gene expression. Physiol. Rev. 2001, 81, 1269–1304. [Google Scholar] [CrossRef]
- Meinsohn, M.C.; Smith, O.E.; Bertolin, K.; Murphy, B.D. The orphan nuclear receptors steroidogenic factor-1 and liver receptor homolog-1: Structure, regulation, and essential roles in mammalian reproduction. Physiol. Rev. 2019, 99, 1249–1279. [Google Scholar] [CrossRef]
- Enmark, E.; Gustafsson, J.A. Orphan nuclear receptors—the first eight years. Mol. Endocrinol. 1996, 10, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.J.; Yang, D.R.; Yang, G.; Lin, C.Y.; Chang, H.C.; Li, G.; Chang, C. TR2 and TR4 Orphan Nuclear Receptors: An Overview. Curr. Top. Dev. Biol. 2017, 125, 357–373. [Google Scholar]
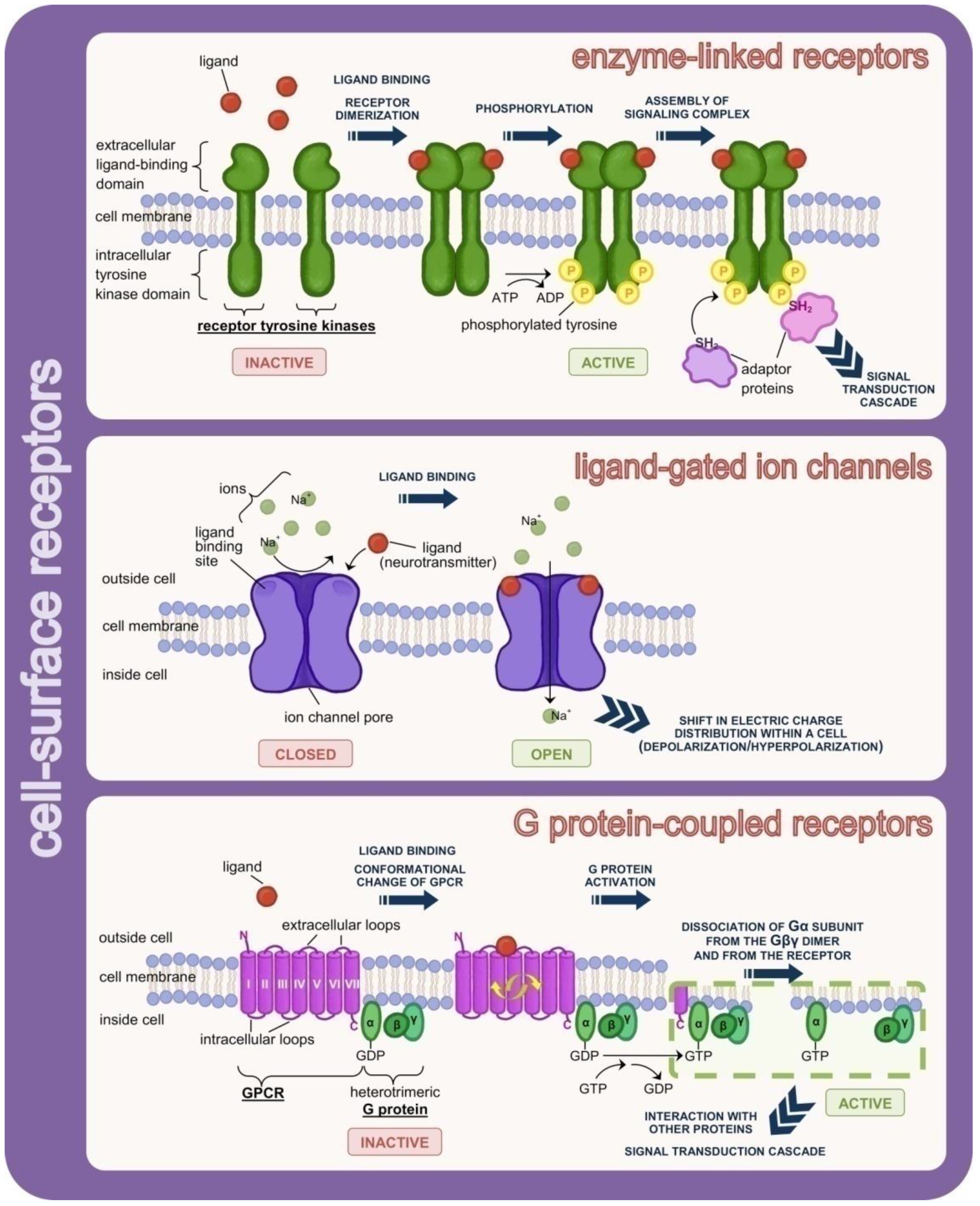
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. Annu. Rev. Biochem. 2015, 84, 739–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trenker, R.; Jura, N. Receptor tyrosine kinase activation: From the ligand perspective. Curr. Opin. Cell Biol. 2020, 63, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Posner, B.I. Insulin Signalling: The Inside Story. Can. J. Diabetes 2017, 41, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotinun, S.; Scott Pearsall, R.; Horne, C.W.; Baron, R. Activin Receptor Signaling: A Potential Therapeutic Target for Osteoporosis. Curr. Mol. Pharmacol. 2012, 5, 195–204. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-Β signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [Green Version]
- Keramidas, A.; Moorhouse, A.J.; Schofield, P.R.; Barry, P.H. Ligand-gated ion channels: Mechanisms underlying ion selectivity. Prog. Biophys. Mol. Biol. 2004, 86, 161–204. [Google Scholar] [CrossRef]
- Pirri, J.K.; Rayes, D.; Alkema, M.J. A Change in the Ion Selectivity of Ligand-Gated Ion Channels Provides a Mechanism to Switch Behavior. PLoS Biol. 2015, 13, e1002238. [Google Scholar] [CrossRef] [Green Version]
- García-Giménez, E.; Alcaraz, A.; Aguilella-Arzo, M.; Aguilella, V.M. Selectivity of Protein Ion Channels and the Role of Buried Charges. Analytical Solutions, Numerical Calculations, and MD Simulations. J. Phys. Chem. B 2015, 119, 8475–8479. [Google Scholar] [CrossRef] [Green Version]
- Weir, C.J. Ion channels, receptors, agonists and antagonists. Anaesth. Intensive Care Med. 2020, 21, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wong, A.H.C.; Liu, F. Ligand-gated ion channel interacting proteins and their role in neuroprotection. Front. Cell. Neurosci. 2014, 8, 125. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.B.; Nigam, A.; Johnson, J.W. Interplay between gating and block of ligand-gated ion channels. Brain Sci. 2020, 10, 928. [Google Scholar] [CrossRef]
- Kim, D.M.; Nimigean, C.M. Voltage-gated potassium channels: A structural examination of selectivity and gating. Cold Spring Harb. Perspect. Biol. 2016, 8, a029231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariev, A.M.; Green, M.E. Voltage gated ion channel function: Gating, conduction, and the role of water and protons. Int. J. Mol. Sci. 2012, 13, 1680–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachs, F. Stretch-activated ion channels: What are they? Physiology 2010, 25, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef]
- Jacoby, E.; Bouhelal, R.; Gerspacher, M.; Seuwen, K. The 7TM G-protein-coupled receptor target family. ChemMedChem 2006, 1, 760–782. [Google Scholar] [CrossRef]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci. 2012, 33, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.Y. Regulation, Signaling, and Physiological Functions of G-Proteins. J. Mol. Biol. 2016, 428, 3850–3868. [Google Scholar] [CrossRef] [Green Version]
- Duc, N.M.; Kim, H.R.; Chung, K.Y. Structural mechanism of G protein activation by G protein-coupled receptor. Eur. J. Pharmacol. 2015, 763, 214–222. [Google Scholar] [CrossRef]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein-Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Nygaard, R.; Frimurer, T.M.; Holst, B.; Rosenkilde, M.M.; Schwartz, T.W. Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol. Sci. 2009, 30, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Oldham, W.M.; Hamm, H.E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2008, 9, 60–71. [Google Scholar] [CrossRef] [PubMed]
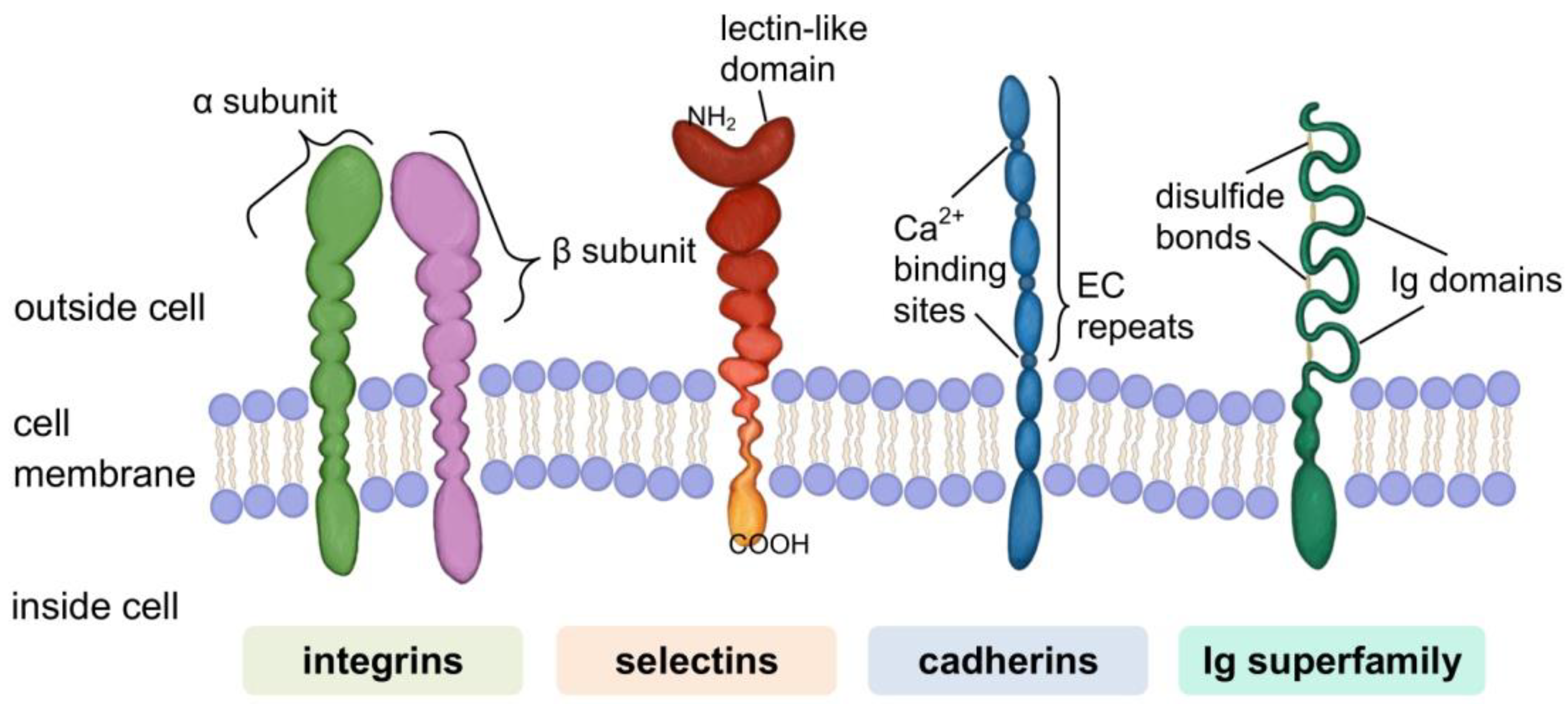
- Janiszewska, M.; Primi, M.C.; Izard, T. Cell adhesion in cancer: Beyond the migration of single cells. J. Biol. Chem. 2020, 295, 2495–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Vinogradova, O.; Plow, E.F. Integrin bidirectional signaling: A molecular view. PLoS Biol. 2004, 2, e169. [Google Scholar] [CrossRef] [Green Version]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [Green Version]
- Bouvard, D.; Pouwels, J.; De Franceschi, N.; Ivaska, J. Integrin inactivators: Balancing cellular functions in vitro and in vivo. Nat. Rev. Mol. Cell Biol. 2013, 14, 432–444. [Google Scholar] [CrossRef]
- Bachmann, M.; Kukkurainen, S.; Hytönen, V.P.; Wehrle-Haller, B. Cell adhesion by integrins. Physiol. Rev. 2019, 99, 1655–1699. [Google Scholar] [CrossRef]
- Revach, O.Y.; Grosheva, I.; Geiger, B. Biomechanical regulation of focal adhesion and invadopodia formation. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Michael, M.; Parsons, M. New perspectives on integrin-dependent adhesions. Curr. Opin. Cell Biol. 2020, 63, 31–37. [Google Scholar] [CrossRef]
- Ma, X.; Hammes, S.R. Paxillin actions in the nucleus. Steroids 2018, 133, 87–92. [Google Scholar] [CrossRef]
- Bays, J.L.; DeMali, K.A. Vinculin in cell–cell and cell–matrix adhesions. Cell. Mol. Life Sci. 2017, 74, 2999–3009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Banerjee, S.; Raina, M.; Haldar, S. Force-Directed “mechanointeractome” of Talin-Integrin. Biochemistry 2019, 58, 4677–4695. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Guan, J.L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Guan, J.L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walko, G.; Castañón, M.J.; Wiche, G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2015, 360, 529–544. [Google Scholar] [CrossRef] [Green Version]
- Turcan, I.; Jonkman, M.F. Blistering disease: Insight from the hemidesmosome and other components of the dermal-epidermal junction. Cell Tissue Res. 2015, 360, 545–569. [Google Scholar] [CrossRef]
- Taylor, M.E.; Drickamer, K. Paradigms for glycan-binding receptors in cell adhesion. Curr. Opin. Cell Biol. 2007, 19, 572–577. [Google Scholar] [CrossRef]
- Brown, G.D.; Willment, J.A.; Whitehead, L. C-type lectins in immunity and homeostasis. Nat. Rev. Immunol. 2018, 18, 374–389. [Google Scholar] [CrossRef]
- Ivetic, A.; Green, H.L.H.; Hart, S.J. L-selectin: A major regulator of leukocyte adhesion, migration and signaling. Front. Immunol. 2019, 10, 1068. [Google Scholar] [CrossRef] [Green Version]
- McEver, R.P. Selectins: Initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 2015, 107, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.; Videira, P.A.; Sackstein, R. E-selectin ligands in the human mononuclear phagocyte system: Implications for infection, inflammation, and immunotherapy. Front. Immunol. 2018, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Springer, T.A. Adhesion receptors of the immune system. Nature 1990, 346, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.A. Structural basis for selectin mechanochemistry. Proc. Natl. Acad. Sci. USA 2009, 106, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEver, R.P.; Zhu, C. Rolling cell adhesion. Annu. Rev. Cell Dev. Biol. 2010, 26, 363–396. [Google Scholar] [CrossRef]
- Elangbam, C.S.; Qualls, C.W.; Dahlgren, R.R. Cell Adhesion Molecules-Update. Vet. Pathol. 1997, 34, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O. Cell adhesion: Old and new questions. Trends Genet. 1999, 15, M33–M37. [Google Scholar] [CrossRef]
- Takeichi, M. Cadherins: A molecular family important in selective cell-cell adhesion. Annu. Rev. Biochem. 1990, 59, 237–252. [Google Scholar] [CrossRef]
- Nollet, F.; Kools, P.; Van Roy, F. Phylogenetic analysis of the cadherin superfamily allows identification of six major subfamilies besides several solitary members. J. Mol. Biol. 2000, 299, 551–572. [Google Scholar] [CrossRef]
- Priest, A.V.; Shafraz, O.; Sivasankar, S. Biophysical basis of cadherin mediated cell-cell adhesion. Exp. Cell Res. 2017, 358, 10–13. [Google Scholar] [CrossRef]
- Brasch, J.; Harrison, O.J.; Honig, B.; Shapiro, L. Thinking outside the cell: How cadherins drive adhesion. Trends Cell Biol. 2012, 22, 299–310. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, L.; Weis, W.I. Structure and biochemistry of cadherins and catenins. Cold Spring Harb. Perspect. Biol. 2009, 1, a003053. [Google Scholar] [CrossRef] [PubMed]
- Yonemura, S. Cadherin-actin interactions at adherens junctions. Curr. Opin. Cell Biol. 2011, 23, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Drees, F.; Pokutta, S.; Yamada, S.; Nelson, W.J.; Weis, W.I. α-catenin is a molecular switch that binds E-cadherin-β-catenin and regulates actin-filament assembly. Cell 2005, 123, 903–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrod, D.; Chidgey, M. Desmosome structure, composition and function. Biochim. Biophys. Acta-Biomembr. 2008, 1778, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Delva, E.; Tucker, D.K.; Kowalczyk, A.P. The desmosome. Cold Spring Harb. Perspect. Biol. 2009, 1, a002543. [Google Scholar] [CrossRef] [Green Version]
- Bork, P.; Holm, L.; Sander, C. The immunoglobulin fold: Structural classification, sequence patterns and common core. J. Mol. Biol. 1994, 242, 309–320. [Google Scholar] [CrossRef]
- Dermody, T.S.; Kirchner, E.; Guglielmi, K.M.; Stehle, T. Immunoglobulin superfamily virus receptors and the evolution of adaptive immunity. PLoS Pathog. 2009, 5, e1000481. [Google Scholar] [CrossRef] [Green Version]
- Wai Wong, C.; Dye, D.E.; Coombe, D.R. The role of immunoglobulin superfamily cell adhesion molecules in cancer metastasis. Int. J. Cell Biol. 2012, 2012, 340296. [Google Scholar] [CrossRef]
- Barclay, A.N. Membrane proteins with immunoglobulin-like domains—A master superfamily of interaction molecules. Semin. Immunol. 2003, 15, 215–223. [Google Scholar] [CrossRef]
- Zuo, T.; Gautam, A.; Wesemann, D.R. Affinity war: Forging immunoglobulin repertoires. Curr. Opin. Immunol. 2019, 57, 32–39. [Google Scholar] [CrossRef]
- Marchalonis, J.J.; Schluter, S.F.; Edmundson, A.B. The T-cell receptor as immunoglobulin: Paradigm regained. Proc. Soc. Exp. Biol. Med. 1997, 216, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Schmid, E.M.; Bakalar, M.H.; Choudhuri, K.; Weichsel, J.; Ann, H.S.; Geissler, P.L.; Dustin, M.L.; Fletcher, D.A. Size-dependent protein segregation at membrane interfaces. Nat. Phys. 2016, 12, 704–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Chang, H.; Dong, Y.; Guo, L.; Shi, X.; Wu, Y.; Huang, Y.; He, Y. Architecture of cell–cell adhesion mediated by sidekicks. Proc. Natl. Acad. Sci. USA 2018, 115, 9246–9251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finegan, T.M.; Bergstralh, D.T. Neuronal immunoglobulin superfamily cell adhesion molecules in epithelial morphogenesis: Insights from Drosophila. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190553. [Google Scholar] [CrossRef] [PubMed]
- Furley, A.J.; Morton, S.B.; Manalo, D.; Karagogeos, D.; Dodd, J.; Jessell, T.M. The axonal glycoprotein TAG-1 is an immunoglobulin superfamily member with neurite outgrowth-promoting activity. Cell 1990, 61, 157–170. [Google Scholar] [CrossRef]
- Maness, P.F.; Schachner, M. Neural recognition molecules of the immunoglobulin superfamily: Signaling transducers of axon guidance and neuronal migration. Nat. Neurosci. 2007, 10, 19–26. [Google Scholar] [CrossRef]
- Cameron, S.; McAllister, A.K. Immunoglobulin-like receptors and their impact on wiring of brain synapses. Annu. Rev. Genet. 2018, 52, 567–590. [Google Scholar] [CrossRef]
- Mohammad Reza, N. Tissue Engineering, Past, Present and Future; Imperialcollege Press: London, UK, 2014; Volume 1. [Google Scholar]
- Mhanna, R.; Hasan, A. Introduction to Tissue Engineering. Tissue Eng. Artif. Organs Regen. Med. Smart Diagnostics Pers. Med. 2016, 1–2, 1–34. [Google Scholar]
- Kim, H.J.; Kim, K.K.; Park, I.K.; Choi, B.S.; Kim, J.H.; Kim, M.S. Hybrid scaffolds composed of hyaluronic acid and collagen for cartilage regeneration. Tissue Eng. Regen. Med. 2012, 9, 57–62. [Google Scholar] [CrossRef]
- Polak, J.M.; Bishop, A.E. Stem cells and tissue engineering: Past, present, and future. Ann. N. Y. Acad. Sci. 2006, 1068, 352–366. [Google Scholar] [CrossRef]
- Kumar Meena, L.; Rather, H.; Kedaria, D.; Vasita, R. Polymeric microgels for bone tissue engineering applications–a review. Int. J. Polym. Mater. Polym. Biomater. 2020, 69, 381–397. [Google Scholar] [CrossRef]
- Amini, A.R.; Laurencin, C.T.; Nukavarapu, S.P. Bone tissue engineering: Recent advances and challenges. Crit. Rev. Biomed. Eng. 2012, 40, 363–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Choi, J.S.; Park, K.S.; Khang, G.; Lee, Y.M.; Lee, H.B. Response of MG63 osteoblast-like cells onto polycarbonate membrane surfaces with different micropore sizes. Biomaterials 2004, 25, 4699–4707. [Google Scholar] [CrossRef] [PubMed]
- Młotek, M.; Gadomska-Gajadhur, A.; Sobczak, A.; Kruk, A.; Perron, M.; Krawczyk, K. Modification of pla scaffold surface for medical applications. Appl. Sci. 2021, 11, 1815. [Google Scholar] [CrossRef]
- Vogler, E.A. Structure and reactivity of water at biomaterial surfaces. Adv. Colloid Interface Sci. 1998, 74, 69–117. [Google Scholar] [CrossRef]
- Kasemo, B.; Gold, J. Implant surfaces and interface processes. Adv. Dent. Res. 1999, 13, 8–20. [Google Scholar] [CrossRef]
- Rabe, M.; Verdes, D.; Seeger, S. Understanding protein adsorption phenomena at solid surfaces. Adv. Colloid Interface Sci. 2011, 162, 87–106. [Google Scholar] [CrossRef] [Green Version]
- Andrade, J.D.; Hlady, V.; Wei, A.P. Adsorption of complex proteins at interfaces. Pure Appl. Chem. 1992, 64, 1777–1781. [Google Scholar] [CrossRef] [Green Version]
- Felgueiras, H.P.; Antunes, J.C.; Martins, M.C.L.; Barbosa, M.A. Fundamentals of protein and cell interactions in biomaterials. In Peptides and Proteins as Biomaterials for Tissue Regeneration and Repair; Barbosa, M.A., Martins, M.C., Eds.; Woodhead Publishing: Thurston, UK, 2018; pp. 1–27. ISBN 9780081008522. [Google Scholar]
- Norde, W.; Giacomelli, C.E. Conformational changes in proteins at interfaces: From solution to the interface, and back. Macromol. Symp. 1999, 145, 125–136. [Google Scholar] [CrossRef]
- Norde, W.; Giacomelli, C.E. BSA structural changes during homomolecular exchange between the adsorbed and the dissolved states. J. Biotechnol. 2000, 79, 259–268. [Google Scholar] [CrossRef]
- Lundstrom, I. Models of Protein Adsorption on Solid Surfaces. Prog. Colloid Polym. Sci. 1985, 70, 76–82. [Google Scholar] [CrossRef]
- Mariani, E.; Lisignoli, G.; Borzì, R.M.; Pulsatelli, L. Biomaterials: Foreign bodies or tuners for the immune response? Int. J. Mol. Sci. 2019, 20, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ode Boni, B.O.; Lamboni, L.; Souho, T.; Gauthier, M.; Yang, G. Immunomodulation and cellular response to biomaterials: The overriding role of neutrophils in healing. Mater. Horizons 2019, 6, 1122–1137. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Aresta-DaSilva, S.; Tang, K.; Alvarez, D.; Webber, M.J.; Tang, B.C.; Lavin, D.M.; Veiseh, O.; Doloff, J.C.; Bose, S.; et al. Neutrophil responses to sterile implant materials. PLoS ONE 2015, 10, e0137550. [Google Scholar] [CrossRef] [Green Version]
- Desalegn, G.; Pabst, O. Inflammation triggers immediate rather than progressive changes in monocyte differentiation in the small intestine. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Mesure, L.; de Visscher, G.; Vranken, I.; Lebacq, A.; Flameng, W. Gene expression study of monocytes/macrophages during early foreign body reaction and identification of potential precursors of myofibroblasts. PLoS ONE 2010, 5, e12949. [Google Scholar] [CrossRef]
- Christenson, E.M.; Dadsetan, M.; Anderson, J.M.; Hiltner, A. Biostability and macrophage-mediated foreign body reaction of silicone-modified polyurethanes. J. Biomed. Mater. Res. Part A 2005, 74, 141–155. [Google Scholar] [CrossRef]
- Sheikh, Z.; Brooks, P.J.; Barzilay, O.; Fine, N.; Glogauer, M. Macrophages, foreign body giant cells and their response to implantable biomaterials. Materials 2015, 8, 5671–5701. [Google Scholar] [CrossRef] [Green Version]
- Ward, W.K.; Slobodzian, E.P.; Tiekotter, K.L.; Wood, M.D. The effect of microgeometry, implant thickness and polyurethane chemistry on the foreign body response to subcutaneous implants. Biomaterials 2002, 23, 4185–4192. [Google Scholar] [CrossRef]
- Hulbert, S.F.; Morrison, S.J.; Klawitter, J.J. Tissue reaction to three ceramics of porous and non-porous structures. J. Biomed. Mater. Res. 1972, 6, 347–374. [Google Scholar] [CrossRef]
- Zdolsek, J.; Eaton, J.W.; Tang, L. Histamine release and fibrinogen adsorption mediate acute inflammatory responses to biomaterial implants in humans. J. Transl. Med. 2007, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Theoharides, T.C.; Tsilioni, I.; Conti, P. Mast cells may regulate the anti-inflammatory activity of IL-37. Int. J. Mol. Sci. 2019, 20, 3701. [Google Scholar] [CrossRef] [Green Version]
- Thevenot, P.T.; Nair, A.M.; Shen, J.; Lotfi, P.; Ko, C.Y.; Tang, L. The effect of incorporation of SDF-1α into PLGA scaffolds on stem cell recruitment and the inflammatory response. Biomaterials 2010, 31, 3997–4008. [Google Scholar] [CrossRef] [Green Version]
- Zachman, A.L.; Crowder, S.W.; Ortiz, O.; Zienkiewicz, K.J.; Bronikowski, C.M.; Yu, S.S.; Giorgio, T.D.; Guelcher, S.A.; Kohn, J.; Sung, H.J. Pro-angiogenic and anti-inflammatory regulation by functional peptides loaded in polymeric implants for soft tissue regeneration. Tissue Eng.-Part A 2013, 19, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Hosoyama, K.; Ahumada, M.; Goel, K.; Ruel, M.; Suuronen, E.J.; Alarcon, E.I. Electroconductive materials as biomimetic platforms for tissue regeneration. Biotechnol. Adv. 2019, 37, 444–458. [Google Scholar] [CrossRef] [PubMed]
- Mason, T.O.; Shimanovich, U. Fibrous Protein Self-Assembly in Biomimetic Materials. Adv. Mater. 2018, 30, 1706462. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.; Michiardi, A.; Castaño, O.; Planell, J.A. Biomaterials in orthopaedics. J. R. Soc. Interface 2008, 5, 1137–1158. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.C.; Siedlecki, C.A. Effects of surface wettability and contact time on protein adhesion to biomaterial surfaces. Biomaterials 2007, 28, 3273–3283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parhi, P.; Golas, A.; Barnthip, N.; Noh, H.; Vogler, E.A. Volumetric interpretation of protein adsorption: Capacity scaling with adsorbate molecular weight and adsorbent surface energy. Biomaterials 2009, 30, 6814–6824. [Google Scholar] [CrossRef] [Green Version]
- Thevenot, P.; Hu, W.; Tang, L. Surface chemistry influences implant biocompatibility. Curr. Top. Med. Chem. 2008, 8, 270–280. [Google Scholar] [CrossRef]
- Roach, P.; Farrar, D.; Perry, C.C. Interpretation of protein adsorption: Surface-induced conformational changes. J. Am. Chem. Soc. 2005, 127, 8168–8173. [Google Scholar] [CrossRef] [PubMed]
- Roach, P.; Parker, T.; Gadegaard, N.; Alexander, M.R. Surface strategies for control of neuronal cell adhesion: A review. Surf. Sci. Rep. 2010, 65, 145–173. [Google Scholar] [CrossRef]
- Chen, S.; Guo, Y.; Liu, R.; Wu, S.; Fang, J.; Huang, B.; Li, Z.; Chen, Z.; Chen, Z. Tuning surface properties of bone biomaterials to manipulate osteoblastic cell adhesion and the signaling pathways for the enhancement of early osseointegration. Colloids Surfaces B Biointerfaces 2018, 164, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Zelzer, M.; Albutt, D.; Alexander, M.R.; Russell, N.A. The Role of Albumin and Fibronectin in the Adhesion of Fibroblasts to Plasma Polymer Surfaces. Plasma Process. Polym. 2012, 9, 149–156. [Google Scholar] [CrossRef]
- Guo, B.; Anzai, J.I.; Osa, T. Adsorption behavior of serum albumin on electrode surfaces and the effects of electrode potential. Chem. Pharm. Bull. 1996, 44, 800–803. [Google Scholar] [CrossRef] [Green Version]
- Faucheux, N.; Schweiss, R.; Lützow, K.; Werner, C.; Groth, T. Self-assembled monolayers with different terminating groups as model substrates for cell adhesion studies. Biomaterials 2004, 25, 2721–2730. [Google Scholar] [CrossRef]
- Keselowsky, B.G.; Collard, D.M.; García, A.J. Integrin binding specificity regulates biomaterial surface chemistry effects on cell differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 5953–5957. [Google Scholar] [CrossRef] [Green Version]
- Lan, M.A.; Gersbach, C.A.; Michael, K.E.; Keselowsky, B.G.; García, A.J. Myoblast proliferation and differentiation on fibronectin-coated self assembled monolayers presenting different surface chemistries. Biomaterials 2005, 26, 4523–4531. [Google Scholar] [CrossRef]
- McClary, K.B.; Ugarova, T.; Grainger, D.W. Modulating fibroblast adhesion, spreading, and proliferation using self- assembled monolayer films of alkylthiolates on gold. J. Biomed. Mater. Res. 2000, 50, 428–439. [Google Scholar] [CrossRef]
- Aiyelabegan, H.T.; Sadroddiny, E. Fundamentals of protein and cell interactions in biomaterials. Biomed. Pharmacother. 2017, 88, 956–970. [Google Scholar] [CrossRef]
- Ruoslahti, E.; Pierschbacher, M.D. Arg-Gly-Asp: A versatile cell recognition signal. Cell 1986, 44, 517–518. [Google Scholar] [CrossRef]
- Karimi, F.; O’Connor, A.J.; Qiao, G.G.; Heath, D.E. Integrin Clustering Matters: A Review of Biomaterials Functionalized with Multivalent Integrin-Binding Ligands to Improve Cell Adhesion, Migration, Differentiation, Angiogenesis, and Biomedical Device Integration. Adv. Healthc. Mater. 2018, 7, 1701324. [Google Scholar] [CrossRef] [PubMed]
- Reyes, C.D.; García, A.J. Engineering integrin-specific surfaces with a triple-helical collagen-mimetic peptide. J. Biomed. Mater. Res.-Part A 2003, 65, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Nomizu, M.; Weeks, B.S.; Weston, C.A.; Kim, W.H.; Kleinman, H.K.; Yamada, Y. Structure-activity study of a laminin α1 chain active peptide segment Ile-Lys-Val-Ala-Val (IKVAV). FEBS Lett. 1995, 365, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Wang, W.; Shao, Q.; Li, B.; Yu, D.; Zhou, X.; Parajuli, J.; Xu, H.; Qiu, T.; Yetisen, A.K.; et al. Pentapeptide IKVAV-engineered hydrogels for neural stem cell attachment. Biomater. Sci. 2021, 9, 2887–2892. [Google Scholar] [CrossRef] [PubMed]
- Boateng, S.Y.; Lateef, S.S.; Mosley, W.; Hartman, T.J.; Hanley, L.; Russell, B. RGD and YIGSR synthetic peptides facilitate cellular adhesion identical to that of laminin and fibronectin but alter the physiology of neonatal cardiac myocytes. Am. J. Physiol.-Cell Physiol. 2005, 288, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Aota, S.I.; Nomizu, M.; Yamada, K.M. The short amino acid sequence Pro-His-Ser-Arg-Asn in human fibronectin enhances cell-adhesive function. J. Biol. Chem. 1994, 269, 24756–24761. [Google Scholar] [CrossRef]
- Cutler, S.M.; García, A.J. Engineering cell adhesive surfaces that direct integrin α5β1 binding using a recombinant fragment of fibronectin. Biomaterials 2003, 24, 1759–1770. [Google Scholar] [CrossRef]
- Lebaron, R.G.; Athanasiou, K.A. Extracellular matrix cell adhesion peptides: Functional applications in orthopedic materials. Tissue Eng. 2000, 6, 85–103. [Google Scholar] [CrossRef]
- Song, W.; Mano, J.F. Interactions between cells or proteins and surfaces exhibiting extreme wettabilities. Soft Matter 2013, 9, 2985–2999. [Google Scholar] [CrossRef] [Green Version]
- Siebers, M.C.; Ter Brugge, P.J.; Walboomers, X.F.; Jansen, J.A. Integrins as linker proteins between osteoblasts and bone replacing materials. A critical review. Biomaterials 2005, 26, 137–146. [Google Scholar] [CrossRef]
- Ponche, A.; Bigerelle, M.; Anselme, K. Relative influence of surface topography and surface chemistry on cell response to bone implant materials. Part 1: Physico-chemical effects. J. Eng. Med. 2010, 224, 1471–1486. [Google Scholar] [CrossRef] [PubMed]
- Ayala, P.; Desai, T.A. Integrin α3 blockade enhances microtopographical down-regulation of α-smooth muscle actin: Role of microtopography in ECM regulation. Integr. Biol. 2011, 3, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Jäger, M.; Zilkens, C.; Zanger, K.; Krauspe, R. Significance of nano- and microtopography for cell-surface interactions in orthopaedic implants. J. Biomed. Biotechnol. 2007, 2007, 69036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulander, M.; Lundgren, A.; Berglin, M.; Ohrlander, M.; Lausmaa, J.; Elwing, H. Immune complement activation is attenuated by surface nanotopography. Int. J. Nanomed. 2011, 6, 2653–2666. [Google Scholar] [CrossRef] [Green Version]
- Draghi, L.; Cigada, A. Nanostructured surfaces for biomedical applications. Part I: Nanotopography. J. Appl. Biomater. Biomech. 2007, 5, 61–69. [Google Scholar]
- Folch, A.; Toner, M. Microengineering of cellular interactions. Annu. Rev. Biomed. Eng. 2000, 2, 227–256. [Google Scholar] [CrossRef] [Green Version]
- Poellmann, M.J.; Harrell, P.A.; King, W.P.; Johnson, A.J.W. Geometric microenvironment directs cell morphology on topographically patterned hydrogel substrates. Acta Biomater. 2010, 6, 3514–3523. [Google Scholar] [CrossRef]
- Solanki, A.; Shah, S.; Memoli, K.A.; Park, S.Y.; Hong, S.; Lee, K.B. Controlling differentiation of neural stem cells using extracellular matrix protein patterns. Small 2010, 6, 2509–2513. [Google Scholar] [CrossRef] [Green Version]
- Carthew, J.; Abdelmaksoud, H.H.; Hodgson-Garms, M.; Aslanoglou, S.; Ghavamian, S.; Elnathan, R.; Spatz, J.P.; Brugger, J.; Thissen, H.; Voelcker, N.H.; et al. Precision Surface Microtopography Regulates Cell Fate via Changes to Actomyosin Contractility and Nuclear Architecture. Adv. Sci. 2021, 8, 2003186. [Google Scholar] [CrossRef]
- Anselme, K.; Ploux, L.; Ponche, A. Cell/material interfaces: Influence of surface chemistry and surface topography on cell adhesion. J. Adhes. Sci. Technol. 2010, 24, 831–852. [Google Scholar] [CrossRef]
- Majhy, B.; Priyadarshini, P.; Sen, A.K. Effect of surface energy and roughness on cell adhesion and growth-facile surface modification for enhanced cell culture. RSC Adv. 2021, 11, 15467–15476. [Google Scholar] [CrossRef]
- Hallab, N.J.; Bundy, K.J.; O’Connor, K.; Moses, R.L.; Jacobs, J.J. Evaluation of metallic and polymeric biomaterial surface energy and surface roughness characteristics for directed cell adhesion. Tissue Eng. 2001, 7, 55–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Xie, W.; Yu, L.; Camacho, L.C.; Nie, C.; Zhang, M.; Haag, R.; Wei, Q. Surface Roughness Gradients Reveal Topography-Specific Mechanosensitive Responses in Human Mesenchymal Stem Cells. Small 2020, 16, 1905422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunetti, V.; Maiorano, G.; Rizzello, L.; Sorce, B.; Sabella, S.; Cingolani, R.; Pompa, P.P. Neurons sense nanoscale roughness with nanometer sensitivity. Proc. Natl. Acad. Sci. USA 2010, 107, 6264–6269. [Google Scholar] [CrossRef] [Green Version]
- Łopacińska, J.M.; Grǎdinaru, C.; Wierzbicki, R.; Købler, C.; Schmidt, M.S.; Madsen, M.T.; Skolimowski, M.; Dufva, M.; Flyvbjerg, H.; Mølhave, K. Cell motility, morphology, viability and proliferation in response to nanotopography on silicon black. Nanoscale 2012, 4, 3739–3745. [Google Scholar] [CrossRef]
- Lange, R.; Lüthen, F.; Beck, U.; Rychly, J.; Baumann, A.; Nebe, B. Cell-extracellular matrix interaction and physico-chemical characteristics of titanium surfaces depend on the roughness of the material. In Proceedings of the Biomolecular Engineering; Elsevier: Amsterdam, The Netherlands, 2002; Volume 19, pp. 255–261. [Google Scholar]
- Martinez, M.A.F.; de Balderrama, Í.F.; Karam, P.S.B.H.; de Oliveira, R.C.; de Oliveira, F.A.; Grandini, C.R.; Vicente, F.B.; Stavropoulos, A.; Zangrando, M.S.R.; Sant’Ana, A.C.P. Surface roughness of titanium disks influences the adhesion, proliferation and differentiation of osteogenic properties derived from human. Int. J. Implant Dent. 2020, 6, 46. [Google Scholar] [CrossRef]
- Ter Brugge, P.J.; Torensma, R.; De Ruijter, J.E.; Figdor, C.G.; Jansen, J.A. Modulation of integrin expression on rat bone marrow cells by substrates with different surface characteristics. Tissue Eng. 2002, 8, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Ricci, J.L.; Grew, J.C.; Alexander, H. Connective-tissue responses to defined biomaterial surfaces. I. Growth of rat fibroblast and bone marrow cell colonies on microgrooved substrates. J. Biomed. Mater. Res.-Part A 2008, 85, 313–325. [Google Scholar] [CrossRef] [PubMed]

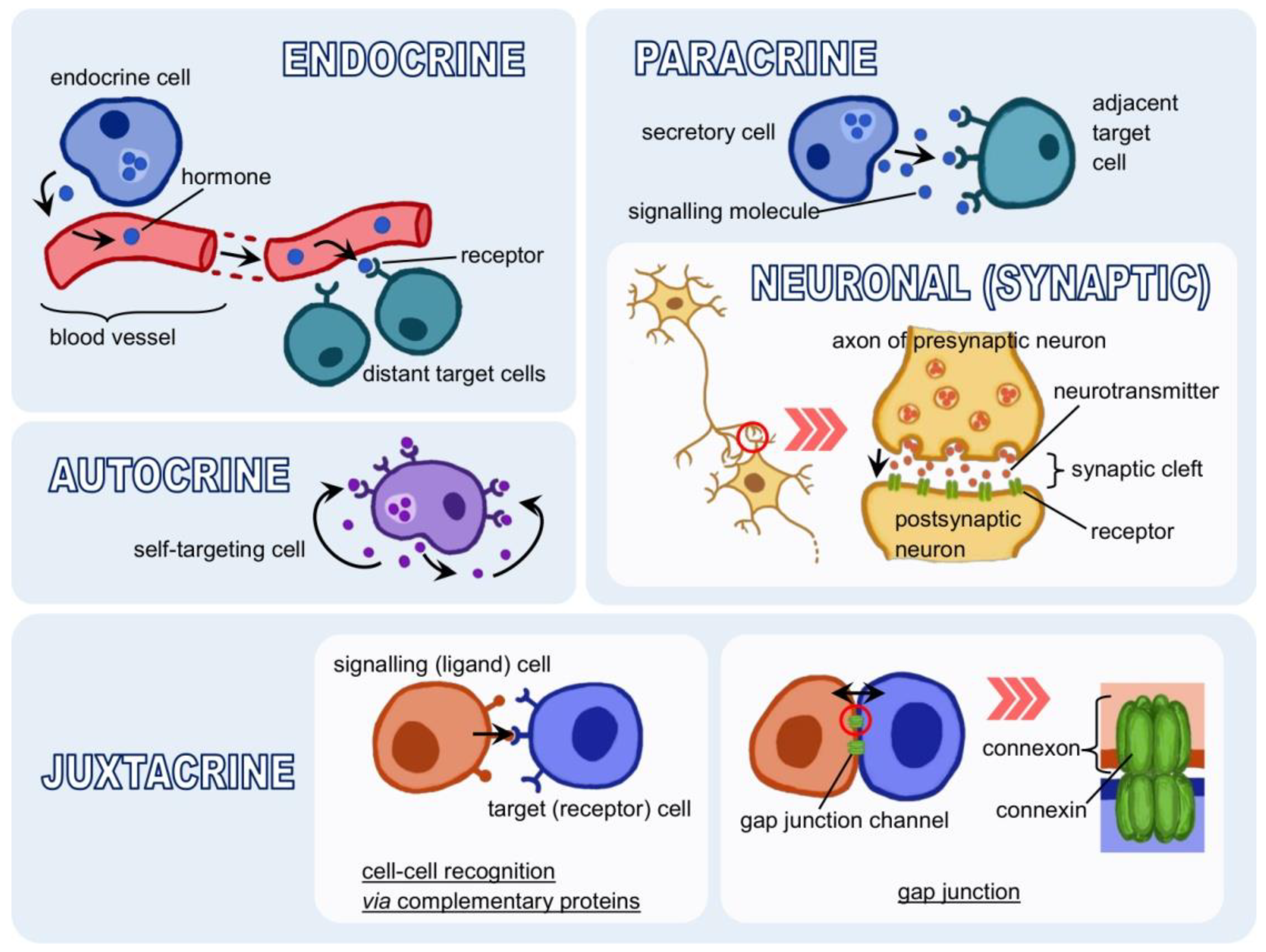

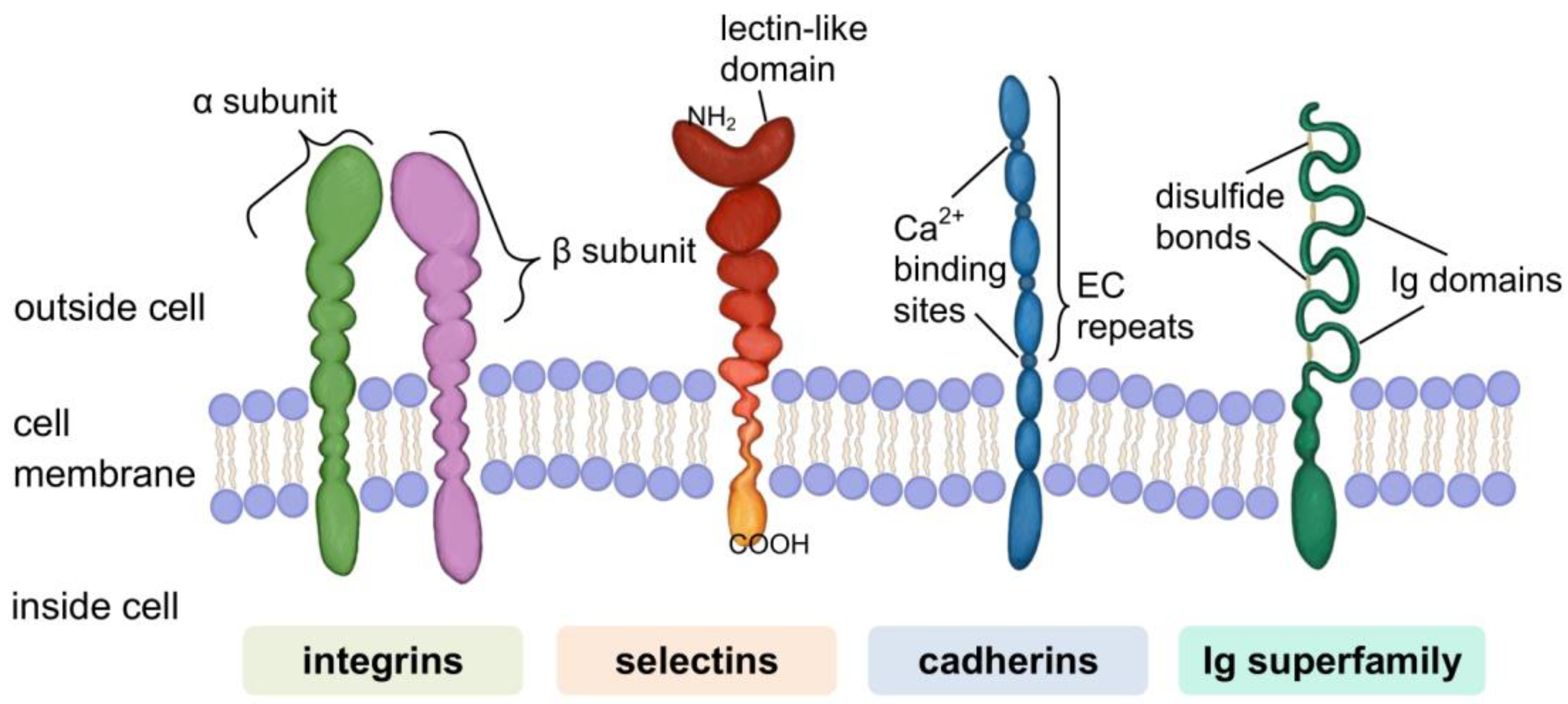
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bandzerewicz, A.; Gadomska-Gajadhur, A. Into the Tissues: Extracellular Matrix and Its Artificial Substitutes: Cell Signalling Mechanisms. Cells 2022, 11, 914. https://doi.org/10.3390/cells11050914
Bandzerewicz A, Gadomska-Gajadhur A. Into the Tissues: Extracellular Matrix and Its Artificial Substitutes: Cell Signalling Mechanisms. Cells. 2022; 11(5):914. https://doi.org/10.3390/cells11050914
Chicago/Turabian StyleBandzerewicz, Aleksandra, and Agnieszka Gadomska-Gajadhur. 2022. "Into the Tissues: Extracellular Matrix and Its Artificial Substitutes: Cell Signalling Mechanisms" Cells 11, no. 5: 914. https://doi.org/10.3390/cells11050914
APA StyleBandzerewicz, A., & Gadomska-Gajadhur, A. (2022). Into the Tissues: Extracellular Matrix and Its Artificial Substitutes: Cell Signalling Mechanisms. Cells, 11(5), 914. https://doi.org/10.3390/cells11050914