
Artificial Intelligent Deep Learning Molecular Generative Modeling of Scaffold-Focused and Cannabinoid CB2 Target-Specific Small-Molecule Sublibraries


Abstract
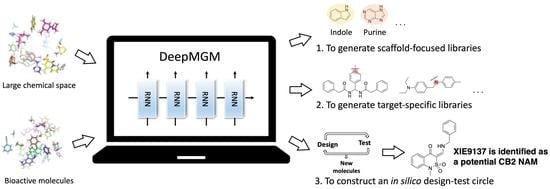
:
1. Introduction
2. Materials and Methods
2.1. Dataset Preparation
2.2. Molecular Representation and One-Hot Encoding
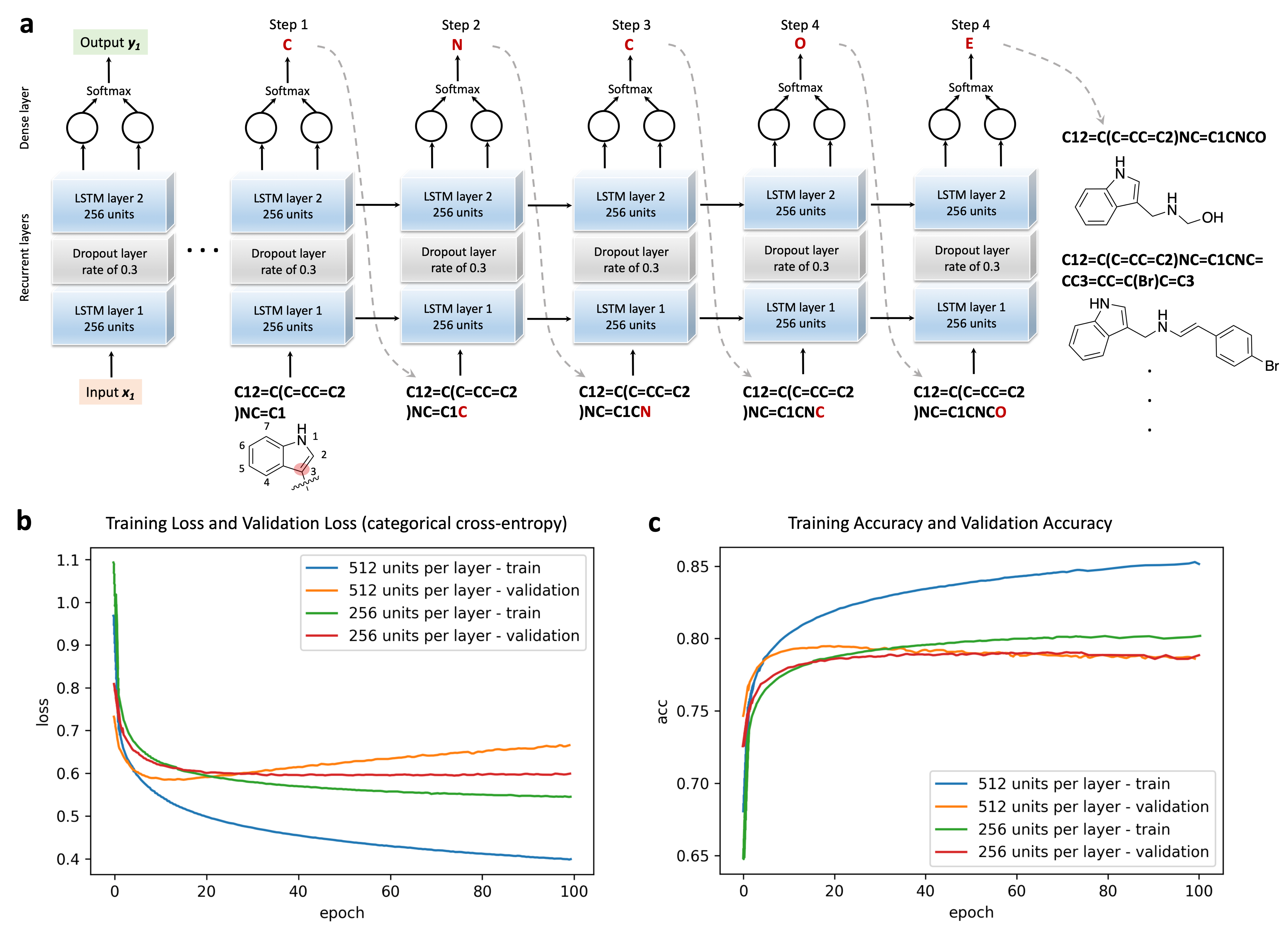
2.3. Neural Network Implementation
2.4. Transfer Learning
2.5. Model Evaluation
2.6. MLP Discriminator for Cannabinoid Receptor 2
2.7. Chemistry
2.8. CB2 [35S]-GTPγS Functional Assay
3. Results and Discussion
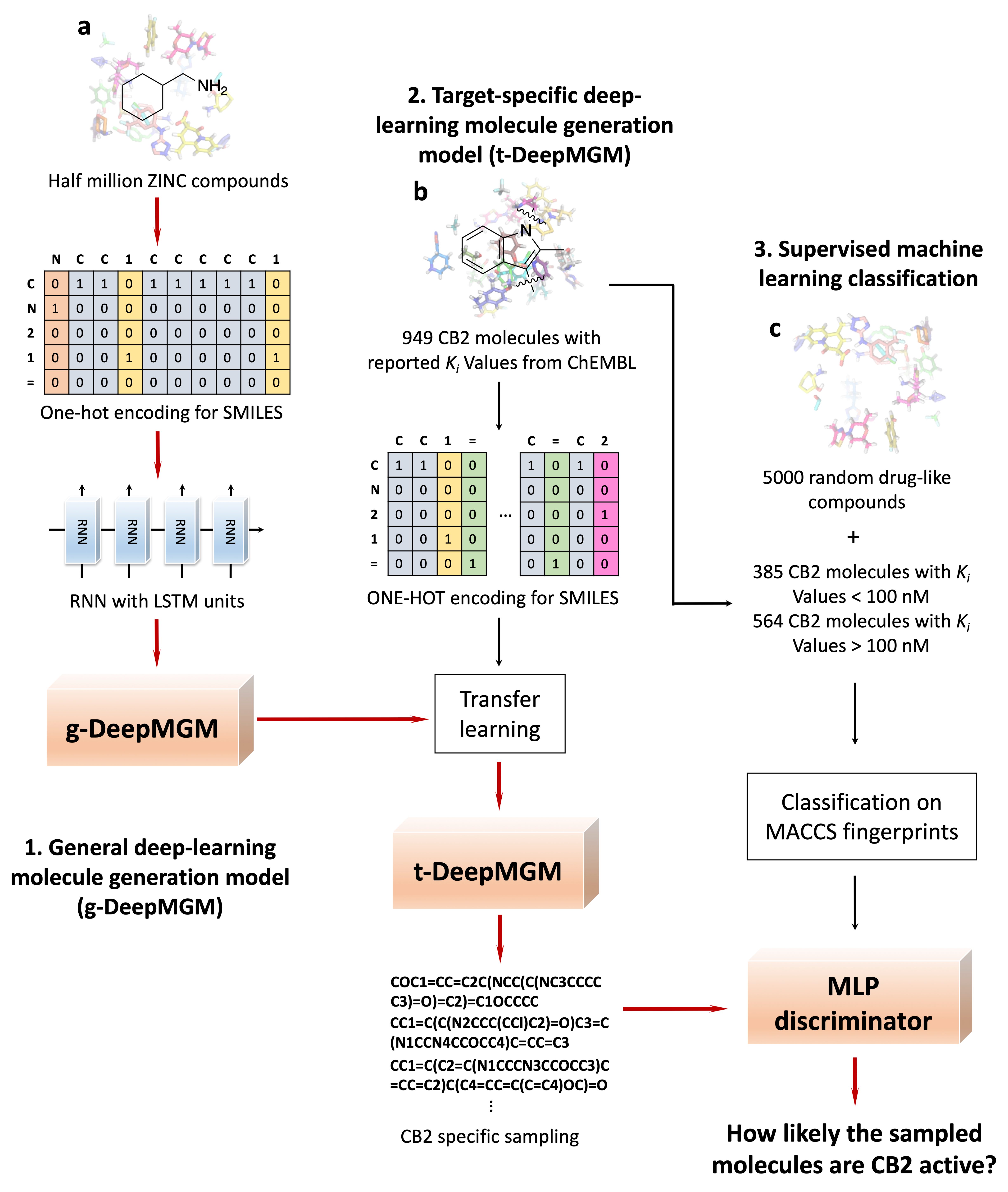
3.1. The Overview of General/Target-Specific Molecule Generation Models (g-DeepMGM and t-DeepMGM)
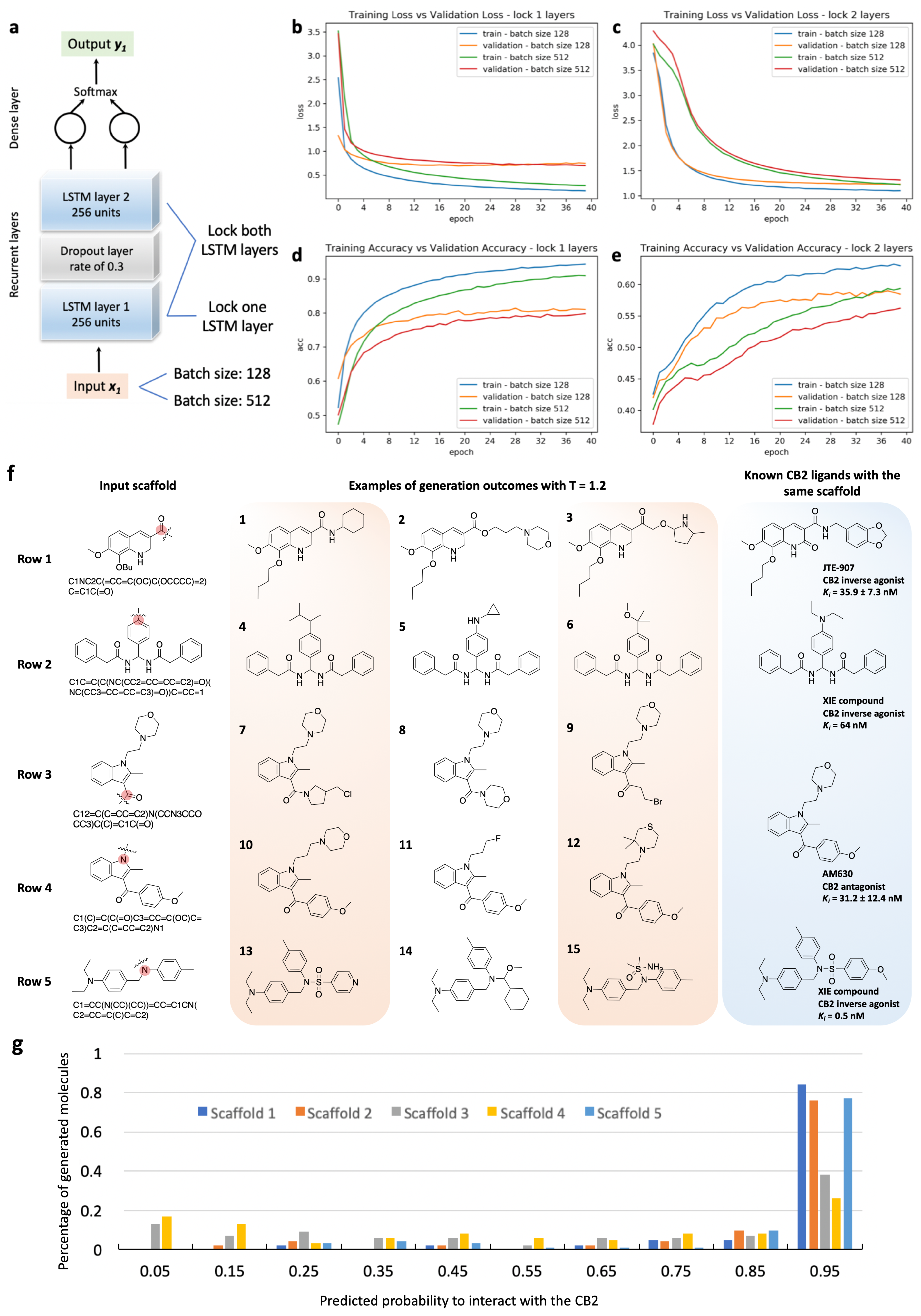
3.2. RNN Model Training and Sampling
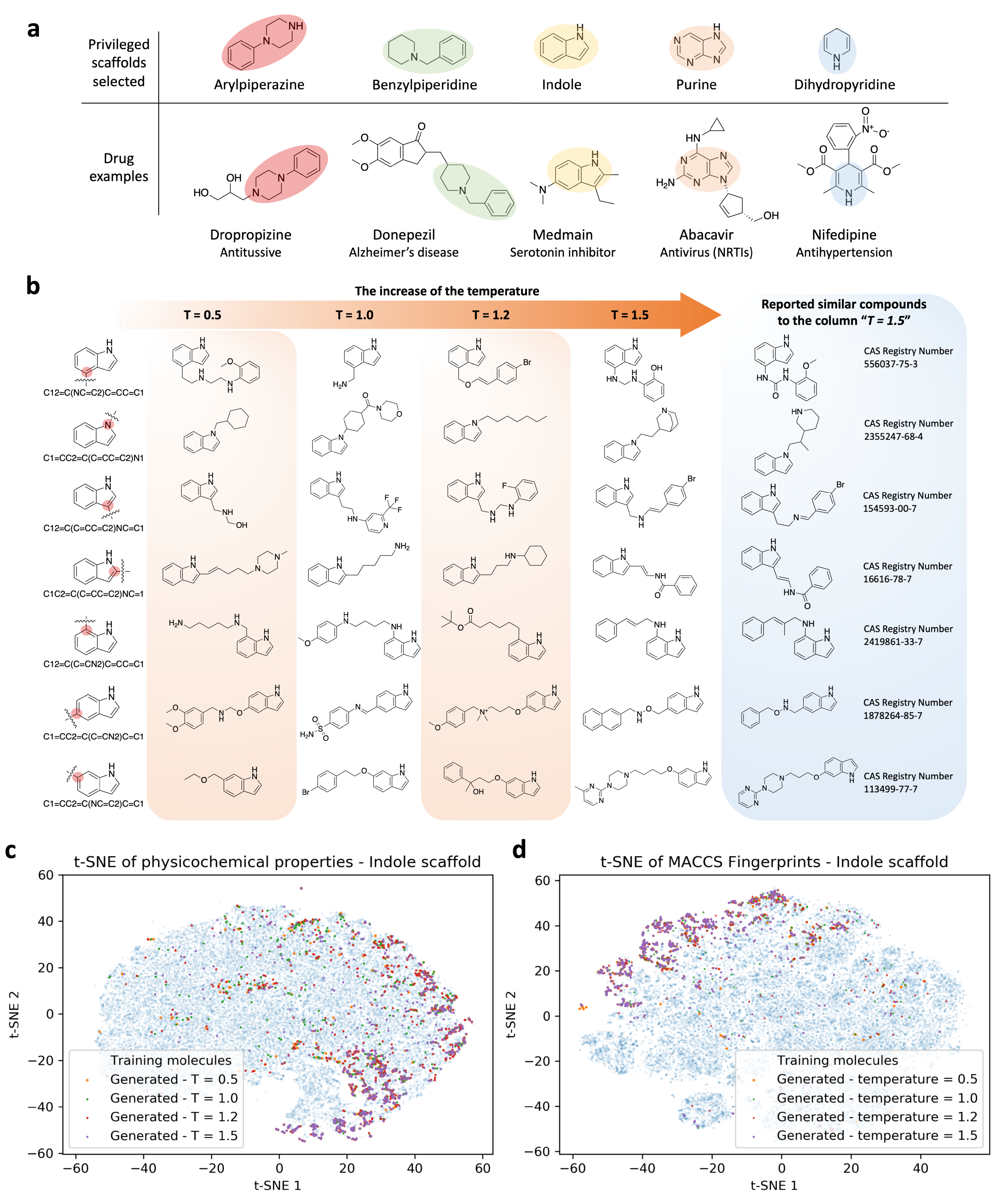
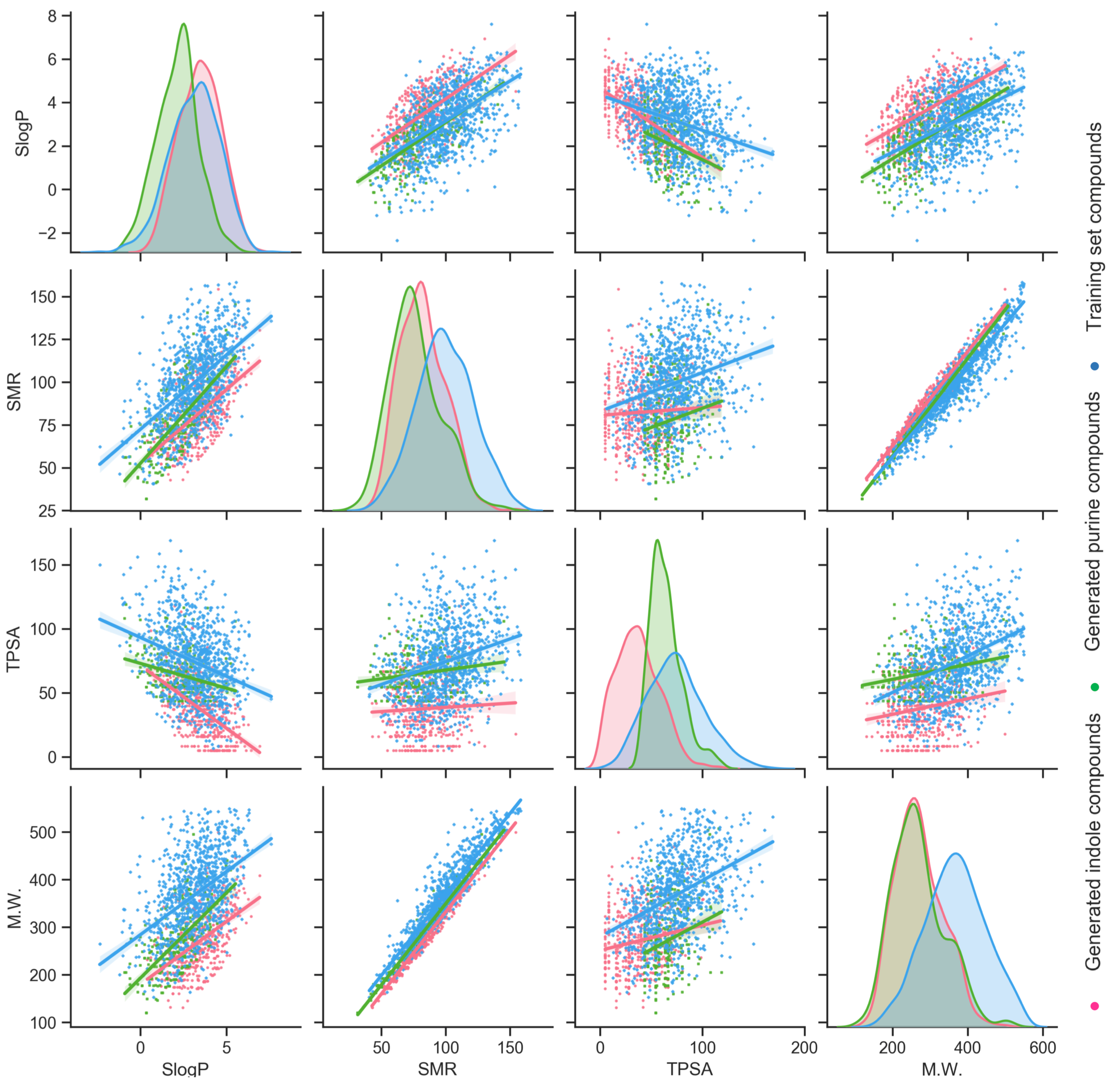
3.3. Indole Scaffold Compounds Generation
3.4. Transfer Learning for Cannabinoid Receptor 2
3.5. Classification of Generated Molecules with a Discriminator
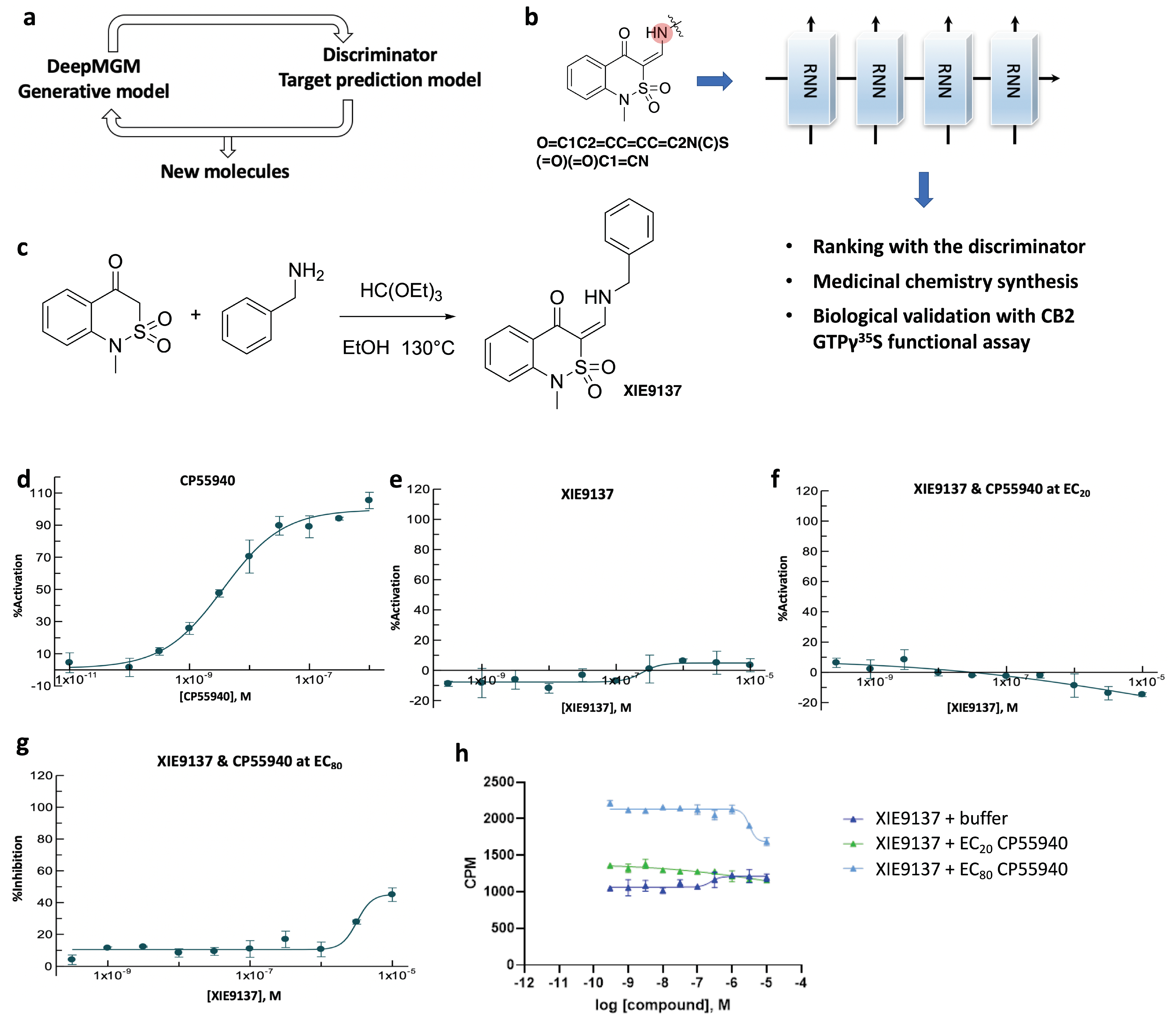
3.6. Proof-of-Evidence in Discovering a Potential CB2 Negative Allosteric Modulator, XIE9137
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated research and development investment needed to bring a new medicine to market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef]
- Blay, V.; Tolani, B.; Ho, S.P.; Arkin, M.R. High-Throughput Screening: Today’s biochemical and cell-based approaches. Drug Discov. Today 2020, 25, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Reymond, J.L.; Ruddigkeit, L.; Blum, L.; van Deursen, R. The enumeration of chemical space. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 717–733. [Google Scholar] [CrossRef]
- Schneider, G.; Baringhaus, K.-H. Molecular Design: Concepts and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Evans, B.; Rittle, K.; Bock, M.; DiPardo, R.; Freidinger, R.; Whitter, W.; Lundell, G.; Veber, D.; Anderson, P.; Chang, R. Methods for drug discovery: Development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Welsch, M.E.; Snyder, S.A.; Stockwell, B.R. Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 2010, 14, 347–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakubczyk, D.; Pfau, R.; Encinas, A.; Rösch, E.; Gil, C.; Masters, K.; Gläser, F.; Kramer, C.S.; Newman, D.; Albericio, F. Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation; Royal Society of Chemistry: Cambridge, UK, 2015. [Google Scholar]
- Zhao, H.; Dietrich, J. Privileged scaffolds in lead generation. Expert Opin. Drug Discov. 2015, 10, 781–790. [Google Scholar] [CrossRef]
- Jing, Y.; Bian, Y.; Hu, Z.; Wang, L.; Xie, X.-Q.S. Deep learning for drug design: An artificial intelligence paradigm for drug discovery in the big data era. AAPS J. 2018, 20, 58. [Google Scholar] [CrossRef]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design—A review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef] [Green Version]
- Bian, Y.; Xie, X.-Q. Generative chemistry: Drug discovery with deep learning generative models. J. Mol. Model. 2021, 27, 71. [Google Scholar] [CrossRef]
- Bian, Y.; Jing, Y.; Wang, L.; Ma, S.; Jun, J.J.; Xie, X.-Q. Prediction of orthosteric and allosteric regulations on cannabinoid receptors using supervised machine learning classifiers. Mol. Pharm. 2019, 16, 2605–2615. [Google Scholar] [CrossRef]
- Hou, T.; Bian, Y.; McGuire, T.; Xie, X.-Q. Integrated Multi-Class Classification and Prediction of GPCR Allosteric Modulators by Machine Learning Intelligence. Biomolecules 2021, 11, 870. [Google Scholar] [CrossRef]
- Moret, M.; Friedrich, L.; Grisoni, F.; Merk, D.; Schneider, G. Generative molecular design in low data regimes. Nat. Mach. Intell. 2020, 2, 171–180. [Google Scholar] [CrossRef]
- Zheng, S.; Yan, X.; Gu, Q.; Yang, Y.; Du, Y.; Lu, Y.; Xu, J. QBMG: Quasi-biogenic molecule generator with deep recurrent neural network. J. Cheminform. 2019, 11, 5. [Google Scholar] [CrossRef] [Green Version]
- Imrie, F.; Bradley, A.R.; van der Schaar, M.; Deane, C.M. Deep generative models for 3d linker design. J. Chem. Inf. Model. 2020, 60, 1983–1995. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Méndez-Lucio, O.; Baillif, B.; Clevert, D.-A.; Rouquié, D.; Wichard, J. De novo generation of hit-like molecules from gene expression signatures using artificial intelligence. Nat. Commun. 2020, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Segler, M.H.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Shayakhmetov, R.; Kuznetsov, M.; Zhebrak, A.; Kadurin, A.; Nikolenko, S.; Aliper, A.; Polykovskiy, D. Molecular Generation for Desired Transcriptome Changes With Adversarial Autoencoders. Front. Pharmacol. 2020, 11, 269. [Google Scholar] [CrossRef]
- Bian, Y.; Wang, J.; Jun, J.J.; Xie, X.-Q. Deep convolutional generative adversarial network (dcGAN) models for screening and design of small molecules targeting cannabinoid receptors. Mol. Pharm. 2019, 16, 4451–4460. [Google Scholar] [CrossRef]
- Maziarka, Ł.; Pocha, A.; Kaczmarczyk, J.; Rataj, K.; Danel, T.; Warchoł, M. Mol-CycleGAN: A generative model for molecular optimization. J. Cheminform. 2020, 12, 2. [Google Scholar] [CrossRef] [Green Version]
- Grebner, C.; Matter, H.; Plowright, A.T.; Hessler, G. Automated De Novo Design in Medicinal Chemistry: Which Types of Chemistry Does a Generative Neural Network Learn? J. Med. Chem. 2020, 63, 8809–8823. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.P.; Murcko, M. Assessing the impact of generative AI on medicinal chemistry. Nat. Biotechnol. 2020, 38, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Vanhaelen, Q.; Lin, Y.-C.; Zhavoronkov, A. The Advent of Generative Chemistry. ACS Med. Chem. Lett. 2020, 11, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Yosinski, J.; Clune, J.; Bengio, Y.; Lipson, H. How transferable are features in deep neural networks? In Proceedings of the Advances in Neural Information Processing Systems, Montreal, QC, Canada, 8 December 2014; pp. 3320–3328. [Google Scholar]
- Xing, C.; Zhuang, Y.; Xu, T.-H.; Feng, Z.; Zhou, X.E.; Chen, M.; Wang, L.; Meng, X.; Xue, Y.; Wang, J. Cryo-EM structure of the human cannabinoid receptor CB2-Gi signaling complex. Cell 2020, 180, 645–654.e13. [Google Scholar] [CrossRef]
- Bian, Y.-m.; He, X.-b.; Jing, Y.-k.; Wang, L.-r.; Wang, J.-m.; Xie, X.-Q. Computational systems pharmacology analysis of cannabidiol: A combination of chemogenomics-knowledgebase network analysis and integrated in silico modeling and simulation. Acta Pharmacol. Sin. 2019, 40, 374–386. [Google Scholar] [CrossRef]
- Mackie, K. Distribution of cannabinoid receptors in the central and peripheral nervous system. In Cannabinoids. Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2005; pp. 299–325. [Google Scholar]
- Svíženská, I.; Dubový, P.; Šulcová, A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures—a short review. Pharmacol. Biochem. Behav. 2008, 90, 501–511. [Google Scholar] [CrossRef]
- Cabral, G.; Marciano-Cabral, F. Cannabinoid receptors in microglia of the central nervous system: Immune functional relevance. J. Leukoc. Biol. 2005, 78, 1192–1197. [Google Scholar] [CrossRef]
- Yang, P.; Wang, L.; Xie, X.-Q. Latest advances in novel cannabinoid CB2 ligands for drug abuse and their therapeutic potential. Future Med. Chem. 2012, 4, 187–204. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey Conn, P.; Christopoulos, A.; Lindsley, C.W. Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 2009, 8, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Bian, Y.; Jun, J.J.; Cuyler, J.; Xie, X.-Q. Covalent allosteric modulation: An emerging strategy for GPCRs drug discovery. Eur. J. Med. Chem. 2020, 206, 112690. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [Green Version]
- Merk, D.; Grisoni, F.; Friedrich, L.; Schneider, G. Tuning artificial intelligence on the de novo design of natural-product-inspired retinoid X receptor modulators. Commun. Chem. 2018, 1, 68. [Google Scholar] [CrossRef] [Green Version]
- Irwin, J.J.; Shoichet, B.K. ZINC− a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today: Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- O’Boyle, N.M. Towards a Universal SMILES representation-A standard method to generate canonical SMILES based on the InChI. J. Cheminform. 2012, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Graves, A. Generating sequences with recurrent neural networks. arXiv 2013, arXiv:1308.0850. [Google Scholar]
- Chollet, F. Deep Learning with Python; Simon and Schuster: Shelter Island, NY, USA, 2021. [Google Scholar]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M. TensorFlow: A System for Large-Scale Machine Learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI 16), Savannah, GA, USA, 2–4 November 2016; pp. 265–283. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org (accessed on 9 September 2018).
- Silicos-it. Available online: https://github.com/bgruening/galaxytools/tree/master/chemicaltoolbox/silicos-it (accessed on 12 May 2020).
- Maaten, L.v.d.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Prykhodko, O.; Johansson, S.V.; Kotsias, P.-C.; Arús-Pous, J.; Bjerrum, E.J.; Engkvist, O.; Chen, H. A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminform. 2019, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- DeSimone, R.; Currie, K.; Mitchell, S.; Darrow, J.; Pippin, D. Privileged structures: Applications in drug discovery. Comb. Chem. High Throughput Screen. 2004, 7, 473–493. [Google Scholar] [CrossRef]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Müller, A.T.; Huisman, B.J.; Fuchs, J.A.; Schneider, P.; Schneider, G. Generative recurrent networks for de novo drug design. Mol. Inform. 2018, 37, 1700111. [Google Scholar] [CrossRef] [Green Version]
- Merk, D.; Friedrich, L.; Grisoni, F.; Schneider, G. De novo design of bioactive small molecules by artificial intelligence. Mol. Inform. 2018, 37, 1700153. [Google Scholar] [CrossRef] [Green Version]
- Iwamura, H.; Suzuki, H.; Ueda, Y.; Kaya, T.; Inaba, T. In vitro and in vivo pharmacological characterization of JTE-907, a novel selective ligand for cannabinoid CB2 receptor. J. Pharmacol. Exp. Ther. 2001, 296, 420–425. [Google Scholar]
- Ueda, Y.; Miyagawa, N.; Matsui, T.; Kaya, T.; Iwamura, H. Involvement of cannabinoid CB2 receptor-mediated response and efficacy of cannabinoid CB2 receptor inverse agonist, JTE-907, in cutaneous inflammation in mice. Eur. J. Pharmacol. 2005, 520, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Myint, K.-Z.; Tong, Q.; Feng, R.; Cao, H.; Almehizia, A.A.; Alqarni, M.H.; Wang, L.; Bartlow, P.; Gao, Y. Lead discovery, chemistry optimization, and biological evaluation studies of novel biamide derivatives as CB2 receptor inverse agonists and osteoclast inhibitors. J. Med. Chem. 2012, 55, 9973–9987. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.; Griffin, G.; Fernando, S.; Li, X.; Hill, A.; Makriyannis, A. AM630, a competitive cannabinoid receptor antagonist. Life Sci. 1995, 56, 1949–1955. [Google Scholar] [CrossRef]
- Ross, R.A.; Brockie, H.C.; Stevenson, L.A.; Murphy, V.L.; Templeton, F.; Makriyannis, A.; Pertwee, R.G. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656 and AM630. Br. J. Pharmacol. 1999, 126, 665. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Wang, L.; Feng, R.; Almehizia, A.A.; Tong, Q.; Myint, K.-Z.; Ouyang, Q.; Alqarni, M.H.; Wang, L.; Xie, X.-Q. Novel triaryl sulfonamide derivatives as selective cannabinoid receptor 2 inverse agonists and osteoclast inhibitors: Discovery, optimization, and biological evaluation. J. Med. Chem. 2013, 56, 2045–2058. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Zhang, Y.; Hua, Y.; Wang, Y.; Zhu, L.; Zhao, J.; Yang, Y.; Chen, X.; Lu, S.; Lu, T. Investigation of Machine Intelligence in Compound Cell Activity Classification. Mol. Pharm. 2019, 16, 4472–4484. [Google Scholar] [CrossRef]
- Minerali, E.; Foil, D.H.; Zorn, K.M.; Lane, T.R.; Ekins, S. Comparing Machine Learning Algorithms for Predicting Drug-Induced Liver Injury (DILI). Mol. Pharm. 2020, 17, 2628–2637. [Google Scholar] [CrossRef]
- Zorn, K.M.; Foil, D.H.; Lane, T.R.; Russo, D.P.; Hillwalker, W.; Feifarek, D.J.; Jones, F.; Klaren, W.D.; Brinkman, A.M.; Ekins, S. Machine Learning Models for Estrogen Receptor Bioactivity and Endocrine Disruption Prediction. Environ. Sci. Technol. 2020, 54, 12202–12213. [Google Scholar] [CrossRef]
- Russo, D.P.; Zorn, K.M.; Clark, A.M.; Zhu, H.; Ekins, S. Comparing multiple machine learning algorithms and metrics for estrogen receptor binding prediction. Mol. Pharm. 2018, 15, 4361–4370. [Google Scholar] [CrossRef]
- Burger, B.; Maffettone, P.M.; Gusev, V.V.; Aitchison, C.M.; Bai, Y.; Wang, X.; Li, X.; Alston, B.M.; Li, B.; Clowes, R. A mobile robotic chemist. Nature 2020, 583, 237–241. [Google Scholar] [CrossRef]
- Maryasin, B.; Marquetand, P.; Maulide, N. Machine learning for organic synthesis: Are robots replacing chemists? Angew. Chem. Int. Ed. 2018, 57, 6978–6980. [Google Scholar] [CrossRef]
- Coley, C.W.; Thomas, D.A.; Lummiss, J.A.; Jaworski, J.N.; Breen, C.P.; Schultz, V.; Hart, T.; Fishman, J.S.; Rogers, L.; Gao, H. A robotic platform for flow synthesis of organic compounds informed by AI planning. Science 2019, 365, eaax1566. [Google Scholar] [CrossRef]
- Steiner, S.; Wolf, J.; Glatzel, S.; Andreou, A.; Granda, J.M.; Keenan, G.; Hinkley, T.; Aragon-Camarasa, G.; Kitson, P.J.; Angelone, D. Organic synthesis in a modular robotic system driven by a chemical programming language. Science 2019, 363, eaav2211. [Google Scholar] [CrossRef] [Green Version]
- Granda, J.M.; Donina, L.; Dragone, V.; Long, D.-L.; Cronin, L. Controlling an organic synthesis robot with machine learning to search for new reactivity. Nature 2018, 559, 377–381. [Google Scholar] [CrossRef]

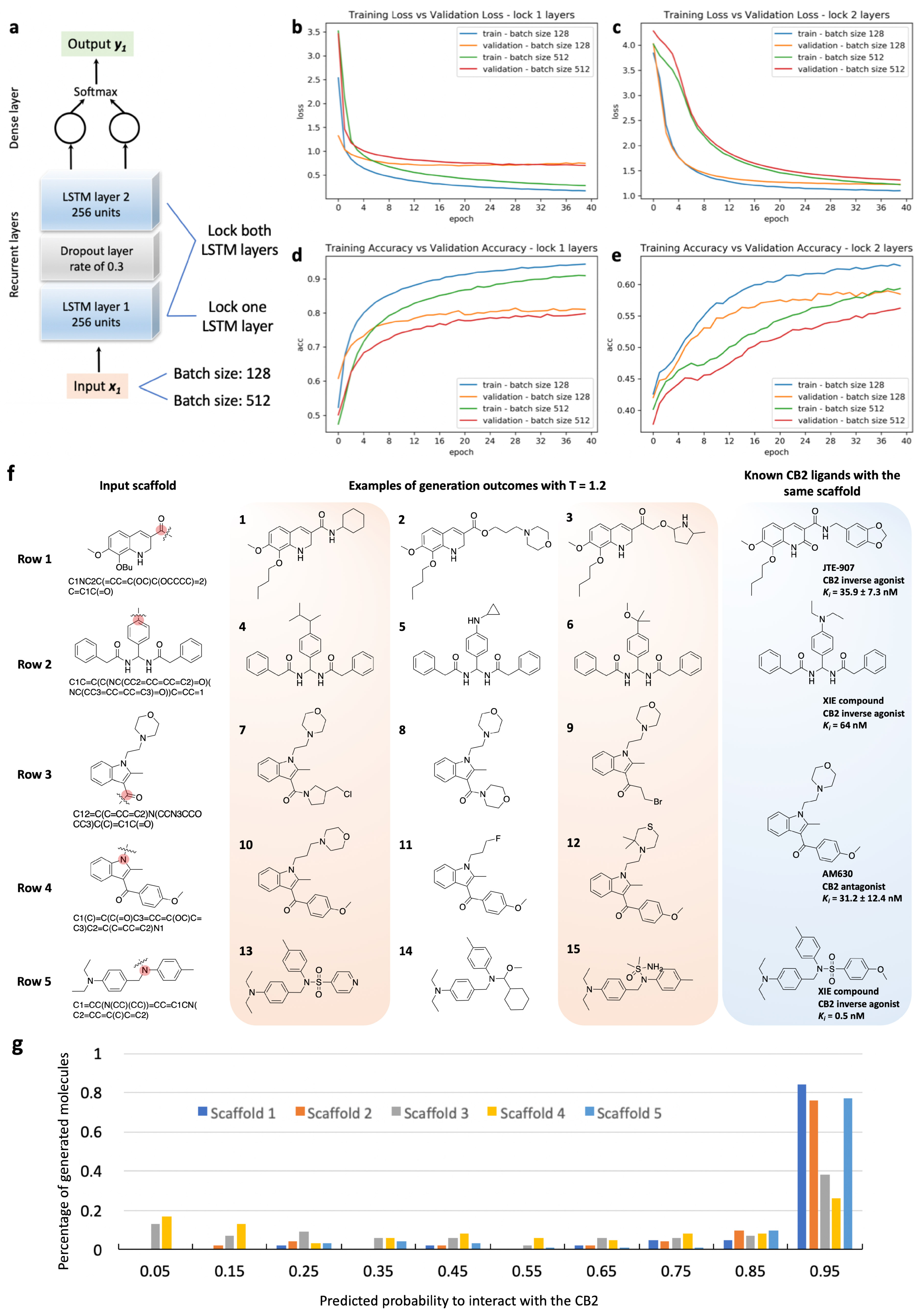

{kind=link}
{kind=link}
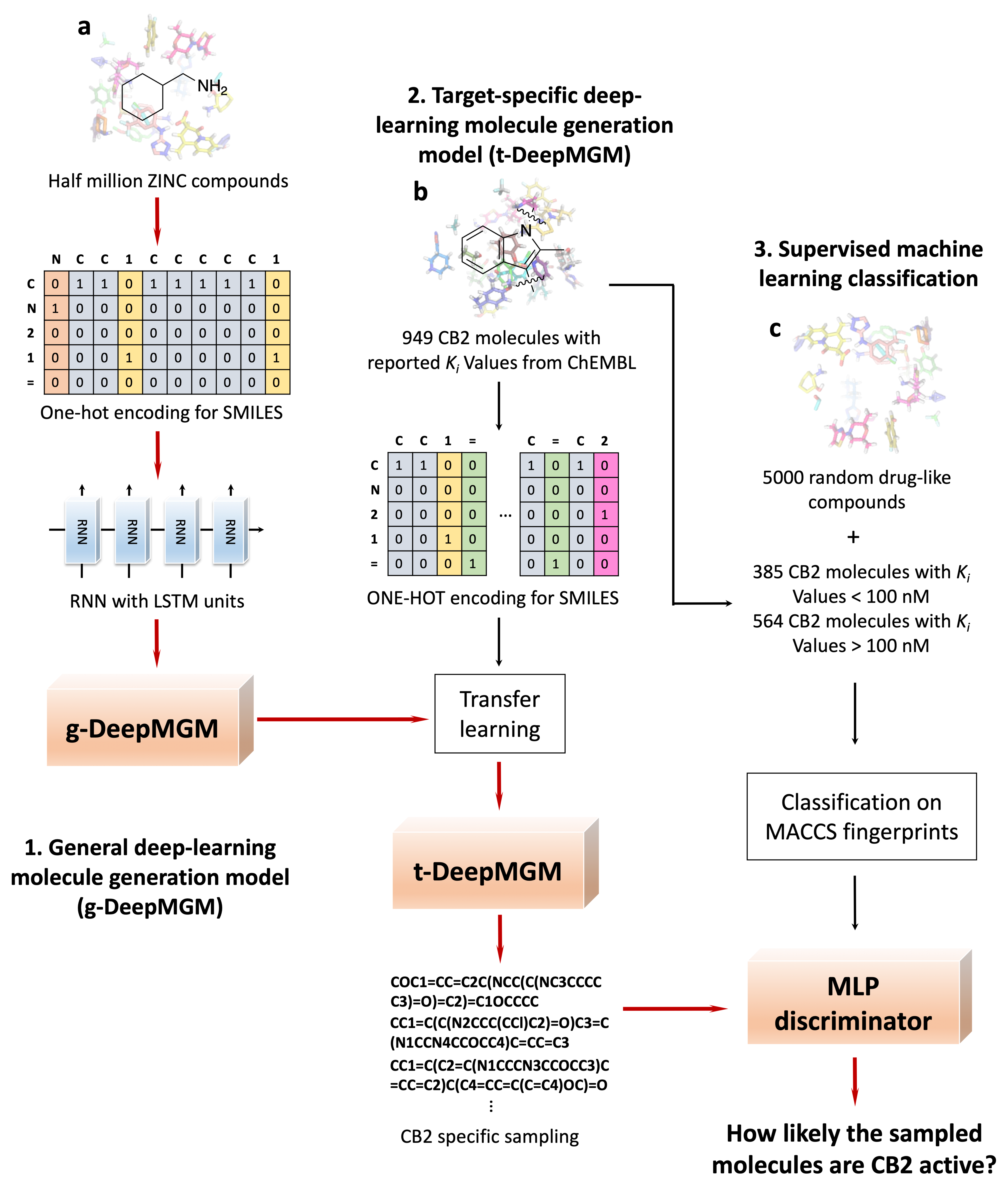
{kind=link}
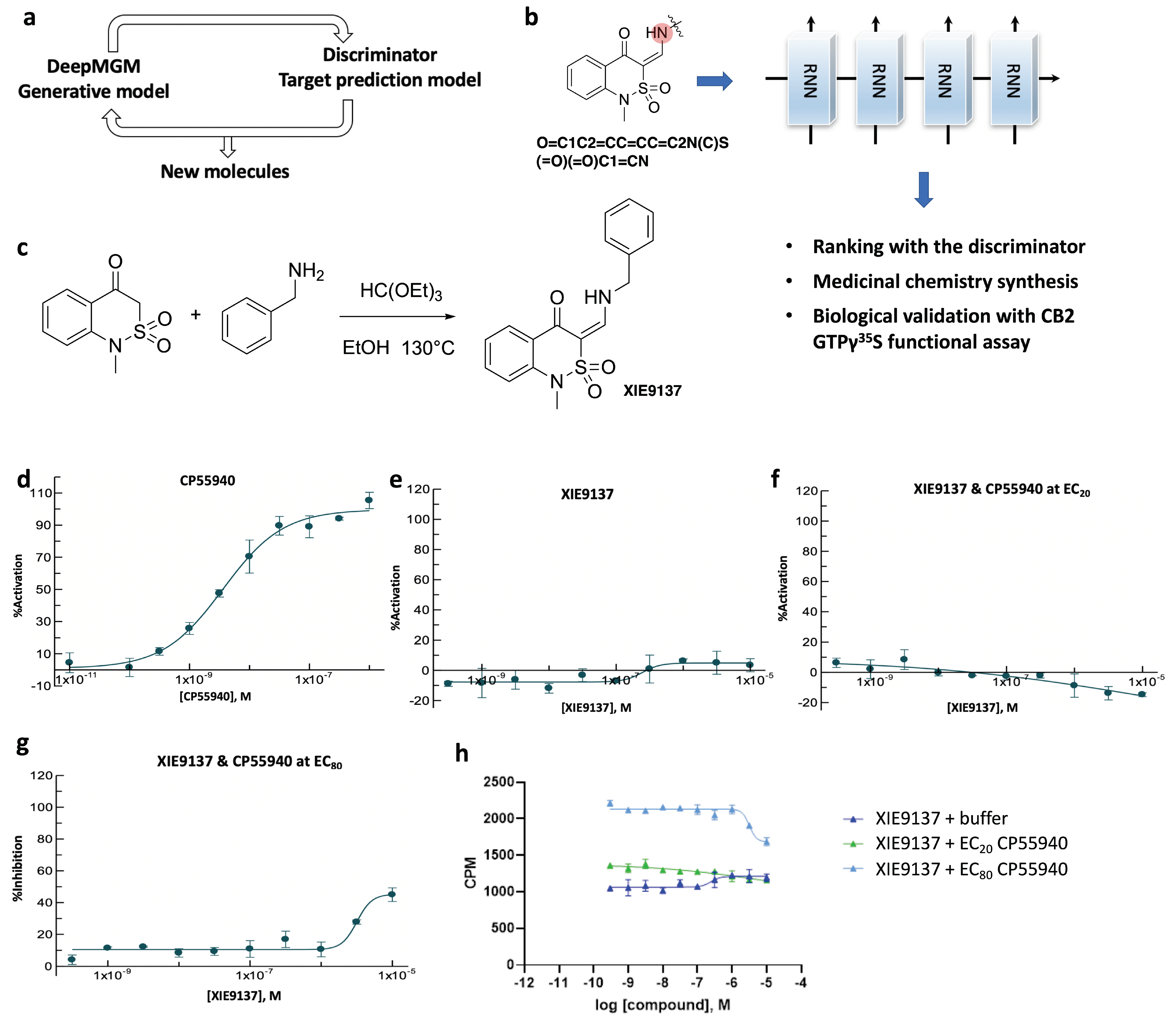
{kind=link}
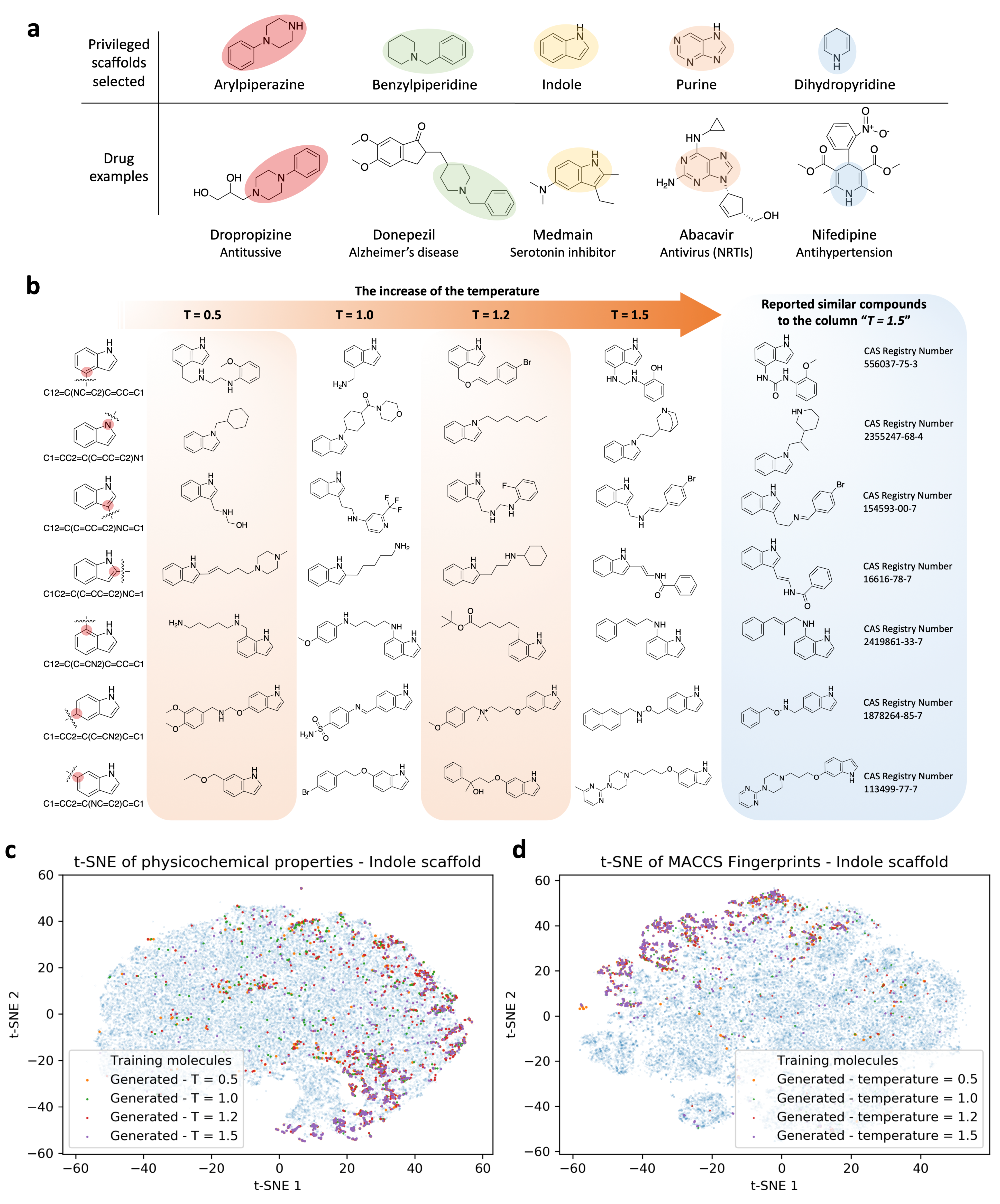
{kind=link}
{kind=link}
{kind=link}
| Epoch | Temperature | Validity | Uniqueness | Novelty |
|---|---|---|---|---|
| 10 (training loss: 0.6309) | 0.5 | 0.867 | 0.092 | 0.271 |
| 1.0 | 0.517 | 0.303 | 0.528 | |
| 1.2 | 0.409 | 0.349 | 0.627 | |
| 1.5 | 0.313 | 0.357 | 0.719 | |
| 20 (training loss: 0.5966) | 0.5 | 0.891 | 0.115 | 0.265 |
| 1.0 | 0.552 | 0.469 | 0.518 | |
| 1.2 | 0.456 | 0.522 | 0.597 | |
| 1.5 | 0.342 | 0.508 | 0.706 | |
| 40 (training loss: 0.5700) | 0.5 | 0.905 | 0.086 | 0.202 |
| 1.0 | 0.672 | 0.350 | 0.409 | |
| 1.2 | 0.553 | 0.433 | 0.527 | |
| 1.5 | 0.399 | 0.476 | 0.653 | |
| 100 (training loss: 0.5444) | 0.5 | 0.789 | 0.069 | 0.234 |
| 1.0 | 0.598 | 0.311 | 0.419 | |
| 1.2 | 0.505 | 0.394 | 0.523 | |
| 1.5 | 0.381 | 0.455 | 0.629 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bian, Y.; Xie, X.-Q. Artificial Intelligent Deep Learning Molecular Generative Modeling of Scaffold-Focused and Cannabinoid CB2 Target-Specific Small-Molecule Sublibraries. Cells 2022, 11, 915. https://doi.org/10.3390/cells11050915
Bian Y, Xie X-Q. Artificial Intelligent Deep Learning Molecular Generative Modeling of Scaffold-Focused and Cannabinoid CB2 Target-Specific Small-Molecule Sublibraries. Cells. 2022; 11(5):915. https://doi.org/10.3390/cells11050915
Chicago/Turabian StyleBian, Yuemin, and Xiang-Qun Xie. 2022. "Artificial Intelligent Deep Learning Molecular Generative Modeling of Scaffold-Focused and Cannabinoid CB2 Target-Specific Small-Molecule Sublibraries" Cells 11, no. 5: 915. https://doi.org/10.3390/cells11050915
APA StyleBian, Y., & Xie, X.-Q. (2022). Artificial Intelligent Deep Learning Molecular Generative Modeling of Scaffold-Focused and Cannabinoid CB2 Target-Specific Small-Molecule Sublibraries. Cells, 11(5), 915. https://doi.org/10.3390/cells11050915