
SWI/SNF Antagonism of PRC2 Mediates Estrogen-Induced Progesterone Receptor Expression

 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Cell Lines
2.2. Transfection and Treatment of 12Z Cells with siRNA
2.3. Western Blotting
2.4. RNA Isolation and qRT-PCR
2.5. Construction and Sequencing of Directional mRNA-seq Libraries
2.6. RNA-seq Analysis
2.7. Analysis of PGR Chromatin from Roadmap
2.8. Bioinformatics and Statistics
2.9. Cell Growth Assay
3. Results
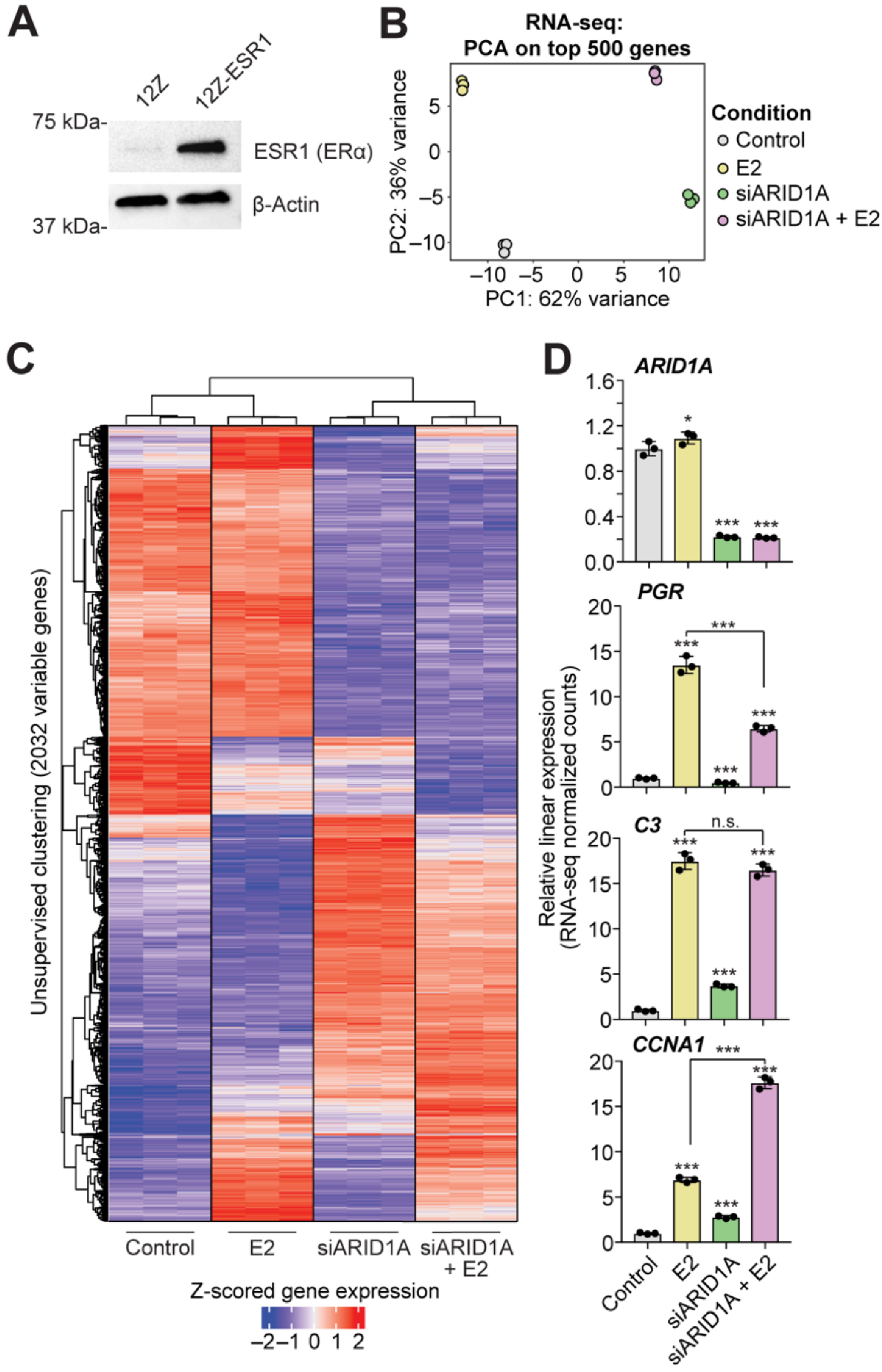
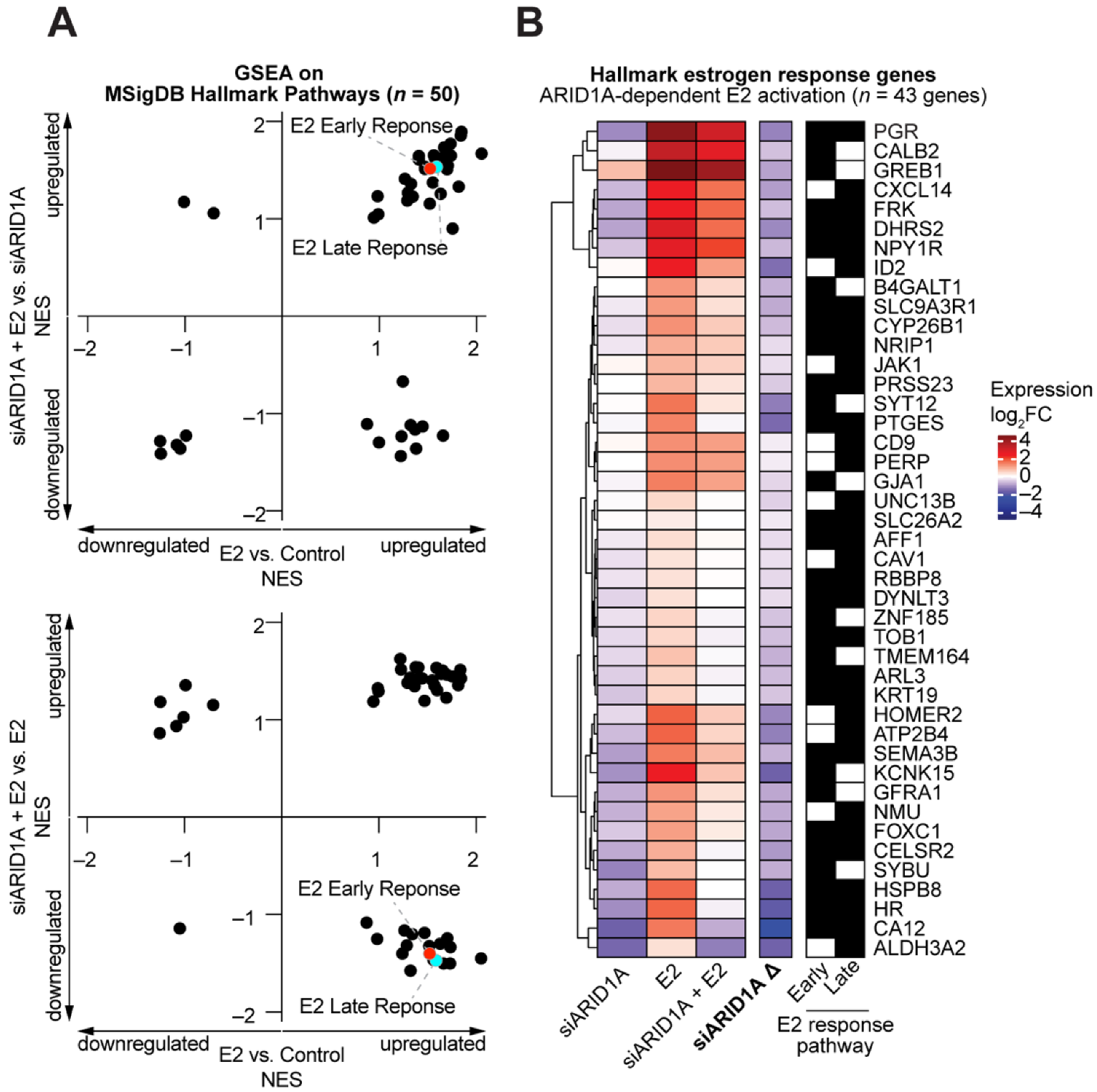
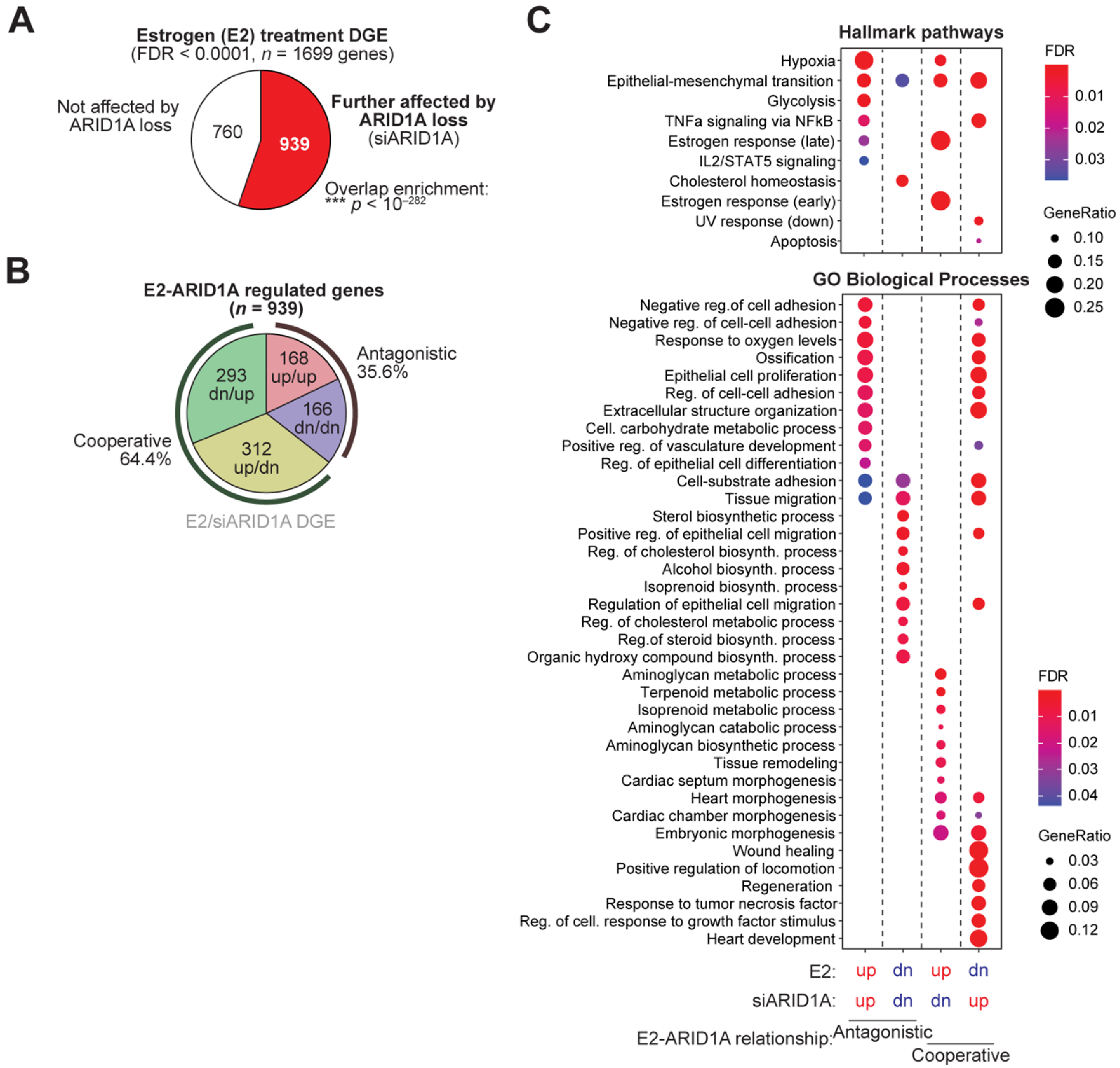
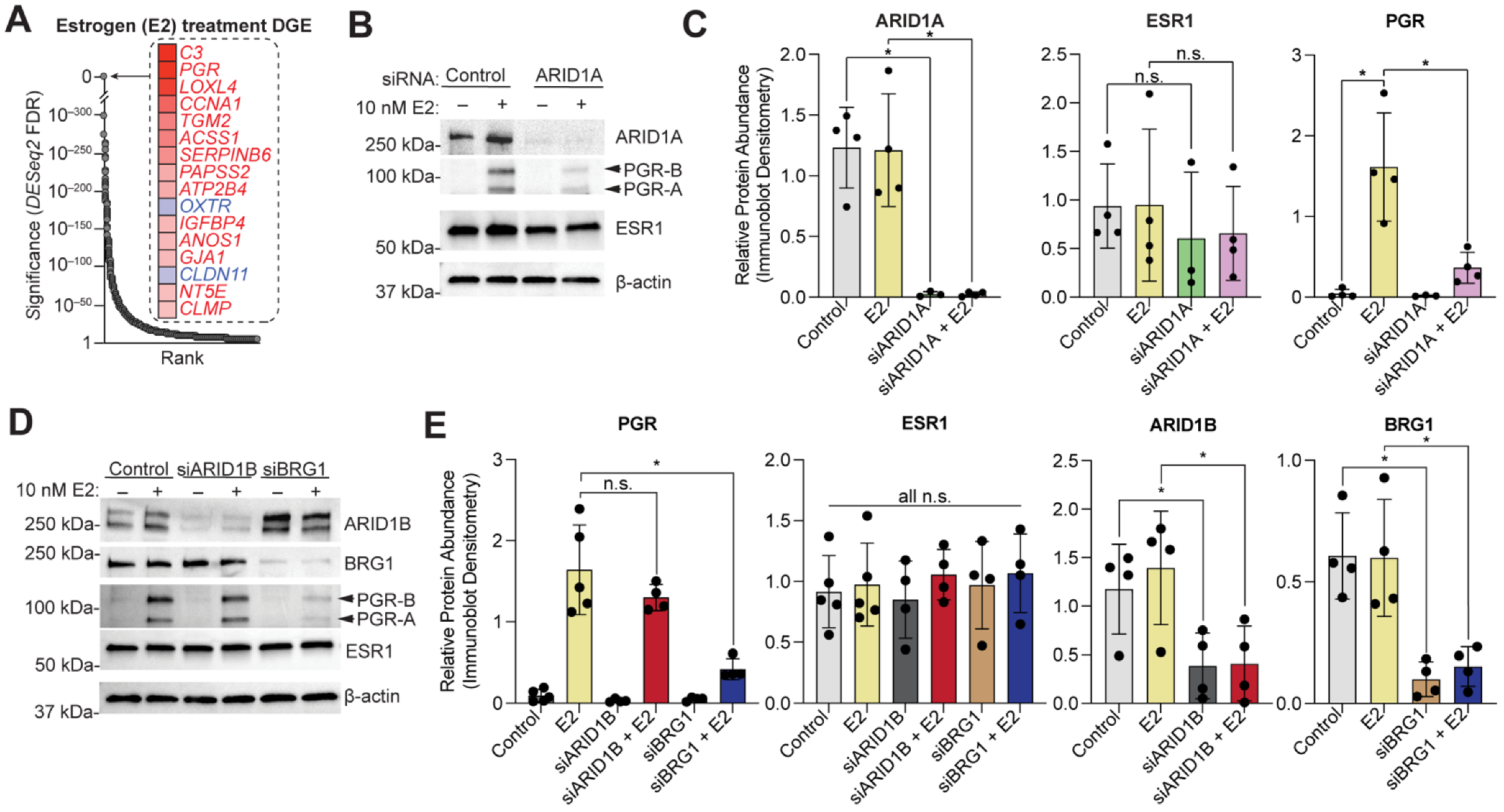
3.1. ARID1A and Estrogen Signaling Cooperatively Regulate Biological Processes in the Endometrial Epithelium
3.2. Cell Identity of the Endometrial Epithelium Depends on ARID1A Expression and Regulation by Estrogen
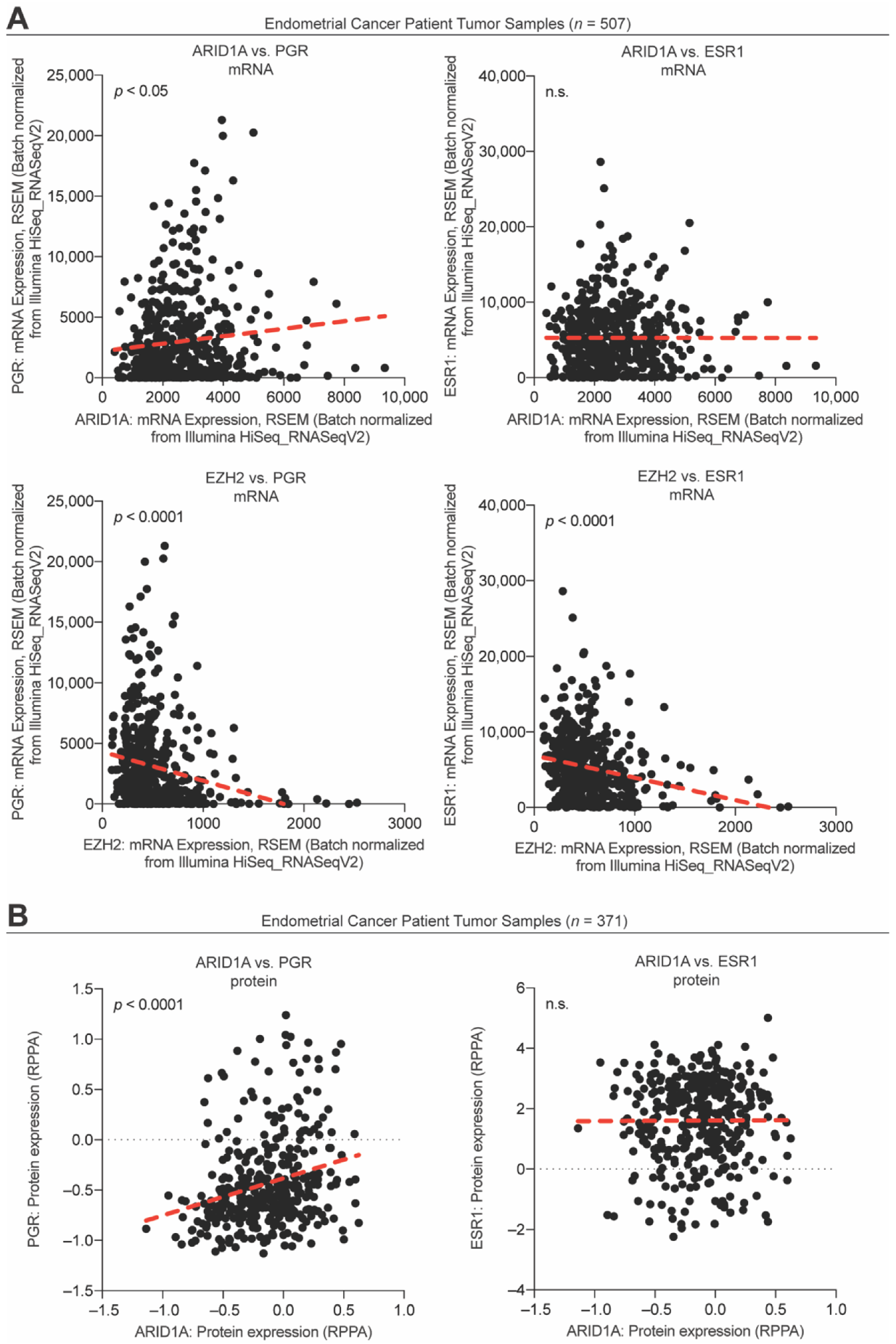
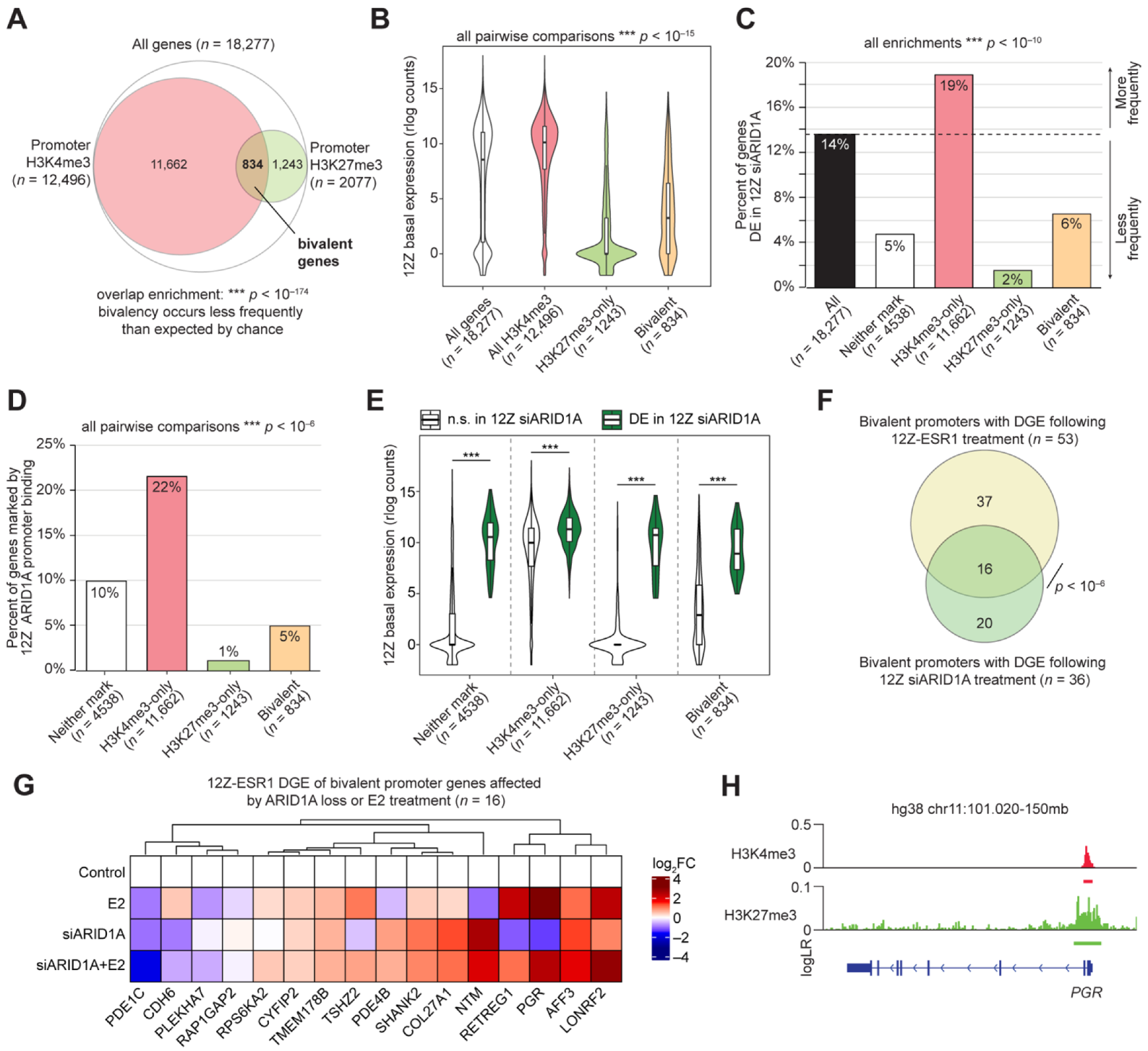
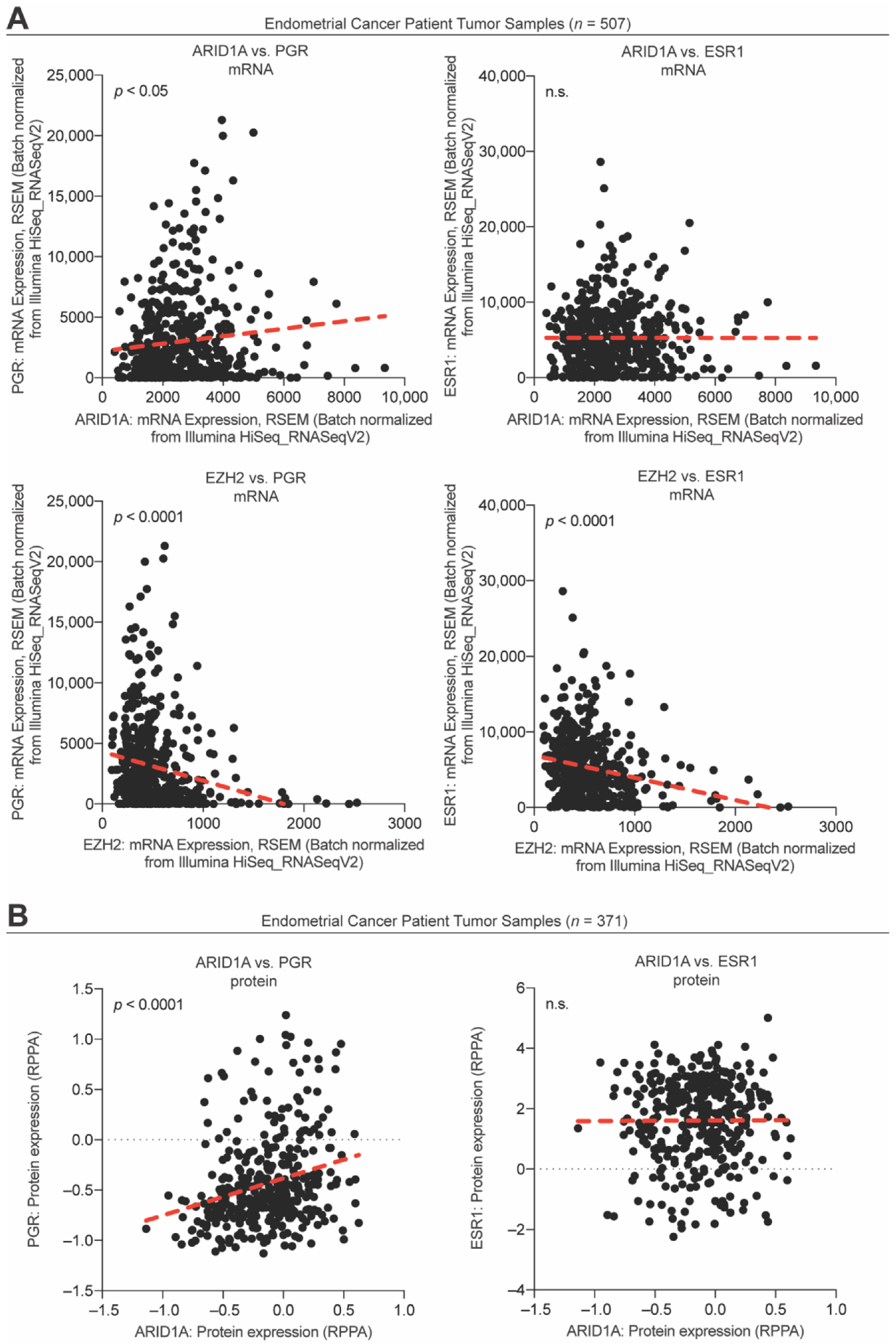
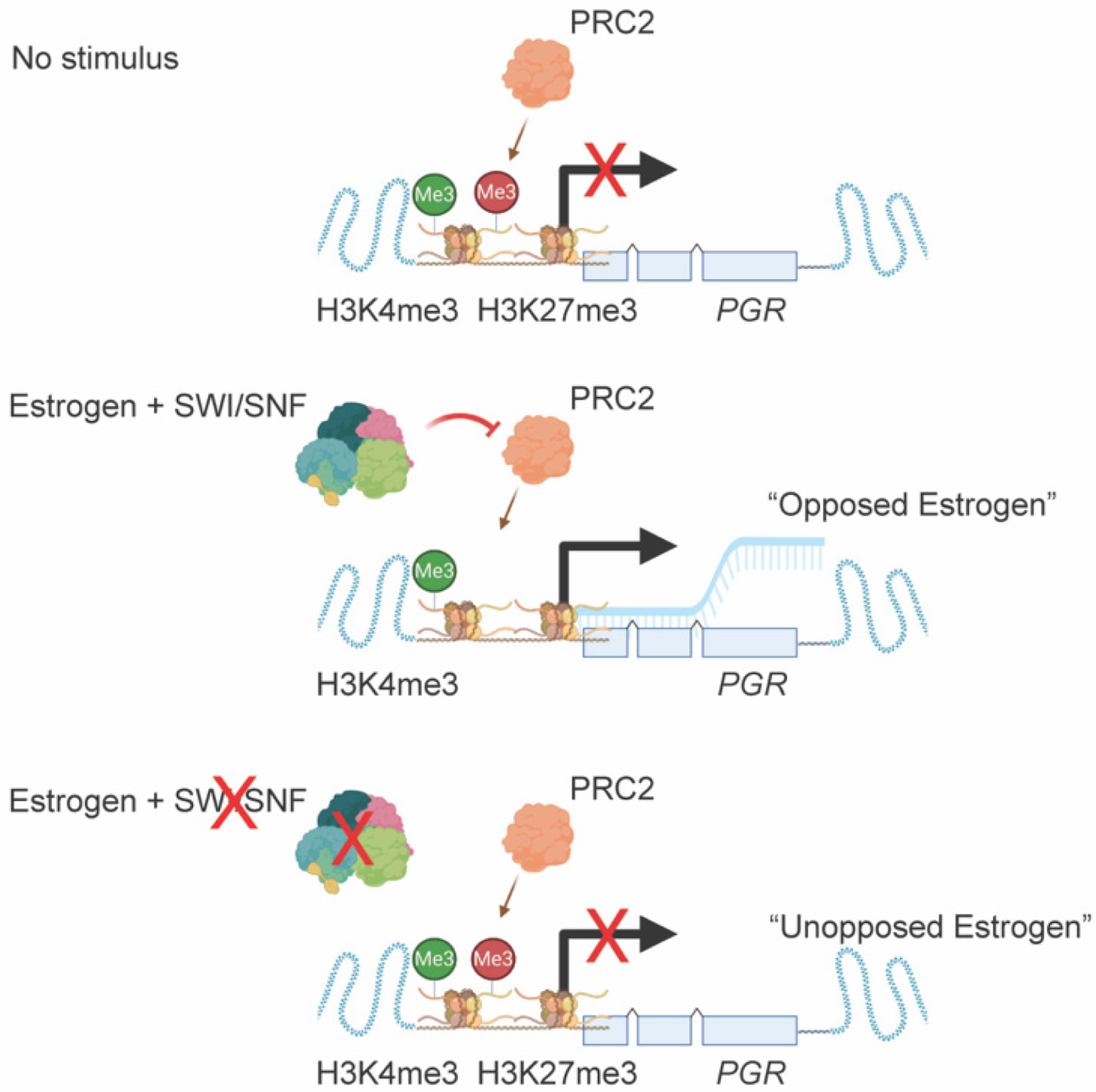
3.3. ARID1A Opposes PRC2-Mediated Repression of Estrogen-Mediated Progesterone Receptor Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Kaaks, R.; Lukanova, A.; Kurzer, M.S. Obesity, endogenous hormones, and endometrial cancer risk: A synthetic review. Cancer Epidemiol. Biomarkers Prev. 2002, 11, 1531–1543. [Google Scholar]
- Bianchini, F.; Kaaks, R.; Vainio, H. Overweight, obesity, and cancer risk. Lancet Oncol. 2002, 3, 565–574. [Google Scholar] [CrossRef]
- Mihm, M.; Gangooly, S.; Muttukrishna, S. The normal menstrual cycle in women. Anim. Reprod. Sci. 2011, 124, 229–236. [Google Scholar] [CrossRef]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef]
- Urick, M.E.; Bell, D.W. Clinical actionability of molecular targets in endometrial cancer. Nat. Rev. Cancer 2019, 19, 510–521. [Google Scholar] [CrossRef]
- Marquardt, R.M.; Kim, T.H.; Shin, J.H.; Jeong, J.W. Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis? Int. J. Mol. Sci. 2019, 20, 3822. [Google Scholar] [CrossRef] [Green Version]
- Moutsatsou, P.; Sekeris, C.E. Estrogen and progesterone receptors in the endometrium. Ann. N. Y. Acad. Sci. 1997, 816, 99–115. [Google Scholar] [CrossRef]
- Al-Sabbagh, M.; Lam, E.W.; Brosens, J.J. Mechanisms of endometrial progesterone resistance. Mol. Cell. Endocrinol. 2012, 358, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Siiteri, P.K. Steroid hormones and endometrial cancer. Cancer Res. 1978, 38, 4360–4366. [Google Scholar] [PubMed]
- Kim, J.J.; Chapman-Davis, E. Role of progesterone in endometrial cancer. Semin. Reprod. Med. 2010, 28, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, C.E.; Young, P.C.; Stehman, F.B.; Sutton, G.P.; Alford, W.M. Steroid receptors and clinical outcome in patients with adenocarcinoma of the endometrium. Am. J. Obstet. Gynecol. 1988, 158, 796–807. [Google Scholar] [CrossRef]
- Nunobiki, O.; Taniguchi, E.; Ishii, A.; Tang, W.; Utsunomiya, H.; Nakamura, Y.; Mori, I.; Kakudo, K. Significance of hormone receptor status and tumor vessels in normal, hyperplastic and neoplastic endometrium. Pathol. Int. 2003, 53, 846–852. [Google Scholar] [CrossRef]
- Uchikawa, J.; Shiozawa, T.; Shih, H.C.; Miyamoto, T.; Feng, Y.Z.; Kashima, H.; Oka, K.; Konishi, I. Expression of steroid receptor coactivators and corepressors in human endometrial hyperplasia and carcinoma with relevance to steroid receptors and Ki-67 expression. Cancer 2003, 98, 2207–2213. [Google Scholar] [CrossRef]
- Flores, V.A.; Vanhie, A.; Dang, T.; Taylor, H.S. Progesterone Receptor Status Predicts Response to Progestin Therapy in Endometriosis. J. Clin. Endocrinol. Metab. 2018, 103, 4561–4568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, H.M.; Berg, A.; Wik, E.; Birkeland, E.; Krakstad, C.; Kusonmano, K.; Petersen, K.; Kalland, K.H.; Oyan, A.M.; Akslen, L.A.; et al. ARID1A loss is prevalent in endometrial hyperplasia with atypia and low-grade endometrioid carcinomas. Mod. Pathol. 2013, 26, 428–434. [Google Scholar] [CrossRef] [Green Version]
- Yen, T.T.; Miyamoto, T.; Asaka, S.; Chui, M.H.; Wang, Y.; Lin, S.F.; Stone, R.L.; Fader, A.N.; Asaka, R.; Kashima, H.; et al. Loss of ARID1A expression in endometrial samplings is associated with the risk of endometrial carcinoma. Gynecol. Oncol. 2018, 150, 426–431. [Google Scholar] [CrossRef]
- Mao, T.L.; Ardighieri, L.; Ayhan, A.; Kuo, K.T.; Wu, C.H.; Wang, T.L.; Shih, I.M. Loss of ARID1A expression correlates with stages of tumor progression in uterine endometrioid carcinoma. Am. J. Surg. Pathol. 2013, 37, 1342–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Guan, B.; Mao, T.L.; Panuganti, P.K.; Kuhn, E.; Kurman, R.J.; Maeda, D.; Chen, E.; Jeng, Y.M.; Wang, T.L.; Shih, I.M. Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. Am. J. Surg. Pathol. 2011, 35, 625–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegand, K.C.; Lee, A.F.; Al-Agha, O.M.; Chow, C.; Kalloger, S.E.; Scott, D.W.; Steidl, C.; Wiseman, S.M.; Gascoyne, R.D.; Gilks, B.; et al. Loss of BAF250a (ARID1A) is frequent in high-grade endometrial carcinomas. J. Pathol. 2011, 224, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Becker, P.B.; Workman, J.L. Nucleosome remodeling and epigenetics. Cold Spring Harb. Perspect. Biol. 2013, 5, a017905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.R.; Holladay, J.; Chandler, R.L. A mouse model of endometriosis mimicking the natural spread of invasive endometrium. Hum. Reprod. 2020, 35, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Reske, J.J.; Holladay, J.; Wilber, G.E.; Rhodes, M.; Koeman, J.; Adams, M.; Johnson, B.; Su, R.W.; Joshi, N.R.; et al. ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat. Commun. 2019, 10, 3554. [Google Scholar] [CrossRef]
- Wilson, M.R.; Reske, J.J.; Holladay, J.; Neupane, S.; Ngo, J.; Cuthrell, N.; Wegener, M.; Rhodes, M.; Adams, M.; Sheridan, R.; et al. ARID1A Mutations Promote P300-Dependent Endometrial Invasion through Super-Enhancer Hyperacetylation. Cell Rep. 2020, 33, 108366. [Google Scholar] [CrossRef] [PubMed]
- Reske, J.J.; Wilson, M.R.; Holladay, J.; Siwicki, R.A.; Skalski, H.; Harkins, S.; Adams, M.; Risinger, J.I.; Hostetter, G.; Lin, K.; et al. Co-existing TP53 and ARID1A mutations promote aggressive endometrial tumorigenesis. PLoS Genet. 2021, 17, e1009986. [Google Scholar] [CrossRef]
- Reske, J.J.; Wilson, M.R.; Holladay, J.; Wegener, M.; Adams, M.; Chandler, R.L. SWI/SNF inactivation in the endometrial epithelium leads to loss of epithelial integrity. Hum. Mol. Genet. 2020, 29, 3412–3430. [Google Scholar] [CrossRef]
- Voigt, P.; Tee, W.W.; Reinberg, D. A double take on bivalent promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef] [Green Version]
- Zeitvogel, A.; Baumann, R.; Starzinski-Powitz, A. Identification of an invasive, N-cadherin-expressing epithelial cell type in endometriosis using a new cell culture model. Am. J. Pathol. 2001, 159, 1839–1852. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. Babraham Institute. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 11 March 2022).
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Anders, S.; Kim, V.; Huber, W. RNA-Seq workflow: Gene-level exploratory analysis and differential expression. F1000Research 2015, 4, 1070. [Google Scholar] [CrossRef]
- Ignatiadis, N.; Klaus, B.; Zaugg, J.B.; Huber, W. Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods 2016, 13, 577–580. [Google Scholar] [CrossRef] [Green Version]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bioconductor Core Team; Bioconductor Package Maintainer. TxDb.Hsapiens.UCSC.hg38.knownGene: Annotation Package for TxDb Object(s), R Package Version 3.4.0; Bioconductor; 2016. Available online: https://bioconductor.org/packages/release/data/annotation/html/TxDb.Hsapiens.UCSC.hg38.knownGene.html (accessed on 11 March 2022).
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.; Huber, W.; Pages, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Larsson, J. eulerr: Area-Proportional Euler and Venn Diagrams with Ellipses. R package version 6.1.0. CRAN 2020. Available online: https://cran.r-project.org/package=eulerr (accessed on 11 March 2022).
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundstrom, S.A.; Komm, B.S.; Xu, Q.; Boundy, V.; Lyttle, C.R. The stimulation of uterine complement component C3 gene expression by antiestrogens. Endocrinology 1990, 126, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Hasty, L.A.; Lambris, J.D.; Lessey, B.A.; Pruksananonda, K.; Lyttle, C.R. Hormonal regulation of complement components and receptors throughout the menstrual cycle. Am. J. Obstet. Gynecol. 1994, 170, 168–175. [Google Scholar] [CrossRef]
- Ace, C.I.; Okulicz, W.C. Differential gene regulation by estrogen and progesterone in the primate endometrium. Mol. Cell. Endocrinol. 1995, 115, 95–103. [Google Scholar] [CrossRef]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Kadoch, C.; Crabtree, G.R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 2015, 1, e1500447. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, P.; Croce, S.; McCluggage, W.G. Loss of expression of SMARCA4 (BRG1), SMARCA2 (BRM) and SMARCB1 (INI1) in undifferentiated carcinoma of the endometrium is not uncommon and is not always associated with rhabdoid morphology. Histopathology 2017, 70, 359–366. [Google Scholar] [CrossRef]
- Kolin, D.L.; Quick, C.M.; Dong, F.; Fletcher, C.D.M.; Stewart, C.J.R.; Soma, A.; Hornick, J.L.; Nucci, M.R.; Howitt, B.E. SMARCA4-deficient Uterine Sarcoma and Undifferentiated Endometrial Carcinoma Are Distinct Clinicopathologic Entities. Am. J. Surg. Pathol. 2020, 44, 263–270. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.E.; Cho, Y.J.; Koellhoffer, E.C.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C.W. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, L.; Miller, E.L.; Ronan, J.L.; Ho, W.Q.; Jothi, R.; Crabtree, G.R. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat. Cell Biol. 2011, 13, 903–913. [Google Scholar] [CrossRef] [Green Version]
- Stanton, B.Z.; Hodges, C.; Calarco, J.P.; Braun, S.M.; Ku, W.L.; Kadoch, C.; Zhao, K.; Crabtree, G.R. Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat. Genet. 2017, 49, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef]
- Varambally, S.; Cao, Q.; Mani, R.S.; Shankar, S.; Wang, X.; Ateeq, B.; Laxman, B.; Cao, X.; Jing, X.; Ramnarayanan, K.; et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 2008, 322, 1695–1699. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagener, N.; Macher-Goeppinger, S.; Pritsch, M.; Husing, J.; Hoppe-Seyler, K.; Schirmacher, P.; Pfitzenmaier, J.; Haferkamp, A.; Hoppe-Seyler, F.; Hohenfellner, M. Enhancer of zeste homolog 2 (EZH2) expression is an independent prognostic factor in renal cell carcinoma. BMC Cancer 2010, 10, 524. [Google Scholar] [CrossRef] [Green Version]
- Takawa, M.; Masuda, K.; Kunizaki, M.; Daigo, Y.; Takagi, K.; Iwai, Y.; Cho, H.S.; Toyokawa, G.; Yamane, Y.; Maejima, K.; et al. Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci. 2011, 102, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cai, Q.; Godwin, A.K.; Zhang, R. Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells. Mol. Cancer Res. 2010, 8, 1610–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadoch, C.; Williams, R.T.; Calarco, J.P.; Miller, E.L.; Weber, C.M.; Braun, S.M.; Pulice, J.L.; Chory, E.J.; Crabtree, G.R. Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet. 2017, 49, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Kennison, J.A.; Tamkun, J.W. Dosage-dependent modifiers of polycomb and antennapedia mutations in Drosophila. Proc. Natl. Acad. Sci. USA 1988, 85, 8136–8140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges, C.; Kirkland, J.G.; Crabtree, G.R. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih, I.M.; Conejo-Garcia, J.R.; et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.; Zhang, Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 2004, 14, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Ruthenburg, A.J.; Allis, C.D.; Wysocka, J. Methylation of lysine 4 on histone H3: Intricacy of writing and reading a single epigenetic mark. Mol. Cell 2007, 25, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Vastenhouw, N.L.; Schier, A.F. Bivalent histone modifications in early embryogenesis. Curr. Opin. Cell Biol. 2012, 24, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Cinghu, S.; Oldfield, A.J.; Yang, P.; Jothi, R. Decoding the function of bivalent chromatin in development and cancer. Genome Res. 2021, 31, 2170–2184. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Porter Scott, M.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knutson, S.K.; Kawano, S.; Minoshima, Y.; Warholic, N.M.; Huang, K.C.; Xiao, Y.; Kadowaki, T.; Uesugi, M.; Kuznetsov, G.; Kumar, N.; et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 2014, 13, 842–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierre, R.S.; Kadoch, C. Mammalian SWI/SNF complexes in cancer: Emerging therapeutic opportunities. Curr. Opin. Genet. Dev. 2017, 42, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, S.; Rao, S.V.; Sutton, J.; Cheeseman, D.; Dunn, S.; Papachristou, E.K.; Prada, J.G.; Couturier, D.L.; Kumar, S.; Kishore, K.; et al. ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat. Genet. 2020, 52, 187–197. [Google Scholar] [CrossRef]
- Kurita, T.; Young, P.; Brody, J.R.; Lydon, J.P.; O’Malley, B.W.; Cunha, G.R. Stromal progesterone receptors mediate the inhibitory effects of progesterone on estrogen-induced uterine epithelial cell deoxyribonucleic acid synthesis. Endocrinology 1998, 139, 4708–4713. [Google Scholar] [CrossRef]
- Li, Q.; Kannan, A.; DeMayo, F.J.; Lydon, J.P.; Cooke, P.S.; Yamagishi, H.; Srivastava, D.; Bagchi, M.K.; Bagchi, I.C. The antiproliferative action of progesterone in uterine epithelium is mediated by Hand2. Science 2011, 331, 912–916. [Google Scholar] [CrossRef] [Green Version]
- Kurita, T.; Wang, Y.Z.; Donjacour, A.A.; Zhao, C.; Lydon, J.P.; O’Malley, B.W.; Isaacs, J.T.; Dahiya, R.; Cunha, G.R. Paracrine regulation of apoptosis by steroid hormones in the male and female reproductive system. Cell Death Differ. 2001, 8, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Pierro, E.; Minici, F.; Alesiani, O.; Miceli, F.; Proto, C.; Screpanti, I.; Mancuso, S.; Lanzone, A. Stromal-epithelial interactions modulate estrogen responsiveness in normal human endometrium. Biol. Reprod. 2001, 64, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Blauer, M.; Heinonen, P.K.; Martikainen, P.M.; Tomas, E.; Ylikomi, T. A novel organotypic culture model for normal human endometrium: Regulation of epithelial cell proliferation by estradiol and medroxyprogesterone acetate. Hum. Reprod. 2005, 20, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Kurita, T.; Lee, K.J.; Cooke, P.S.; Lydon, J.P.; Cunha, G.R. Paracrine regulation of epithelial progesterone receptor and lactoferrin by progesterone in the mouse uterus. Biol. Reprod. 2000, 62, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Franco, H.L.; Rubel, C.A.; Large, M.J.; Wetendorf, M.; Fernandez-Valdivia, R.; Jeong, J.W.; Spencer, T.E.; Behringer, R.R.; Lydon, J.P.; Demayo, F.J. Epithelial progesterone receptor exhibits pleiotropic roles in uterine development and function. FASEB J. 2012, 26, 1218–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Strawn, E.; Basir, Z.; Halverson, G.; Guo, S.W. Promoter hypermethylation of progesterone receptor isoform B (PR-B) in endometriosis. Epigenetics 2006, 1, 106–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha-Junior, C.V.; Da Broi, M.G.; Miranda-Furtado, C.L.; Navarro, P.A.; Ferriani, R.A.; Meola, J. Progesterone Receptor B (PGR-B) Is Partially Methylated in Eutopic Endometrium From Infertile Women With Endometriosis. Reprod. Sci. 2019, 26, 1568–1574. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Strawn, E.; Basir, Z.; Halverson, G.; Guo, S.W. Aberrant expression of deoxyribonucleic acid methyltransferases DNMT1, DNMT3A, and DNMT3B in women with endometriosis. Fertil. Steril. 2007, 87, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, A.; Haendler, B. The histone demethylase JARID1A regulates progesterone receptor expression. FEBS J. 2011, 278, 1458–1469. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.Y.; Smith, R.; Fitter, J.T.; Mitchell, C.; Pan, X.; Ilicic, M.; Maiti, K.; Zakar, T.; Madsen, G. Increased progesterone receptor A expression in labouring human myometrium is associated with decreased promoter occupancy by the histone demethylase JARID1A. Mol. Hum. Reprod. 2014, 20, 442–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, B.; Bouras, T.; Shi, W.; Vaillant, F.; Sheridan, J.M.; Fu, N.; Breslin, K.; Jiang, K.; Ritchie, M.E.; Young, M.; et al. Global changes in the mammary epigenome are induced by hormonal cues and coordinated by Ezh2. Cell Rep. 2013, 3, 411–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, B.; Liang, J.; Yang, X.; Wang, Y.; Zhao, Y.; Wu, H.; Sun, L.; Zhang, Y.; Chen, Y.; Li, R.; et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol. Cell. Biol. 2007, 27, 5105–5119. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, J.B.; Colon-Diaz, M.; Garcia, M.; Gutierrez, S.; Colon, M.; Seto, E.; Laboy, J.; Flores, I. Endometriosis is characterized by a distinct pattern of histone 3 and histone 4 lysine modifications. Reprod. Sci. 2014, 21, 305–318. [Google Scholar] [CrossRef] [Green Version]
- Colon-Caraballo, M.; Monteiro, J.B.; Flores, I. H3K27me3 is an Epigenetic Mark of Relevance in Endometriosis. Reprod. Sci. 2015, 22, 1134–1142. [Google Scholar] [CrossRef] [Green Version]
- Krill, L.; Deng, W.; Eskander, R.; Mutch, D.; Zweizig, S.; Hoang, B.; Ioffe, O.; Randall, L.; Lankes, H.; Miller, D.S.; et al. Overexpression of enhance of Zeste homolog 2 (EZH2) in endometrial carcinoma: An NRG Oncology/Gynecologic Oncology Group Study. Gynecol. Oncol. 2020, 156, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Roh, J.W.; Bandyopadhyay, S.; Chen, Z.; Munkarah, A.R.; Hussein, Y.; Alosh, B.; Jazaerly, T.; Hayek, K.; Semaan, A.; et al. Overexpression of enhancer of zeste homolog 2 (EZH2) and focal adhesion kinase (FAK) in high grade endometrial carcinoma. Gynecol. Oncol. 2013, 128, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Li, Q.; Tao, X.; Wang, J.; Hua, K.; Feng, W. Enhancer of zeste homolog 2 is involved in the proliferation of endometrial carcinoma. Oncol. Lett. 2014, 8, 2049–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oki, S.; Sone, K.; Oda, K.; Hamamoto, R.; Ikemura, M.; Maeda, D.; Takeuchi, M.; Tanikawa, M.; Mori-Uchino, M.; Nagasaka, K.; et al. Oncogenic histone methyltransferase EZH2: A novel prognostic marker with therapeutic potential in endometrial cancer. Oncotarget 2017, 8, 40402–40411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskander, R.N.; Ji, T.; Huynh, B.; Wardeh, R.; Randall, L.M.; Hoang, B. Inhibition of enhancer of zeste homolog 2 (EZH2) expression is associated with decreased tumor cell proliferation, migration, and invasion in endometrial cancer cell lines. Int. J. Gynecol. Cancer 2013, 23, 997–1005. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.W.; Choi, J.E.; Han, H.D.; Hu, W.; Matsuo, K.; Nishimura, M.; Lee, J.S.; Kwon, S.Y.; Cho, C.H.; Kim, J.; et al. Clinical and biological significance of EZH2 expression in endometrial cancer. Cancer Biol. Ther. 2020, 21, 147–156. [Google Scholar] [CrossRef]
- Wang, J.; Ai, Z.; Chen, J.; Teng, Y.; Zhu, J. Enhancer of zeste homolog 2 blockade by RNA interference is implicated with inhibited proliferation, invasion and promoted apoptosis in endometrial carcinoma. Oncol. Lett. 2018, 15, 9429–9435. [Google Scholar] [CrossRef] [Green Version]
- Nanjappa, M.K.; Mesa, A.M.; Medrano, T.I.; Jefferson, W.N.; DeMayo, F.J.; Williams, C.J.; Lydon, J.P.; Levin, E.R.; Cooke, P.S. The histone methyltransferase EZH2 is required for normal uterine development and function in micedagger. Biol. Reprod. 2019, 101, 306–317. [Google Scholar] [CrossRef]
- Soyal, S.M.; Mukherjee, A.; Lee, K.Y.; Li, J.; Li, H.; DeMayo, F.J.; Lydon, J.P. Cre-mediated recombination in cell lineages that express the progesterone receptor. Genesis 2005, 41, 58–66. [Google Scholar] [CrossRef]
- Mesa, A.M.; Mao, J.; Nanjappa, M.K.; Medrano, T.I.; Tevosian, S.; Yu, F.; Kinkade, J.; Lyu, Z.; Liu, Y.; Joshi, T.; et al. Mice lacking uterine enhancer of zeste homolog 2 have transcriptomic changes associated with uterine epithelial proliferation. Physiol. Genom. 2020, 52, 81–95. [Google Scholar] [CrossRef]
- Mesa, A.M.; Mao, J.; Medrano, T.I.; Bivens, N.J.; Jurkevich, A.; Tuteja, G.; Cooke, P.S.; Rosenfeld, C.S. Spatial transcriptomics analysis of uterine gene expression in enhancer of zeste homolog 2 conditional knockout micedagger. Biol. Reprod. 2021, 105, 1126–1139. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Jones, S.; Wang, T.L.; Shih, I.M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitner, K.; Tsibulak, I.; Wieser, V.; Knoll, K.; Reimer, D.; Marth, C.; Fiegl, H.; Zeimet, A.G. Clinical impact of EZH2 and its antagonist SMARCA4 in ovarian cancer. Sci. Rep. 2020, 10, 20412. [Google Scholar] [CrossRef]
- Wu, S.; Fatkhutdinov, N.; Fukumoto, T.; Bitler, B.G.; Park, P.H.; Kossenkov, A.V.; Trizzino, M.; Tang, H.Y.; Zhang, L.; Gardini, A.; et al. SWI/SNF catalytic subunits’ switch drives resistance to EZH2 inhibitors in ARID1A-mutated cells. Nat. Commun. 2018, 9, 4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnezis, A.N.; Cho, K.R.; Gilks, C.B.; Pearce, C.L.; Huntsman, D.G. The disparate origins of ovarian cancers: Pathogenesis and prevention strategies. Nat. Rev. Cancer 2017, 17, 65–74. [Google Scholar] [CrossRef]
- Heidemann, L.N.; Hartwell, D.; Heidemann, C.H.; Jochumsen, K.M. The relation between endometriosis and ovarian cancer—A review. Acta Obstet. Gynecol. Scand. 2014, 93, 20–31. [Google Scholar] [CrossRef]
- Brunty, S.; Clower, L.; Mitchell, B.; Fleshman, T.; Zgheib, N.B.; Santanam, N. Peritoneal Modulators of Endometriosis-Associated Ovarian Cancer. Front. Oncol. 2021, 11, 793297. [Google Scholar] [CrossRef]
- Fan, H.; Atiya, H.I.; Wang, Y.; Pisanic, T.R.; Wang, T.H.; Shih, I.M.; Foy, K.K.; Frisbie, L.; Buckanovich, R.J.; Chomiak, A.A.; et al. Epigenomic Reprogramming toward Mesenchymal-Epithelial Transition in Ovarian-Cancer-Associated Mesenchymal Stem Cells Drives Metastasis. Cell Rep. 2020, 33, 108473. [Google Scholar] [CrossRef]
- Han, Y.; Wei, Y.; Yao, J.; Chu, Y.Y.; Li, C.W.; Hsu, J.L.; Nie, L.; Hung, M.C. Inhibition of CDK2 reduces EZH2 phosphorylation and reactivates ERalpha expression in high-grade serous ovarian carcinoma. Am. J. Cancer Res. 2020, 10, 1194–1206. [Google Scholar] [PubMed]
- Wang, Y.; Chen, S.Y.; Colborne, S.; Lambert, G.; Shin, C.Y.; Santos, N.D.; Orlando, K.A.; Lang, J.D.; Hendricks, W.P.D.; Bally, M.B.; et al. Histone Deacetylase Inhibitors Synergize with Catalytic Inhibitors of EZH2 to Exhibit Antitumor Activity in Small Cell Carcinoma of the Ovary, Hypercalcemic Type. Mol. Cancer Ther. 2018, 17, 2767–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normalized Enrichment Score (NES) | |||
|---|---|---|---|
| Hallmark Pathway | E2 vs. Control | siARID1A + E2 vs. siARID1A | siARID1A + E2 vs. E2 |
| Adipogenesis | 1.73 * | 1.77 * | –1.50 * |
| Allograft Rejection | 1.00 * | –1.30* | 1.29 * |
| Androgen Response | 1.56 * | 1.65 * | –1.46 * |
| Angiogenesis | 1.70 * | 1.65 * | 1.23 |
| Apical Junction | 1.37 * | –1.17 | 1.34 * |
| Apical Surface | 1.45 * | –1.13 | 1.42 * |
| Apoptosis | 1.55 * | 1.38 * | 1.40 * |
| Bile Acid Metabolism | 1.52 * | 1.16 | –1.33 |
| Cholesterol Homeostasis | 1.34 * | 1.23 | –1.20 |
| Coagulation | 1.41 * | 1.61 * | 1.54 * |
| Complement | 1.59 * | 1.62 * | 1.54 * |
| DNA Repair | –1.05 | –1.36 * | –1.14 |
| E2F Targets | –1.25 | –1.28 | 0.86 |
| Epithelial Mesenchymal Transition | 1.38 * | –1.36 * | 1.54 * |
| Estrogen Response Early | 1.52 * | 1.52 * | –1.40 * |
| Estrogen Response Late | 1.59 * | 1.54 * | –1.47 * |
| Fatty Acid Metabolism | 2.05 * | 1.67 * | –1.45 * |
| G2M Checkpoint | –0.70 | 1.06 | 1.15 |
| Glycolysis | 1.57 * | 1.62 * | 1.35 * |
| Hedgehog Signaling | –0.99 | –1.23 * | 1.35 * |
| Heme Metabolism | 1.70 * | 1.55 * | –1.24 |
| Hypoxia | 1.60 * | 1.57 * | 1.30 |
| Il2 Stat5 Signaling | 1.64 * | 1.62 * | 1.47 * |
| Il6 Jak Stat3 Signaling | 1.22 | –1.44 * | 1.63 * |
| Inflammatory Response | 1.23 * | –1.23 | 1.52 * |
| Interferon Alpha Response | 1.32 | –1.12 | 1.43 |
| Interferon Gamma Response | 1.29 | 1.27 * | 1.38 * |
| Kras Signaling Dn | 1.29 * | 1.19 | –1.32 |
| Kras Signaling Up | 1.82 * | 1.33 * | 1.36 * |
| Mitotic Spindle | 0.95 | 1.01 | 1.19 |
| MTORC1 Signaling | 1.63 * | 1.26 | –1.30 |
| Myc Targets V1 | –1.08 | –1.32 * | 0.94 |
| Myc Targets V2 | –1.25 * | –1.41 * | 1.19 * |
| Myogenesis | 1.26 | 1.41 * | –1.16 |
| Notch Signaling | 0.99 | 1.05 | 1.32 * |
| Oxidative Phosphorylation | 0.98 | 1.23 | –1.25 |
| P53 Pathway | 1.85 * | 1.90* | 1.43 * |
| Pancreas Beta Cells | 1.47 | 1.51* | 1.20 |
| Peroxisome | 1.74 * | 1.65* | –1.33 * |
| PI3K Akt MTOR Signaling | 1.33 | 1.36* | –1.58 * |
| Protein Secretion | 1.24 | –0.67 | –1.40 * |
| Oxygen Species Pathway | 0.87 | –1.11 | –1.08 |
| Spermatogenesis | 1.47 * | 1.53 * | –1.19 |
| TGF-Beta Signaling | 1.84 * | 1.86 * | 1.51 * |
| TNF-Alpha Signaling Via NFkB | 1.70 * | 1.51 * | 1.46 * |
| Unfolded Protein Response | 1.75 * | 0.90 | 1.45 * |
| UV Response Dn | 1.66 * | –1.23 * | 1.51 * |
| UV Response Up | 1.41 * | 1.65 * | 1.42 * |
| Wnt Beta Catenin Signaling | –1.01 | 1.17 | 1.03 |
| Xenobiotic Metabolism | 1.67 * | 1.73 * | –1.50 * |
| Genes with Bivalent Promoters Differentially Expressed Upon E2 Treatment of 12Z-ESR1 Cells (n = 53) | |||||
| AFF3 | CYFIP2 | LDHB | PDE4B | RAP1GAP2 | SVEP1 |
| APCDD1 | ERG | LONRF2 | PDE4D | RETREG1 | SYBU |
| ASIC2 | ESR1 | LPIN1 | PDGFD | RNF152 | TGFA |
| BMP6 | GFRA2 | MAST4 | PGR | RPS6KA2 | TMEM178B |
| C4orf19 | GUCY1A2 | MKX | PLCB1 | SHANK2 | TMOD1 |
| C4orf3 | HAND1 | NPY1R | PLEKHA7 | SLC47A1 | TPD52L1 |
| CACNB4 | ISM1 | NRXN3 | PRKAG2 | SLCO5A1 | TSHZ2 |
| CDH6 | ISOC1 | NTM | PRR15 | SNCAIP | WNT16 |
| COL27A1 | KIAA1217 | PDE1C | PTGER4 | SOX5 | |
| Genes withBivalent Promoters Differentially Expressed Upon ARID1A Knockdown in 12Z Cells (n = 36) | |||||
| AFF3 | COL27A1 | FAM131B | MOCS1 | PLAGL1 | RPS6KA2 |
| ANK3 | CYFIP2 | FMN1 | NR3C2 | PLEKHA7 | SHANK2 |
| C17orf51 | DEPDC1B | FOXQ1 | NTM | RAP1GAP2 | STOX2 |
| CDH6 | DLGAP4 | GJB2 | PDE1C | RETREG1 | TMEM178B |
| CELF2 | EIF4E3 | LONRF2 | PDE4B | RNF220 | TSHZ2 |
| CHRM3 | ELOVL7 | MAD2L2 | PGR | RPL38 | VDR |
| Overlapping Genes (n = 16) | |||||
| AFF3 | CYFIP2 | PDE1C | PLEKHA7 | RPS6KA2 | TSHZ2 |
| CDH6 | LONRF2 | PDE4B | RAP1GAP2 | SHANK2 | |
| COL27A1 | NTM | PGR | RETREG1 | TMEM178B | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, M.R.; Reske, J.J.; Koeman, J.; Adams, M.; Joshi, N.R.; Fazleabas, A.T.; Chandler, R.L. SWI/SNF Antagonism of PRC2 Mediates Estrogen-Induced Progesterone Receptor Expression. Cells 2022, 11, 1000. https://doi.org/10.3390/cells11061000
Wilson MR, Reske JJ, Koeman J, Adams M, Joshi NR, Fazleabas AT, Chandler RL. SWI/SNF Antagonism of PRC2 Mediates Estrogen-Induced Progesterone Receptor Expression. Cells. 2022; 11(6):1000. https://doi.org/10.3390/cells11061000
Chicago/Turabian StyleWilson, Mike R., Jake J. Reske, Julie Koeman, Marie Adams, Niraj R. Joshi, Asgerally T. Fazleabas, and Ronald L. Chandler. 2022. "SWI/SNF Antagonism of PRC2 Mediates Estrogen-Induced Progesterone Receptor Expression" Cells 11, no. 6: 1000. https://doi.org/10.3390/cells11061000
APA StyleWilson, M. R., Reske, J. J., Koeman, J., Adams, M., Joshi, N. R., Fazleabas, A. T., & Chandler, R. L. (2022). SWI/SNF Antagonism of PRC2 Mediates Estrogen-Induced Progesterone Receptor Expression. Cells, 11(6), 1000. https://doi.org/10.3390/cells11061000