
Analyzing the Androgen Receptor Interactome in Prostate Cancer: Implications for Therapeutic Intervention

Abstract
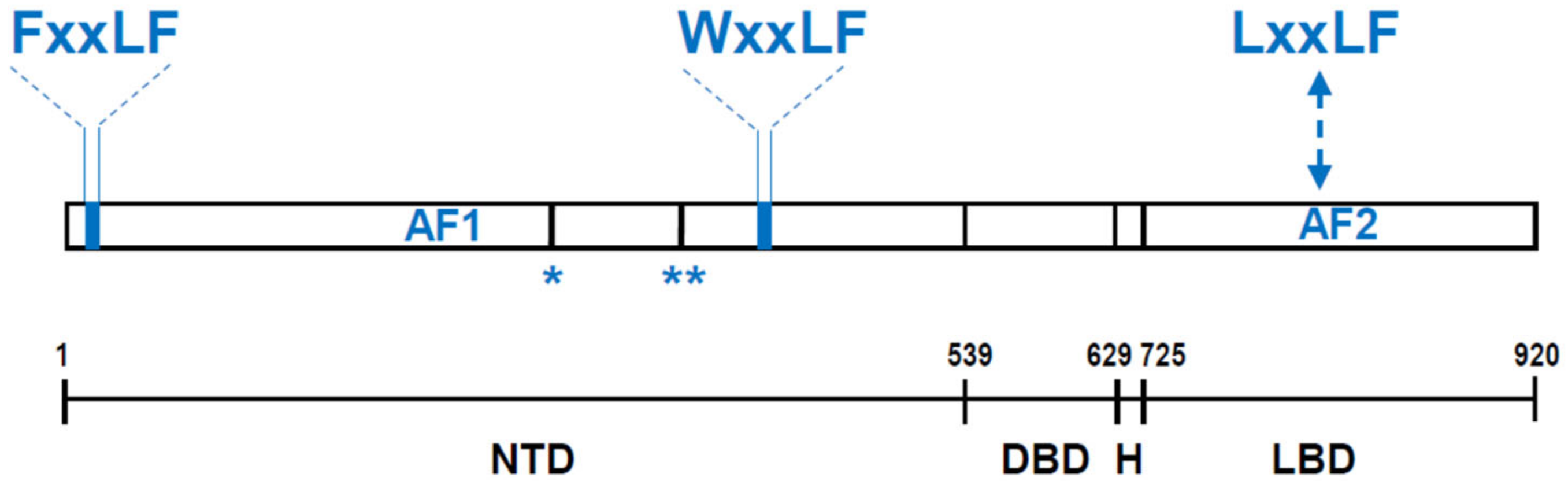
:1. AR Structure and Function
2. AR Action Drives CaP Progression
3. Novel Strategies to Target AR for CaP Treatment
4. Screening for Novel AR Interactors
4.1. 2 Hybrid Assays
4.2. Phage Display Assays
5. Characterizing the Composition of AR Transcriptional Complexes
5.1. Affinity Purification Mass Spectrometry
5.2. Rapid Immunoprecipitation Mass Spectrometry of Endogenous Proteins
5.3. Biotin-Based Proximity Ligation Assay
6. Opportunities, Challenges and Limitations for Effective Therapeutic AR Complex Disruption
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef] [Green Version]
- McKenna, N.J.; Cooney, A.J.; DeMayo, F.J.; Downes, M.; Glass, C.K.; Lanz, R.B.; Lazar, M.A.; Mangelsdorf, D.J.; Moore, D.D.; Qin, J.; et al. Minireview: Evolution of NURSA, the Nuclear Receptor Signaling Atlas. Mol. Endocrinol. 2009, 23, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen receptor (AR) coregulators: An overview. Endocrinol. Rev. 2002, 23, 175–200. [Google Scholar] [CrossRef]
- Heemers, H.V.; Tindall, D.J. Androgen Receptor (AR) Coregulators: A Diversity of Functions Converging on and Regulating the AR Transcriptional Complex. Endocrinol. Rev. 2007, 28, 778–808. [Google Scholar] [CrossRef] [Green Version]
- Helsen, C.; Claessens, F. Looking at nuclear receptors from a new angle. Mol. Cell. Endocrinol. 2014, 382, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Senapati, D.; Heemers, H.V. Rationale for the development of alternative forms of androgen deprivation therapy. Endocr. Relat. Cancer 2017, 24, R275–R295. [Google Scholar] [CrossRef] [Green Version]
- McEwan, I.J.; Brinkmann, A.O. Androgen Physiology: Receptor and Metabolic Disorders. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., New, M., Sahay, R., Levy, M., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Quayle, S.N.; Mawji, N.R.; Wang, J.; Sadar, M.D. Androgen receptor decoy molecules block the growth of prostate cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 1331–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, Y.; Dehm, S.M. Androgen Receptor Rearrangement and Splicing Variants in Resistance to Endocrine Therapies in Prostate Cancer. Endocrinology 2017, 158, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- McEwan, I.J. Intrinsic disorder in the androgen receptor: Identification, characterisation and drugability. Mol. BioSyst. 2012, 8, 82–90. [Google Scholar] [CrossRef]
- Yu, X.; Yi, P.; Hamilton, R.A.; Shen, H.; Chen, M.; Foulds, C.E.; Mancini, M.A.; Ludtke, S.J.; Wang, Z.; O’Malley, B.W. Structural Insights of Transcriptionally Active, Full-Length Androgen Receptor Coactivator Complexes. Mol. Cell 2020, 79, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Davison, S.L.; Bell, R. Androgen physiology. Semin. Reprod. Med. 2006, 24, 71–77. [Google Scholar] [CrossRef]
- Marker, P.C.; Donjacour, A.A.; Dahiya, R.; Cunha, G.R. Hormonal, cellular, and molecular control of prostatic development. Dev. Biol. 2003, 253, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Ceruti, J.M.; Leirós, G.J.; Balañá, M.E. Androgens and androgen receptor action in skin and hair follicles. Mol. Cell. Endocrinol. 2018, 465, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Querin, G.; Bede, P.; Marchand-Pauvert, V.; Pradat, P.-F. Biomarkers of Spinal and Bulbar Muscle Atrophy (SBMA): A Comprehensive Review. Front. Neurol. 2018, 9, 844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denmeade, S.R.; Isaacs, J.T. A history of prostate cancer treatment. Nat. Rev. Cancer 2002, 2, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.J.; Tindall, D.J. Androgen receptor: Past, present and future. Curr. Drug Targets 2013, 14, 401–407. [Google Scholar] [CrossRef]
- Dai, C.; Heemers, H.; Sharifi, N. Androgen Signaling in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a030452. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Huggins, C.S. Studies on prostatic cancer: 2. The effects of castration on advanced carcinoma of the prostate gland. Arch. Surg. 1941, 43, 209–212. [Google Scholar] [CrossRef]
- Huggins, C.; Hodges, C.V. Studies on Prostatic Cancer. I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 2018, 174, 433–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, A.H.; Beltran, H.; Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 2018, 15, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Demichelis, F. Therapy considerations in neuroendocrine prostate cancer: What next? Endocrinol. Relat. Cancer 2021, 28, T67–T78. [Google Scholar] [CrossRef] [PubMed]
- Henzler, C.; Li, Y.; Yang, R.; McBride, T.; Ho, Y.; Sprenger, C.; Liu, G.; Coleman, I.; Lakely, B.; Li, R.; et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat. Commun. 2016, 7, 13668. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.G.; Yegnasubramanian, S. Resistance Emerges to Second-Generation Antiandrogens in Prostate Cancer. Cancer Discov. 2013, 3, 971–974. [Google Scholar] [CrossRef] [Green Version]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid Receptor Confers Resistance to Antiandrogens by Bypassing Androgen Receptor Blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Alyamani, M.; Zhang, A.; Chang, K.-H.; Berk, M.; Li, Z.; Zhu, Z.; Petro, M.; Magi-Galluzzi, C.; Taplin, M.-E.; et al. Aberrant corticosteroid metabolism in tumor cells enables GR takeover in enzalutamide resistant prostate cancer. eLife 2017, 6, 1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Chang, Y.-J.; Yu, I.-C.; Yeh, S.; Wu, C.-C.; Miyamoto, H.; Merry, D.E.; Sobue, G.; Chen, L.-M.; Chang, S.-S.; et al. ASC-J9 ameliorates spinal and bulbar muscular atrophy phenotype via degradation of androgen receptor. Nat. Med. 2007, 13, 348–353. [Google Scholar] [CrossRef]
- Lee, G.T.; Nagaya, N.; Desantis, J.; Madura, K.; Sabaawy, H.E.; Kim, W.-J.; Vaz, R.J.; Cruciani, G.; Kim, I.Y. Effects of MTX-23, a Novel PROTAC of Androgen Receptor Splice Variant-7 and Androgen Receptor, on CRPC Resistant to Second-Line Antiandrogen Therapy. Mol. Cancer Ther. 2020, 20, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.-K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H.; Yang, Y.; Tavakoli, I.; Haile, S.; Watt, K.; et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef] [Green Version]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.-K.; Watt, K.; Tam, T.; Yang, Y.C.; Bañuelos, C.A.; et al. Regression of Castrate-Recurrent Prostate Cancer by a Small-Molecule Inhibitor of the Amino-Terminus Domain of the Androgen Receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Ravindranathan, P.; Lee, T.-K.; Yang, L.; Centenera, M.; Butler, L.; Tilley, W.; Hsieh, J.-T.; Ahn, J.-M.; Raj, G.V. Peptidomimetic targeting of critical androgen receptor–coregulator interactions in prostate cancer. Nat. Commun. 2013, 4, 1923. [Google Scholar] [CrossRef]
- Chang, C.-Y.; McDonnell, D.P. Androgen receptor–cofactor interactions as targets for new drug discovery. Trends Pharmacol. Sci. 2005, 26, 225–228. [Google Scholar] [CrossRef]
- DePriest, A.D.; Fiandalo, M.; Schlanger, S.; Heemers, F.; Mohler, J.L.; Liu, S.; Heemers, H.V. Regulators of Androgen Action Resource: A one-stop shop for the comprehensive study of androgen receptor action. Database 2016, 2016, bav125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 2012, 33, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Chmelar, R.; Buchanan, G.; Need, E.F.; Tilley, W.; Greenberg, N.M. Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int. J. Cancer 2006, 120, 719–733. [Google Scholar] [CrossRef]
- Liu, S.; Kumari, S.; Hu, Q.; Senapati, D.; Venkadakrishnan, V.B.; Wang, D.; DePriest, A.D.; Schlanger, S.E.; Ben-Salem, S.; Valenzuela, M.M.; et al. A comprehensive analysis of coregulator recruitment, androgen receptor function and gene expression in prostate cancer. eLife 2017, 6, e28482. [Google Scholar] [CrossRef]
- Ianculescu, I.; Wu, D.Y.; Siegmund, K.D.; Stallcup, M.R. Selective roles for cAMP response element-binding protein binding protein and p300 protein as coregulators for androgen-regulated gene expression in advanced prostate cancer cells. J. Biol. Chem. 2012, 287, 4000–4013. [Google Scholar] [CrossRef] [Green Version]
- Asangani, I.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Waddell, A.; Huang, H.; Liao, D. CBP/p300: Critical Co-Activators for Nuclear Steroid Hormone Receptors and Emerging Therapeutic Targets in Prostate and Breast Cancers. Cancers 2021, 13, 2872. [Google Scholar] [CrossRef] [PubMed]
- Link, K.A.; Balasubramaniam, S.; Sharma, A.; Comstock, C.; Godoy-Tundidor, S.; Powers, N.; Cao, K.H.; Haelens, A.; Claessens, F.; Revelo, M.P.; et al. Targeting the BAF57 SWI/SNF Subunit in Prostate Cancer: A Novel Platform to Control Androgen Receptor Activity. Cancer Res. 2008, 68, 4551–4558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, S.; Song, O.-K. A novel genetic system to detect protein–protein interactions. Nature 1989, 340, 245–246. [Google Scholar] [CrossRef]
- Rao, M.A.; Cheng, H.; Quayle, A.N.; Nishitani, H.; Nelson, C.C.; Rennie, P.S. RanBPM, a Nuclear Protein That Interacts with and Regulates Transcriptional Activity of Androgen Receptor and Glucocorticoid Receptor. J. Biol. Chem. 2002, 277, 48020–48027. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Bai, S.; Hnat, A.T.; Kalman, R.I.; Minges, J.T.; Patterson, C.; Wilson, E.M. An Androgen Receptor NH2-terminal Conserved Motif Interacts with the COOH Terminus of the Hsp70-interacting Protein (CHIP). J. Biol. Chem. 2004, 279, 30643–30653. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yeh, S.; Wu, G.; Hsu, C.-L.; Wang, L.; Chiang, T.; Yang, Y.; Guo, Y.; Chang, C. Identification and Characterization of a Novel Androgen Receptor Coregulator ARA267-α in Prostate Cancer Cells. J. Biol. Chem. 2001, 276, 40417–40423. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Zarnegar, M.; Li, X.; Lim, B.; Sun, Z. Androgen Receptor Interacts with a Novel MYST Protein, HBO1. J. Biol. Chem. 2000, 275, 35200–35208. [Google Scholar] [CrossRef] [Green Version]
- Grötsch, H.; Kunert, M.; Mooslehner, K.A.; Gao, Z.; Struve, D.; Hughes, I.A.; Hiort, O.; Werner, R. RWDD1 interacts with the ligand binding domain of the androgen receptor and acts as a coactivator of androgen-dependent transactivation. Mol. Cell. Endocrinol. 2012, 358, 53–62. [Google Scholar] [CrossRef]
- Obendorf, M.; Meyer, R.; Henning, K.; Mitev, Y.A.; Schröder, J.; Patchev, V.K.; Wolf, S.S. FoxG1, a member of the forkhead family, is a corepressor of the androgen receptor. J. Steroid Biochem. Mol. Biol. 2007, 104, 195–207. [Google Scholar] [CrossRef]
- Clark, E.L.; Coulson, A.; Dalgliesh, C.; Rajan, P.; Nicol, S.M.; Fleming, S.; Heer, R.; Gaughan, L.; Leung, H.Y.; Elliott, D.; et al. The RNA Helicase p68 Is a Novel Androgen Receptor Coactivator Involved in Splicing and Is Overexpressed in Prostate Cancer. Cancer Res. 2008, 68, 7938–7946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, N.; Takagi, T.; Nakano, Y.; Yamaji, R.; Inui, H. Protein arginine methyltransferase 10 is required for androgen-dependent proliferation of LNCaP prostate cancer cells. Biosci. Biotechnol. Biochem. 2015, 79, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Wafa, L.A.; Cheng, H.; Rao, M.A.; Nelson, C.C.; Cox, M.; Hirst, M.; Sadowski, I.; Rennie, P.S. Isolation and identification of l-dopa decarboxylase as a protein that binds to and enhances transcriptional activity of the androgen receptor using the repressed transactivator yeast two-hybrid system. Biochem. J. 2003, 375 Pt 2, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Hu, H.; Yin, M.; Sun, Z.; Chen, X.; Li, Y.; Sun, Z.; Liu, C.; Li, L.; Qiu, Y. Sertad1 promotes prostate cancer progression through binding androgen receptor ligand binding domain. Int. J. Cancer 2018, 144, 558–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavassoli, P.; Wafa, L.A.; Cheng, H.; Zoubeidi, A.; Fazli, L.; Gleave, M.; Snoek, R.; Rennie, P.S. TAF1 Differentially Enhances Androgen Receptor Transcriptional Activity via Its N-Terminal Kinase and Ubiquitin-Activating and -Conjugating Domains. Mol. Endocrinol. 2010, 24, 696–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, K.H. Yeast two-hybrid: So many interactions, (in) so little time. Biol. Reprod. 1998, 58, 302–311. [Google Scholar] [CrossRef] [Green Version]
- Stagljar, I.; Korostensky, C.; Johnsson, N.; Heesen, S.T. A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 5187–5192. [Google Scholar] [CrossRef] [Green Version]
- Ray, M.R.; Wafa, L.A.; Cheng, H.; Snoek, R.; Fazli, L.; Gleave, M.; Rennie, P.S. Cyclin G-associated kinase: A novel androgen receptor-interacting transcriptional coactivator that is overexpressed in hormone refractory prostate cancer. Int. J. Cancer 2006, 118, 1108–1119. [Google Scholar] [CrossRef]
- Yeh, S.; Chang, C. Cloning and characterization of a specific coactivator, ARA70, for the androgen receptor in human prostate cells. Proc. Natl. Acad. Sci. USA 1996, 93, 5517–5521. [Google Scholar] [CrossRef] [Green Version]
- Sundell, G.N.; Ivarsson, Y. Interaction Analysis through Proteomic Phage Display. BioMed Res. Int. 2014, 2014, 176172. [Google Scholar] [CrossRef]
- Sidhu, S.S.; Koide, S. Phage display for engineering and analyzing protein interaction interfaces. Curr. Opin. Struct. Biol. 2007, 17, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Danner, S.; Belasco, J.G. T7 phage display: A novel genetic selection system for cloning RNA-binding proteins from cDNA libraries. Proc. Natl. Acad. Sci. USA 2001, 98, 12954–12959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, J.; Goodson, B.; Winter, J. T7 displayed peptides as targets for selecting peptide specific scFvs from M13 scFv display libraries. J. Immunol. Methods 2001, 257, 117–122. [Google Scholar] [CrossRef]
- Müller, J.M.; Isele, U.; Metzger, E.; Rempel, A.; Moser, M.; Pscherer, A.; Breyer, T.; Holubarsch, C.; Buettner, R.; Schüle, R. FHL2, a novel tissue-specific coactivator of the androgen receptor. EMBO J. 2000, 19, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heery, D.; Kalkhoven, E.; Hoare, S.; Parker, M.G. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 1997, 387, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Kemppainen, J.A.; Wilson, E.M. FXXLF and WXXLF Sequences Mediate the NH2-terminal Interaction with the Ligand Binding Domain of the Androgen Receptor. J. Biol. Chem. 2000, 275, 22986–22994. [Google Scholar] [CrossRef] [Green Version]
- Dehm, S.M.; Regan, K.M.; Schmidt, L.J.; Tindall, D.J. Selective Role of an NH2-Terminal WxxLF Motif for Aberrant Androgen Receptor Activation in Androgen Depletion–Independent Prostate Cancer Cells. Cancer Res. 2007, 67, 10067–10077. [Google Scholar] [CrossRef] [Green Version]
- Hur, E.; Pfaff, S.J.; Payne, E.S.; Grøn, H.; Buehrer, B.; Fletterick, R.J. Recognition and Accommodation at the Androgen Receptor Coactivator Binding Interface. PLoS Biol. 2004, 2, e274. [Google Scholar] [CrossRef]
- Kazmin, D.; Prytkova, T.; Cook, C.E.; Wolfinger, R.; Chu, T.-M.; Beratan, D.; Norris, J.; Chang, C.-Y.; McDonnell, D.P. Linking Ligand-Induced Alterations in Androgen Receptor Structure to Differential Gene Expression: A First Step in the Rational Design of Selective Androgen Receptor Modulators. Mol. Endocrinol. 2006, 20, 1201–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-Y.; Abdo, J.; Hartney, T.; McDonnell, D.P. Development of Peptide Antagonists for the Androgen Receptor Using Combinatorial Peptide Phage Display. Mol. Endocrinol. 2005, 19, 2478–2490. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-L.; Liu, J.-S.; Wu, P.-L.; Guan, H.-H.; Chen, Y.-L.; Lin, A.-C.; Ting, H.-J.; Pang, S.-T.; Yeh, S.-D.; Ma, W.-L.; et al. Identification of a new androgen receptor (AR) co-regulator BUD31 and related peptides to suppress wild-type and mutated AR-mediated prostate cancer growth via peptide screening and X-ray structure analysis. Mol. Oncol. 2014, 8, 1575–1587. [Google Scholar] [CrossRef]
- Ozers, M.S.; Marks, B.D.; Gowda, K.; Kupcho, K.R.; Ervin, K.M.; De Rosier, T.; Qadir, N.; Eliason, H.C.; Riddle, A.S.M.; Shekhani, M.S. The Androgen Receptor T877A Mutant Recruits LXXLL and FXXLF Peptides Differently than Wild-Type Androgen Receptor in a Time-Resolved Fluorescence Resonance Energy Transfer Assay. Biochemistry 2007, 46, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Blessing, A.; Ganesan, S.; Rajapakshe, K.; Sung, Y.Y.; Bollu, L.; Shi, Y.; Cheung, E.; Coarfa, C.; Chang, J.T.; McDonnell, D.P.; et al. Identification of a Novel Coregulator, SH3YL1, That Interacts with the Androgen Receptor N-Terminus. Mol. Endocrinol. 2015, 29, 1426–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senapati, D.; Kumari, S.; Heemers, H.V. Androgen receptor co-regulation in prostate cancer. Asian J. Urol. 2020, 7, 219–232. [Google Scholar] [CrossRef]
- Norris, J.D.; Joseph, J.D.; Sherk, A.B.; Juzumiene, D.; Turnbull, P.S.; Rafferty, S.W.; Cui, H.; Anderson, E.; Fan, D.; Dye, D.A.; et al. Differential Presentation of Protein Interaction Surfaces on the Androgen Receptor Defines the Pharmacological Actions of Bound Ligands. Chem. Biol. 2009, 16, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Groner, A.C.; Cato, L.; de Tribolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.C.; Tschopp, P.; Gu, L.; et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 2016, 29, 846–858. [Google Scholar] [CrossRef] [Green Version]
- He, H.-J.; Gu, X.-F.; Xu, W.-H.; Yang, D.-J.; Wang, X.-M.; Su, Y. Krüppel-like factor 8 is a novel androgen receptor co-activator in human prostate cancer. Acta Pharmacol. Sin. 2013, 34, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Mayeur, G.L.; Kung, W.-J.; Martinez, A.; Izumiya, C.; Chen, D.J.; Kung, H.-J. Ku Is a Novel Transcriptional Recycling Coactivator of the Androgen Receptor in Prostate Cancer Cells. J. Biol. Chem. 2005, 280, 10827–10833. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-T.; Okada, M.; Nakato, R.; Izumi, K.; Bando, M.; Shirahige, K. The Deubiquitinating Enzyme USP7 Regulates Androgen Receptor Activity by Modulating Its Binding to Chromatin. J. Biol. Chem. 2015, 290, 21713–21723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, J.M.; Graham, N.A.; Lee, J.K.; Stoyanova, T.; Faltermeier, C.M.; Sud, S.; Titz, B.; Huang, J.; Pienta, K.J.; Graeber, T.G.; et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc. Natl. Acad. Sci. USA 2013, 110, E4762–E4769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, J.; Graham, N.A.; Stoyanova, T.; Sedghi, A.; Goldstein, A.; Cai, H.; Smith, D.A.; Zhang, H.; Komisopoulou, E.; Huang, J.; et al. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc. Natl. Acad. Sci. USA 2012, 109, 1643–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.-L.; Wen, X.-F.; Li, R.-B.; Liu, B.; Qiu, G.-M.; Wen, J.-L.; Wang, Y.-M. Chrebp regulates the transcriptional activity of androgen receptor in prostate cancer. Tumor Biol. 2014, 35, 8143–8148. [Google Scholar] [CrossRef]
- Mohammed, H.; D’Santos, C.; Serandour, A.A.; Ali, R.; Brown, G.D.; Atkins, A.; Palacio, O.R.; Holmes, K.-A.; Theodorou, V.; Robinson, J.L.; et al. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013, 3, 342–349. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, H.; Taylor, C.; Brown, G.D.; Papachristou, E.; Carroll, J.; D’Santos, C.S. Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) for analysis of chromatin complexes. Nat. Protoc. 2016, 11, 316–326. [Google Scholar] [CrossRef]
- Paltoglou, S.; Das, R.; Townley, S.L.; Hickey, T.; Tarulli, G.; Coutinho, I.; Fernandes, R.; Hanson, A.R.; Denis, I.; Carroll, J.; et al. Novel Androgen Receptor Coregulator GRHL2 Exerts Both Oncogenic and Antimetastatic Functions in Prostate Cancer. Cancer Res. 2017, 77, 3417–3430. [Google Scholar] [CrossRef] [Green Version]
- Augello, M.A.; Liu, D.; Deonarine, L.D.; Robinson, B.D.; Huang, D.; Stelloo, S.; Blattner, M.; Doane, A.S.; Wong, E.W.; Chen, Y. CHD1 Loss Alters AR Binding at Lineage-Specific Enhancers and Modulates Distinct Transcriptional Programs to Drive Prostate Tumorigenesis. Cancer Cell. 2019, 35, 603–617. [Google Scholar] [CrossRef] [Green Version]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887, Correction in 2020, 38, 108. [Google Scholar] [CrossRef]
- Cho, K.F.; Branon, T.C.; Udeshi, N.D.; Myers, S.A.; Carr, S.A.; Ting, A.Y. Proximity labeling in mammalian cells with TurboID and split-TurboID. Nat. Protoc. 2020, 15, 3971–3999. [Google Scholar] [CrossRef]
- Vélot, L.; Lessard, F.; Bérubé-Simard, F.-A.; Tav, C.; Neveu, B.; Teyssier, V.; Boudaoud, I.; Dionne, U.; Lavoie, N.; Bilodeau, S.; et al. Proximity-dependent Mapping of the Androgen Receptor Identifies Kruppel-like Factor 4 as a Functional Partner. Mol. Cell. Proteom. 2021, 20, 100064. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, J.J.; Ng, B.H.; Smits, M.M.; Martinez, H.D.; Jasavala, R.J.; Hinkson, I.V.; Fermin, D.; Eng, J.; Nesvizhskii, A.I.; Wright, M.E. Research Resource: Androgen Receptor Activity Is Regulated Through the Mobilization of Cell Surface Receptor Networks. Mol. Endocrinol. 2015, 29, 1195–1218. [Google Scholar] [CrossRef] [Green Version]
- Geng, C.; He, B.; Xu, L.; Barbieri, C.; Eedunuri, V.K.; Chew, S.A.; Zimmermann, M.; Bond, R.; Shou, J.; Li, C.; et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc. Natl. Acad. Sci. USA 2013, 110, 6997–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.-P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification–mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holding, A.N. XL-MS: Protein cross-linking coupled with mass spectrometry. Methods 2015, 89, 54–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dotzlaw, H.; Papaioannou, M.; Moehren, U.; Claessens, F.; Baniahmad, A. Agonist–antagonist induced coactivator and corepressor interplay on the human androgen receptor. Mol. Cell. Endocrinol. 2003, 213, 79–85. [Google Scholar] [CrossRef]
- Stelloo, S.; Nevedomskaya, E.; Kim, Y.; Hoekman, L.; Bleijerveld, O.B.; Mirza, T.; Wessels, L.F.A.; Van Weerden, W.M.; Altelaar, A.F.M.; Bergman, A.M.; et al. Endogenous androgen receptor proteomic profiling reveals genomic subcomplex involved in prostate tumorigenesis. Oncogene 2018, 37, 313–322. [Google Scholar] [CrossRef]
- Stelloo, S.; Bergman, A.M.; Zwart, W. Androgen receptor enhancer usage and the chromatin regulatory landscape in human prostate cancers. Endocr. Relat. Cancer 2019, 26, R267–R285. [Google Scholar] [CrossRef] [Green Version]
- Fletterick, R.J. Molecular modelling of the androgen receptor axis: Rational basis for androgen receptor intervention in androgen-independent prostate cancer. Br. J. Urol. 2005, 96 (Suppl. S2), 2–9. [Google Scholar] [CrossRef]
- Chang, C.Y.; McDonnell, D.P. Evaluation of ligand-dependent changes in AR structure using peptide probes. Mol. Endocrinol. 2002, 16, 647–660. [Google Scholar] [CrossRef]
- Yang, W.; Briegel, A. Use of Cryo-EM to Study the Structure of Chemoreceptor Arrays In Vivo. Methods Mol. Biol. 2018, 1729, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, C.; Piotrowski, C.; Aebersold, R.; Amaral, B.C.; Andrews, P.; Bernfur, K.; Borchers, C.; Brodie, N.I.; Bruce, J.E.; Cao, Y.; et al. First Community-Wide, Comparative Cross-Linking Mass Spectrometry Study. Anal. Chem. 2019, 91, 6953–6961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Corzo, C.; Chang, M.R.; Shang, J.; Lam, V.Q.; Brust, R.; Blayo, A.L.; Bruning, J.B.; Kamenecka, T.M.; Kojetin, D.J.; et al. Chemical Crosslinking Mass Spectrometry Reveals the Conformational Landscape of the Activation Helix of PPARgamma; a Model for Ligand-Dependent Antagonism. Structure 2018, 26, 1431–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Wang, J.; Lampert, E.; Schlanger, S.; DePriest, A.D.; Hu, Q.; Gomez, E.C.; Murakam, M.; Glenn, S.T.; Conroy, J.; et al. Intratumoral and Intertumoral Genomic Heterogeneity of Multifocal Localized Prostate Cancer Impacts Molecular Classifications and Genomic Prognosticators. Eur. Urol. 2017, 71, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malovannaya, A.; Lanz, R.B.; Jung, S.Y.; Bulynko, Y.; Le, N.T.; Chan, D.W.; Ding, C.; Shi, Y.; Yucer, N.; Krenciute, G.; et al. Analysis of the Human Endogenous Coregulator Complexome. Cell 2011, 145, 787–799. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| AR Interactor | Bait | AR Interacting Domain | Library | Validation Assays | Reference |
|---|---|---|---|---|---|
| ARA267-α | aa 595-918 | DBD + LBD | human brain | in vitro coIP, transient tranfection | [48] |
| CHIP | aa 220-270 | NTD | human testis | in vitro coIP, mammalian 2 hybrid, immunocytochemistry, transient transfection | [47] |
| DDC | aa 1-559, aa 233-559, aa 1-646 | NTD, LBD | LNCaP cells | in vitro and in vivo coIP, transient transfection | [54] |
| FOXG1 | aa 325-919 | DBD + hinge + LBD | human fetal brain | 1 and 2 hybrid, in vitro and in vivo coIP, transient transfection | [51] |
| HBO1 | aa 505-919 | DBD + LBD | human prostate | in vitro and in vitro coIP, transient transfection | [49] |
| p68 | DBD + LBD | human prostate | in vivo coIP, immunofluorescence, ChIP, transient transfection | [52] | |
| PRMT10 | aa 1-98 | NTD | universal human | in vitro and in vivo coIP, mammalian 2 hybrid, AR-dependent cell proliferation | [53] |
| RanBPM | aa 1-232 | NTD, DBD | human prostate | in vitro and in vivo coIP, transient transfection | [46] |
| RWDD1 | aa 555-920 | LBD | genital tubercle of male mice | in vitro and in vivo coIP, transient transfection | [50] |
| Sertad1 | LBD | LBD | male peripheral blood monocytes | in vitro and in vivo CoIP, immunocytochemistry | [55] |
| TAF1 | aa 1-559, aa 233-559, aa 1-646 | NTD | LNCaP cells | in vitro and in vivo coIP, ChIP, transient transfection | [56] |
| AR Interactor | Method 1 | Ref 1 | Method 2 | Ref 2 | Cellular Location | Function |
|---|---|---|---|---|---|---|
| TAF1 | Y2H | [56] | PLA | [92] | nucleus | transcriptional regulator |
| NCOR1 | PLA | [92] | RIME | [88] | nucleus | coregulator |
| USP7 | AP-MS | [82] | PLA | [92] | cytoplasm, nucleus | ubiquitinyl hydrolase |
| SRC1 | Y2H | [97] | PLA | [92] | cell junction, cell membrane, mitochondrion, perinuclear region | coregulator |
| RBM10 | PLA | [92] | RIME | [98] | nucleus | RNA binding |
| SSBP2 | PLA | [92] | RIME | [88] | cytoplasm, nucleus | splicing |
| SMARCE1 | RIME | [98] | PLA | [92] | nucleus | chromatin remodeller |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahiya, U.R.; Heemers, H.V. Analyzing the Androgen Receptor Interactome in Prostate Cancer: Implications for Therapeutic Intervention. Cells 2022, 11, 936. https://doi.org/10.3390/cells11060936
Dahiya UR, Heemers HV. Analyzing the Androgen Receptor Interactome in Prostate Cancer: Implications for Therapeutic Intervention. Cells. 2022; 11(6):936. https://doi.org/10.3390/cells11060936
Chicago/Turabian StyleDahiya, Ujjwal R., and Hannelore V. Heemers. 2022. "Analyzing the Androgen Receptor Interactome in Prostate Cancer: Implications for Therapeutic Intervention" Cells 11, no. 6: 936. https://doi.org/10.3390/cells11060936
APA StyleDahiya, U. R., & Heemers, H. V. (2022). Analyzing the Androgen Receptor Interactome in Prostate Cancer: Implications for Therapeutic Intervention. Cells, 11(6), 936. https://doi.org/10.3390/cells11060936