
Senescence-Associated Cell Transition and Interaction (SACTAI): A Proposed Mechanism for Tissue Aging, Repair, and Degeneration



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
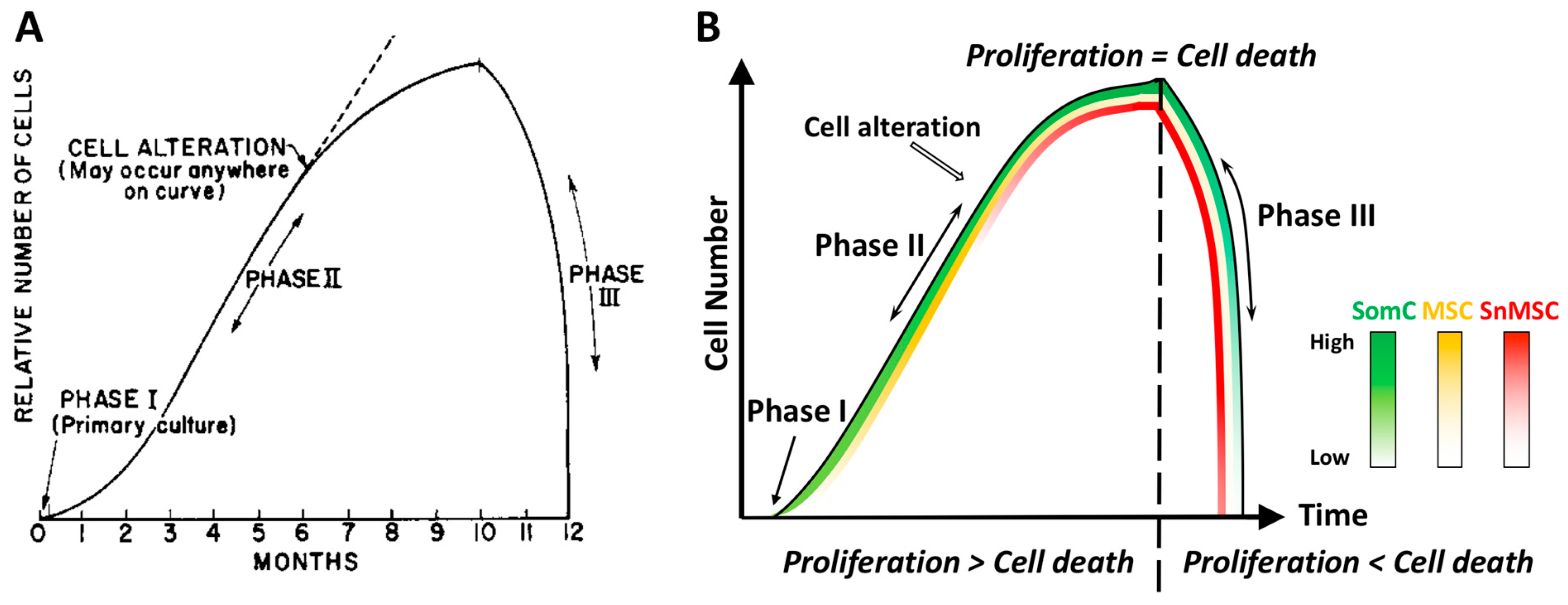
2. Cell Senescence Is Cell-Autonomous: Telomere Shortening, Replicative Stress and SASP Manifestation
3. Cell Senescence-Associated Apoptosis and Degeneration Is Senescent Cell Non-Autonomous: The SACTAI Mechanism for Aging
4. Two-Way Communications between Senescent and Non-Senescent Cells: Altered Intercellular Signaling Mechanisms during Aging
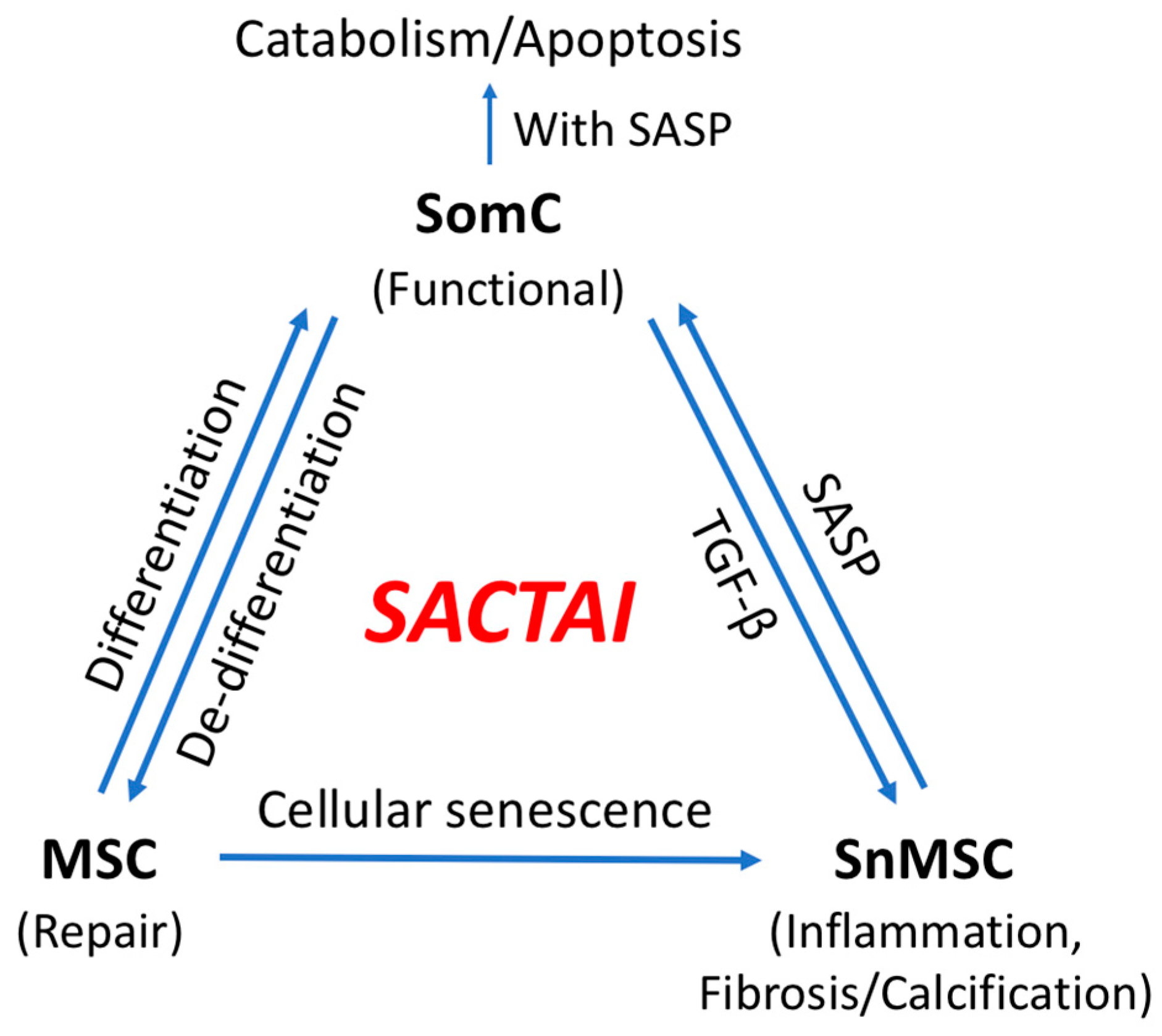
5. SACTAI in Adult Tissues
5.1. Dedifferentiation: SomC to MSC Transition
5.2. MSC Senescence
5.3. BM-MSC Senescence
6. SACTAI: A Mechanism of Balancing Tissue Repair and Degeneration
7. SACTAI in Age-Related Diseases
7.1. Physical Dysfunction
7.2. Cardiovascular Disease
7.3. Neurodegenerative Disorder
7.4. Idiopathic Pulmonary Fibrosis
7.5. Bone and Joint Degeneration
8. Conclusions
8.1. Recent Finding
8.2. Summary
8.3. Implication
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Beard, J.R.; Officer, A.; de Carvalho, I.A.; Sadana, R.; Pot, A.M.; Michel, J.P.; Lloyd-Sherlock, P.; Epping-Jordan, J.E.; Peeters, G.; Mahanani, W.R.; et al. The World report on ageing and health: A policy framework for healthy ageing. Lancet 2016, 387, 2145–2154. [Google Scholar] [CrossRef] [Green Version]
- Melzer, D.; Pilling, L.C.; Ferrucci, L. The genetics of human ageing. Nat. Rev. Genet. 2020, 21, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Jaul, E.; Barron, J. Age-related diseases and clinical and public health implications for the 85 years old and over population. Front. Public Health 2017, 5, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Gall, J.G. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J. Mol. Biol. 1978, 120, 33–53. [Google Scholar] [CrossRef]
- Moyzis, R.K.; Buckingham, J.M.; Cram, L.S.; Dani, M.; Deaven, L.L.; Jones, M.D.; Meyne, J.; Ratliff, R.L.; Wu, J.R. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc. Natl. Acad. Sci. USA 1988, 85, 6622–6626. [Google Scholar] [CrossRef] [Green Version]
- Watson, J.D. Origin of concatemeric T7 DNA. Nat. New Biol. 1972, 239, 197–201. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Reitinger, S.; Schimke, M.; Klepsch, S.; de Sneeuw, S.; Yani, S.L.; Gaßner, R.; Ertl, P.; Lepperdinger, G. Systemic impact molds mesenchymal stromal/stem cell aging. Transfus. Apher. Sci. 2015, 52, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Chen, Q. Adipokines: New therapeutic target for osteoarthritis? Curr. Rheumatol. Rep. 2019, 21, 71. [Google Scholar] [CrossRef] [PubMed]
- Akyurekli, C.; Le, Y.; Richardson, R.B.; Fergusson, D.; Tay, J.; Allan, D.S. A systematic review of preclinical studies on the therapeutic potential of mesenchymal stromal cell-derived microvesicles. Stem Cell Rev. Rep. 2015, 11, 150–160. [Google Scholar] [CrossRef]
- Xie, H.; Wang, Z.; Zhang, L.; Lei, Q.; Zhao, A.; Wang, H.; Li, Q.; Chen, Z.; Zhang, W. Development of an angiogenesis-promoting microvesicle-alginate-polycaprolactone composite graft for bone tissue engineering applications. PeerJ 2016, 4, e2040. [Google Scholar] [CrossRef]
- Yuan, Q.L.; Zhang, Y.G.; Chen, Q. Mesenchymal stem cell (MSC)-derived extracellular vesicles: Potential therapeutics as MSC trophic mediators in regenerative medicine. Anat. Rec. 2020, 303, 1735–1742. [Google Scholar] [CrossRef]
- Lei, Q.; Liu, T.; Gao, F.; Xie, H.; Sun, L.; Zhao, A.; Ren, W.; Guo, H.; Zhang, L.; Wang, H.; et al. Microvesicles as potential biomarkers for the identification of senescence in human mesenchymal stem cells. Theranostics 2017, 7, 2673–2689. [Google Scholar] [CrossRef]
- Hayflick, L. Theories of biological aging. Exp. Gerontol. 1985, 20, 145–159. [Google Scholar] [CrossRef]
- Wang, E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 1995, 55, 2284–2292. [Google Scholar]
- Liu, W.; Brodsky, A.S.; Feng, M.; Liu, Y.; Ding, J.; Jayasuriya, C.T.; Chen, Q. Senescent tissue-resident mesenchymal stromal cells are an internal source of inflammation in human osteoarthritic cartilage. Front. Cell Dev. Biol. 2021, 9, 2395. [Google Scholar] [CrossRef]
- Jayasuriya, C.T.; Hu, N.; Li, J.; Lemme, N.; Terek, R.; Ehrlich, M.G.; Chen, Q. Molecular characterization of mesenchymal stem cells in human osteoarthritis cartilage reveals contribution to the OA phenotype. Sci. Rep. 2018, 8, 7044. [Google Scholar] [CrossRef] [PubMed]
- Alsalameh, S.; Amin, R.; Gemba, T.; Lotz, M. Identification of mesenchymal progenitor cells in normal and osteoarthritic human articular cartilage. Arthritis Rheum. 2004, 50, 1522–1532. [Google Scholar] [CrossRef]
- Hattori, S.; Oxford, C.; Reddi, A.H. Identification of superficial zone articular chondrocyte stem/progenitor cells. Biochem. Biophys. Res. Commun. 2007, 358, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grogan, S.P.; Miyaki, S.; Asahara, H.; D’Lima, D.D.; Lotz, M.K. Mesenchymal progenitor cell markers in human articular cartilage: Normal distribution and changes in osteoarthritis. Arthritis Res. Ther. 2009, 11, R85. [Google Scholar] [CrossRef] [Green Version]
- Fickert, S.; Fiedler, J.; Brenner, R.E. Identification of subpopulations with characteristics of mesenchymal progenitor cells from human osteoarthritic cartilage using triple staining for cell surface markers. Arthritis Res. Ther. 2004, 6, R422–R432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pretzel, D.; Linss, S.; Rochler, S.; Endres, M.; Kaps, C.; Alsalameh, S.; Kinne, R.W. Relative percentage and zonal distribution of mesenchymal progenitor cells in human osteoarthritic and normal cartilage. Arthritis Res. Ther. 2011, 13, R64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, N.; Gao, Y.; Jayasuriya, C.T.; Liu, W.; Du, H.; Ding, J.; Feng, M.; Chen, Q. Chondrogenic induction of human osteoarthritic cartilage-derived mesenchymal stem cells activates mineralization and hypertrophic and osteogenic gene expression through a mechanomiR. Arthritis Res. Ther. 2019, 21, 167. [Google Scholar] [CrossRef] [Green Version]
- Fellows, C.R.; Williams, R.; Davies, I.R.; Gohil, K.; Baird, D.M.; Fairclough, J.; Rooney, P.; Archer, C.W.; Khan, I.M. Characterisation of a divergent progenitor cell sub-populations in human osteoarthritic cartilage: The role of telomere erosion and replicative senescence. Sci. Rep. 2017, 7, 41421. [Google Scholar] [CrossRef] [Green Version]
- Feng, M.; Liu, W.; Ding, J.; Qiu, Y.; Chen, Q. Sonic hedgehog induces mesenchymal stromal cell senescence-associated secretory phenotype and chondrocyte apoptosis in human osteoarthritic cartilage. Front. Cell Dev. Biol. 2021, 9, 716610. [Google Scholar] [CrossRef]
- Qu, F.; Guilak, F.; Mauck, R.L. Cell migration: Implications for repair and regeneration in joint disease. Nat. Rev. Rheumatol. 2019, 15, 167–179. [Google Scholar] [CrossRef]
- Liu, W.; Feng, M.; Jayasuriya, C.T.; Peng, H.; Zhang, L.; Guan, Y.; Froehlich, J.A.; Terek, R.M.; Chen, Q. Human osteoarthritis cartilage-derived stromal cells activate joint degeneration through TGF-beta lateral signaling. FASEB J. 2020, 34, 16552–16566. [Google Scholar] [CrossRef] [PubMed]
- Alman, B.A. The role of hedgehog signalling in skeletal health and disease. Nat. Rev. Rheumatol. 2015, 11, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Ye, Y.; Shi, Y.; Dang, J.; Feng, X.; Chen, Y.; Liu, F.; Olsen, N.; Huang, J.; Zheng, S.G. Sonic hedgehog regulates proliferation, migration and invasion of synoviocytes in rheumatoid arthritis via JNK signaling. Front. Immunol. 2020, 11, 1300. [Google Scholar] [CrossRef] [PubMed]
- Bishop, C.L.; Bergin, A.M.; Fessart, D.; Borgdorff, V.; Hatzimasoura, E.; Garbe, J.C.; Stampfer, M.R.; Koh, J.; Beach, D.H. Primary cilium-dependent and -independent Hedgehog signaling inhibits p16(INK4A). Mol. Cell. 2010, 40, 533–547. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.C.; Seeto, B.L.; Bartoszko, J.M.; Khoury, M.A.; Whetstone, H.; Ho, L.; Hsu, C.; Ali, S.A.; Alman, B.A. Modulating hedgehog signaling can attenuate the severity of osteoarthritis. Nat. Med. 2009, 15, 1421–1425. [Google Scholar] [CrossRef]
- Tan, Z.; Niu, B.; Tsang, K.Y.; Melhado, I.G.; Ohba, S.; He, X.; Huang, Y.; Wang, C.; McMahon, A.P.; Jauch, R.; et al. Synergistic co-regulation and competition by a SOX9-GLI-FOXA phasic transcriptional network coordinate chondrocyte differentiation transitions. PLoS Genet. 2018, 14, e1007346. [Google Scholar] [CrossRef]
- Buttitta, L.A.; Edgar, B.A. Mechanisms controlling cell cycle exit upon terminal differentiation. Curr. Opin. Cell Biol. 2007, 19, 697–704. [Google Scholar] [CrossRef] [Green Version]
- Merrell, A.J.; Stanger, B.Z. Adult cell plasticity in vivo: De-differentiation and transdifferentiation are back in style. Nat. Rev. Mol. Cell Biol. 2016, 17, 413–425. [Google Scholar] [CrossRef]
- Gurdon, J.B. The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J. Embryol. Exp. Morphol. 1962, 10, 622–640. [Google Scholar] [CrossRef]
- Jopling, C.; Boue, S.; Izpisua Belmonte, J.C. Dedifferentiation, transdifferentiation and reprogramming: Three routes to regeneration. Nat. Rev. Mol. Cell Biol. 2011, 12, 79–89. [Google Scholar] [CrossRef]
- Burclaff, J.; Mills, J.C. Plasticity of differentiated cells in wound repair and tumorigenesis, part I: Stomach and pancreas. Dis. Models Mech. 2018, 11, dmm033373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burclaff, J.; Mills, J.C. Plasticity of differentiated cells in wound repair and tumorigenesis, part II: Skin and intestine. Dis. Models Mech. 2018, 11, dmm035071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Cells Tissues Organs 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Goodell, M.A.; Nguyen, H.; Shroyer, N. Somatic stem cell heterogeneity: Diversity in the blood, skin and intestinal stem cell compartments. Nat. Rev. Mol. Cell Biol. 2015, 16, 299–309. [Google Scholar] [CrossRef]
- Gurusamy, N.; Alsayari, A.; Rajasingh, S.; Rajasingh, J. Adult stem cells for regenerative therapy. Prog. Mol. Biol. Transl. Sci. 2018, 160, 1–22. [Google Scholar] [CrossRef]
- Urbán, N.; Cheung, T.H. Stem cell quiescence: The challenging path to activation. Development 2021, 148, dev165084. [Google Scholar] [CrossRef]
- Li, L.; Clevers, H. Coexistence of quiescent and active adult stem cells in mammals. Science 2010, 327, 542–545. [Google Scholar] [CrossRef] [Green Version]
- Pittenger, M.F.; Discher, D.E.; Péault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Sveiven, S.N.; Nordgren, T.M. Lung-resident mesenchymal stromal cells are tissue-specific regulators of lung homeostasis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L197–L210. [Google Scholar] [CrossRef] [PubMed]
- Fellows, C.R.; Matta, C.; Zakany, R.; Khan, I.M.; Mobasheri, A. Adipose, bone marrow and synovial joint-derived mesenchymal stem cells for cartilage repair. Front. Genet. 2016, 7, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rady, D.; Abbass, M.M.S.; El-Rashidy, A.A.; El Moshy, S.; Radwan, I.A.; Dörfer, C.E.; Fawzy El-Sayed, K.M. Mesenchymal stem/progenitor cells: The prospect of human clinical translation. Stem Cells Int. 2020, 2020, 8837654. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Gronthos, S.; Zhao, M.; Lu, B.; Fisher, L.W.; Robey, P.G.; Shi, S. SHED: Stem cells from human exfoliated deciduous teeth. Proc. Natl. Acad. Sci. USA 2003, 100, 5807–5812. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Hong, Y.; Zhang, H.; Li, X. Mesenchymal stem cell senescence and rejuvenation: Current status and challenges. Front. Cell Dev. Biol. 2020, 8, 364. [Google Scholar] [CrossRef]
- Saeedi, P.; Halabian, R.; Imani Fooladi, A.A. A revealing review of mesenchymal stem cells therapy, clinical perspectives and Modification strategies. Stem Cell Investig. 2019, 6, 34. [Google Scholar] [CrossRef]
- Dimmeler, S.; Leri, A. Aging and disease as modifiers of efficacy of cell therapy. Circ. Res. 2008, 102, 1319–1330. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Q.; Wang, Y.; Li, L.; Bu, H.; Bao, J. Senescence of mesenchymal stem cells (Review). Int. J. Mol. Med. 2017, 39, 775–782. [Google Scholar] [CrossRef] [Green Version]
- Schimke, M.M.; Marozin, S.; Lepperdinger, G. Patient-specific age: The other side of the coin in advanced mesenchymal stem cell therapy. Front. Physiol. 2015, 6, 362. [Google Scholar] [CrossRef]
- Lukomska, B.; Stanaszek, L.; Zuba-Surma, E.; Legosz, P.; Sarzynska, S.; Drela, K. Challenges and controversies in human mesenchymal stem cell therapy. Stem Cells Int. 2019, 2019, 9628536. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.J.; Uberti, J.P.; Soiffer, R.J.; Klingemann, H.; Waller, E.K.; Daly, A.S.; Herrmann, R.P.; Kebriaei, P. Prochymal improves response rates in patients with steroid-refractory acute graft versus host disease (SR-GVHD) involving the liver and gut: Results of a randomized, placebo-controlled, multicenter phase III trial in GVHD. Biol. Blood Marrow Transplant. 2010, 16, S169–S170. [Google Scholar] [CrossRef] [Green Version]
- Bartunek, J.; Behfar, A.; Dolatabadi, D.; Vanderheyden, M.; Ostojic, M.; Dens, J.; El Nakadi, B.; Banovic, M.; Beleslin, B.; Vrolix, M.; et al. Cardiopoietic stem cell therapy in heart failure: The C-CURE (Cardiopoietic stem Cell therapy in heart failURE) multicenter randomized trial with lineage-specified biologics. J. Am. Coll. Cardiol. 2013, 61, 2329–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartunek, J.; Davison, B.; Sherman, W.; Povsic, T.; Henry, T.D.; Gersh, B.; Metra, M.; Filippatos, G.; Hajjar, R.; Behfar, A.; et al. Congestive heart failure cardiopoietic regenerative therapy (CHART-1) trial design. Eur. J. Heart Fail. 2016, 18, 160–168. [Google Scholar] [CrossRef]
- Yu, Y.; Park, Y.S.; Kim, H.S.; Kim, H.Y.; Jin, Y.M.; Jung, S.C.; Ryu, K.H.; Jo, I. Characterization of long-term in vitro culture-related alterations of human tonsil-derived mesenchymal stem cells: Role for CCN1 in replicative senescence-associated increase in osteogenic differentiation. J. Anat. 2014, 225, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Q. Senescent mesenchymal stem cells: Disease mechanism and treatment strategy. Curr. Mol. Biol. Rep. 2020, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular senescence in aging primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [Green Version]
- Franzen, J.; Zirkel, A.; Blake, J.; Rath, B.; Benes, V.; Papantonis, A.; Wagner, W. Senescence-associated DNA methylation is stochastically acquired in subpopulations of mesenchymal stem cells. Aging Cell 2017, 16, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Andrzejewska, A.; Lukomska, B.; Janowski, M. Concise review: Mesenchymal stem cells: From roots to boost. Stem Cells 2019, 37, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Greenberger, J.S.; Epperly, M.W.; Goff, J.P.; Adler, C.; Leboff, M.S.; Glowacki, J. Age-related intrinsic changes in human bone-marrow-derived mesenchymal stem cells and their differentiation to osteoblasts. Aging Cell 2008, 7, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Bianco, P.; Robey, P.G. Skeletal stem cells. Development 2015, 142, 1023–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, A.; da Silva, C.L.; dos Santos, F.; Camafeita, E.; Cabral, J.M.; Sá-Correia, I. Human mesenchymal stem cell expression program upon extended ex-vivo cultivation, as revealed by 2-DE-based quantitative proteomics. PLoS ONE 2012, 7, e43523. [Google Scholar] [CrossRef] [PubMed]
- Bertolo, A.; Mehr, M.; Janner-Jametti, T.; Graumann, U.; Aebli, N.; Baur, M.; Ferguson, S.J.; Stoyanov, J.V. An in vitro expansion score for tissue-engineering applications with human bone marrow-derived mesenchymal stem cells. J. Tissue Eng. Regen. Med. 2016, 10, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Horn, P.; Castoldi, M.; Diehlmann, A.; Bork, S.; Saffrich, R.; Benes, V.; Blake, J.; Pfister, S.; Eckstein, V.; et al. Replicative senescence of mesenchymal stem cells: A continuous and organized process. PLoS ONE 2008, 3, e2213. [Google Scholar] [CrossRef] [Green Version]
- Neri, S.; Borzì, R.M. Molecular mechanisms contributing to mesenchymal stromal cell aging. Biomolecules 2020, 10, 340. [Google Scholar] [CrossRef] [Green Version]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Santamaria, P.G.; Moreno-Bueno, G.; Portillo, F.; Cano, A. EMT: Present and future in clinical oncology. Mol. Oncol. 2017, 11, 718–738. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.L.; Zhu, R.T.; Sun, Y.L. Epithelial-mesenchymal transition in liver fibrosis. Biomed. Rep. 2016, 4, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.P.; Watson, R.W.; Mulsow, J.J.; Docherty, N.G.; Coffey, J.C.; O’Connell, P.R. Endoglin negatively regulates transforming growth factor beta1-induced profibrotic responses in intestinal fibroblasts. Br. J. Surg. 2010, 97, 892–901. [Google Scholar] [CrossRef]
- Davids, J.S.; Carothers, A.M.; Damas, B.C.; Bertagnolli, M.M. Chronic cyclooxygenase-2 inhibition promotes myofibroblast-associated intestinal fibrosis. Cancer Prev. Res. 2010, 3, 348–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saika, S.; Yamanaka, O.; Flanders, K.C.; Okada, Y.; Miyamoto, T.; Sumioka, T.; Shirai, K.; Kitano, A.; Miyazaki, K.; Tanaka, S.; et al. Epithelial-mesenchymal transition as a therapeutic target for prevention of ocular tissue fibrosis. Endocr. Metab. Immune Disord.-Drug Targets 2008, 8, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Josephson, A.M.; Bradaschia-Correa, V.; Lee, S.; Leclerc, K.; Patel, K.S.; Muinos Lopez, E.; Litwa, H.P.; Neibart, S.S.; Kadiyala, M.; Wong, M.Z.; et al. Age-related inflammation triggers skeletal stem/progenitor cell dysfunction. Proc. Natl. Acad. Sci. USA 2019, 116, 6995–7004. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Liu, Y.; Liu, Y.; Li, C.; Wan, Q.; Yang, L.; Su, Y.; Cheng, Y.; Liu, C.; Wang, X.; et al. Reversed senescence of retinal pigment epithelial cell by coculture with embryonic stem cell via the TGFβ and PI3K pathways. Front. Cell Dev. Biol. 2020, 8, 588050. [Google Scholar] [CrossRef] [PubMed]
- Crimmins, E.M. Lifespan and healthspan: Past, present, and promise. Gerontologist 2015, 55, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Park, Y.-M.; Myers, M.; Vieira-Potter, V.J. Adipose tissue inflammation and metabolic dysfunction: Role of exercise. Mo. Med. 2014, 111, 65–72. [Google Scholar]
- Wall, M.E.; Bernacki, S.H.; Loboa, E.G. Effects of serial passaging on the adipogenic and osteogenic differentiation potential of adipose-derived human mesenchymal stem cells. Tissue Eng. 2007, 13, 1291–1298. [Google Scholar] [CrossRef]
- Vicinanza, C.; Aquila, I.; Scalise, M.; Cristiano, F.; Marino, F.; Cianflone, E.; Mancuso, T.; Marotta, P.; Sacco, W.; Lewis, F.C.; et al. Adult cardiac stem cells are multipotent and robustly myogenic: C-kit expression is necessary but not sufficient for their identification. Cell Death Differ. 2017, 24, 2101–2116. [Google Scholar] [CrossRef] [Green Version]
- Ellison, G.M.; Vicinanza, C.; Smith, A.J.; Aquila, I.; Leone, A.; Waring, C.D.; Henning, B.J.; Stirparo, G.G.; Papait, R.; Scarfò, M.; et al. Adult c-kit(pos) cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell 2013, 154, 827–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, P.C.; Segers, V.F.; Davis, M.E.; MacGillivray, C.; Gannon, J.; Molkentin, J.D.; Robbins, J.; Lee, R.T. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat. Med. 2007, 13, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Broughton, K.M.; Wang, B.J.; Firouzi, F.; Khalafalla, F.; Dimmeler, S.; Fernandez-Aviles, F.; Sussman, M.A. Mechanisms of cardiac repair and regeneration. Circ. Res. 2018, 122, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Trial, J.; Entman, M.L.; Cieslik, K.A. Mesenchymal stem cell-derived inflammatory fibroblasts mediate interstitial fibrosis in the aging heart. J. Mol. Cell. Cardiol. 2016, 91, 28–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaldi, A.; Dodia, R.M.; Orogo, A.M.; Zambrano, C.M.; Najor, R.H.; Gustafsson, Å.B.; Heller Brown, J.; Purcell, N.H. Decline in cellular function of aged mouse c-kit(+) cardiac progenitor cells. J. Physiol. 2017, 595, 6249–6262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef] [Green Version]
- Budoff, M.J.; Shaw, L.J.; Liu, S.T.; Weinstein, S.R.; Mosler, T.P.; Tseng, P.H.; Flores, F.R.; Callister, T.Q.; Raggi, P.; Berman, D.S. Long-term prognosis associated with coronary calcification: Observations from a registry of 25,253 patients. J. Am. Coll. Cardiol. 2007, 49, 1860–1870. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.C.; Leopold, J.A.; Loscalzo, J. Vascular calcification: Pathobiological mechanisms and clinical implications. Circ. Res. 2006, 99, 1044–1059. [Google Scholar] [CrossRef] [Green Version]
- Nakano-Kurimoto, R.; Ikeda, K.; Uraoka, M.; Nakagawa, Y.; Yutaka, K.; Koide, M.; Takahashi, T.; Matoba, S.; Yamada, H.; Okigaki, M.; et al. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1673–H1684. [Google Scholar] [CrossRef]
- Salvadores, N.; Sanhueza, M.; Manque, P.; Court, F.A. Axonal degeneration during aging and its functional role in neurodegenerative disorders. Front. Neurosci. 2017, 11, 451. [Google Scholar] [CrossRef] [Green Version]
- Si, Z.; Sun, L.; Wang, X. Evidence and perspectives of cell senescence in neurodegenerative diseases. Biomed. Pharmacother. 2021, 137, 111327. [Google Scholar] [CrossRef] [PubMed]
- Nicaise, A.M.; Wagstaff, L.J.; Willis, C.M.; Paisie, C.; Chandok, H.; Robson, P.; Fossati, V.; Williams, A.; Crocker, S.J. Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9030–9039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Saraswat, D.; Sinha, A.K.; Polanco, J.; Dietz, K.; O’Bara, M.A.; Pol, S.U.; Shayya, H.J.; Sim, F.J. Paired related homeobox protein 1 regulates quiescence in human oligodendrocyte progenitors. Cell Rep. 2018, 25, 3435–3450.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Back, S.A.; Tuohy, T.M.; Chen, H.; Wallingford, N.; Craig, A.; Struve, J.; Luo, N.L.; Banine, F.; Liu, Y.; Chang, A.; et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat. Med. 2005, 11, 966–972. [Google Scholar] [CrossRef]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef]
- Liu, R.M.; Liu, G. Cell senescence and fibrotic lung diseases. Exp. Gerontol. 2020, 132, 110836. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Yanai, H.; Shteinberg, A.; Porat, Z.; Budovsky, A.; Braiman, A.; Ziesche, R.; Fraifeld, V.E. Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging 2015, 7, 664–672. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, D.; Cárdenes, N.; Sellarés, J.; Bueno, M.; Corey, C.; Hanumanthu, V.S.; Peng, Y.; D’Cunha, H.; Sembrat, J.; Nouraie, M.; et al. IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L1164–L1173. [Google Scholar] [CrossRef]
- Cárdenes, N.; Álvarez, D.; Sellarés, J.; Peng, Y.; Corey, C.; Wecht, S.; Nouraie, S.M.; Shanker, S.; Sembrat, J.; Bueno, M.; et al. Senescence of bone marrow-derived mesenchymal stem cells from patients with idiopathic pulmonary fibrosis. Stem Cell Res. Ther. 2018, 9, 257. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, Y.; Lu, L.; Liu, L.; Yu, X.; Pei, F. Cellular senescence in knee osteoarthritis: Molecular mechanisms and therapeutic implications. Ageing Res. Rev. 2021, 70, 101413. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B. Articular cartilage degradation in osteoarthritis. HSS J. 2012, 8, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Yin, J.; Gao, J.; Cheng, T.S.; Pavlos, N.J.; Zhang, C.; Zheng, M.H. Subchondral bone in osteoarthritis: Insight into risk factors and microstructural changes. Arthritis Res. Ther. 2013, 15, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.; Khan, I.M.; Richardson, K.; Nelson, L.; McCarthy, H.E.; Analbelsi, T.; Singhrao, S.K.; Dowthwaite, G.P.; Jones, R.E.; Baird, D.M.; et al. Identification and clonal characterisation of a progenitor cell sub-population in normal human articular cartilage. PLoS ONE 2010, 5, e13246. [Google Scholar] [CrossRef] [Green Version]
- Dowthwaite, G.P.; Bishop, J.C.; Redman, S.N.; Khan, I.M.; Rooney, P.; Evans, D.J.; Haughton, L.; Bayram, Z.; Boyer, S.; Thomson, B.; et al. The surface of articular cartilage contains a progenitor cell population. J. Cell Sci. 2004, 117, 889–897. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.K.F.; Gulati, G.S.; Sinha, R.; Tompkins, J.V.; Lopez, M.; Carter, A.C.; Ransom, R.C.; Reinisch, A.; Wearda, T.; Murphy, M.; et al. Identification of the human skeletal stem cell. Cell 2018, 175, 43–56.e21. [Google Scholar] [CrossRef] [Green Version]
- Gulati, G.S.; Murphy, M.P.; Marecic, O.; Lopez, M.; Brewer, R.E.; Koepke, L.S.; Manjunath, A.; Ransom, R.C.; Salhotra, A.; Weissman, I.L.; et al. Isolation and functional assessment of mouse skeletal stem cell lineage. Nat. Protoc. 2018, 13, 1294–1309. [Google Scholar] [CrossRef]
- Malaise, O.; Tachikart, Y.; Constantinides, M.; Mumme, M.; Ferreira-Lopez, R.; Noack, S.; Krettek, C.; Noël, D.; Wang, J.; Jorgensen, C.; et al. Mesenchymal stem cell senescence alleviates their intrinsic and seno-suppressive paracrine properties contributing to osteoarthritis development. Aging 2019, 11, 9128–9146. [Google Scholar] [CrossRef]
- Kiel, J.; Kaiser, K. Stress Reaction and Fractures. In StatPearls; StatPearls Publishing Copyright© 2021; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Bahney, C.S.; Zondervan, R.L.; Allison, P.; Theologis, A.; Ashley, J.W.; Ahn, J.; Miclau, T.; Marcucio, R.S.; Hankenson, K.D. Cellular biology of fracture healing. J. Orthop. Res. 2019, 37, 35–50. [Google Scholar] [CrossRef] [Green Version]
- Colnot, C.; Zhang, X.; Knothe Tate, M.L. Current insights on the regenerative potential of the periosteum: Molecular, cellular, and endogenous engineering approaches. J. Orthop. Res. 2012, 30, 1869–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Fateh, A.; Salem, D.M.; Intini, G. Periosteum: Biology and applications in craniofacial bone regeneration. J. Dent. Res. 2014, 93, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vozzi, G.; Lucarini, G.; Dicarlo, M.; Andreoni, C.; Salvolini, E.; Ferretti, C.; Mattioli-Belmonte, M. In Vitro lifespan and senescent behaviour of human periosteal derived stem cells. Bone 2016, 88, 1–12. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Schwam, J.; Chen, Q. Senescence-Associated Cell Transition and Interaction (SACTAI): A Proposed Mechanism for Tissue Aging, Repair, and Degeneration. Cells 2022, 11, 1089. https://doi.org/10.3390/cells11071089
Liu Y, Schwam J, Chen Q. Senescence-Associated Cell Transition and Interaction (SACTAI): A Proposed Mechanism for Tissue Aging, Repair, and Degeneration. Cells. 2022; 11(7):1089. https://doi.org/10.3390/cells11071089
Chicago/Turabian StyleLiu, Yajun, Jonah Schwam, and Qian Chen. 2022. "Senescence-Associated Cell Transition and Interaction (SACTAI): A Proposed Mechanism for Tissue Aging, Repair, and Degeneration" Cells 11, no. 7: 1089. https://doi.org/10.3390/cells11071089
APA StyleLiu, Y., Schwam, J., & Chen, Q. (2022). Senescence-Associated Cell Transition and Interaction (SACTAI): A Proposed Mechanism for Tissue Aging, Repair, and Degeneration. Cells, 11(7), 1089. https://doi.org/10.3390/cells11071089