
Molecular and Functional Signatures Associated with CAR T Cell Exhaustion and Impaired Clinical Response in Patients with B Cell Malignancies


 and
and
Abstract
:1. Introduction
2. Methods
2.1. Patients and CAR-T Protocol
2.2. Peripheral Blood Mononuclear Cell Separation and Cryopreservation
2.3. RNA Extraction
2.4. Gene Expression Analysis
2.5. RT-PCR Analysis
2.6. Phenotype and Activation Status
2.7. Cell Lines and Cell Culture
2.8. CAR T Ex Vivo Proliferation
2.9. Cytotoxic Activity
2.10. Statistical Analysis
2.11. Data Availability
3. Results
3.1. Patients’ Characteristics and Clinical Response
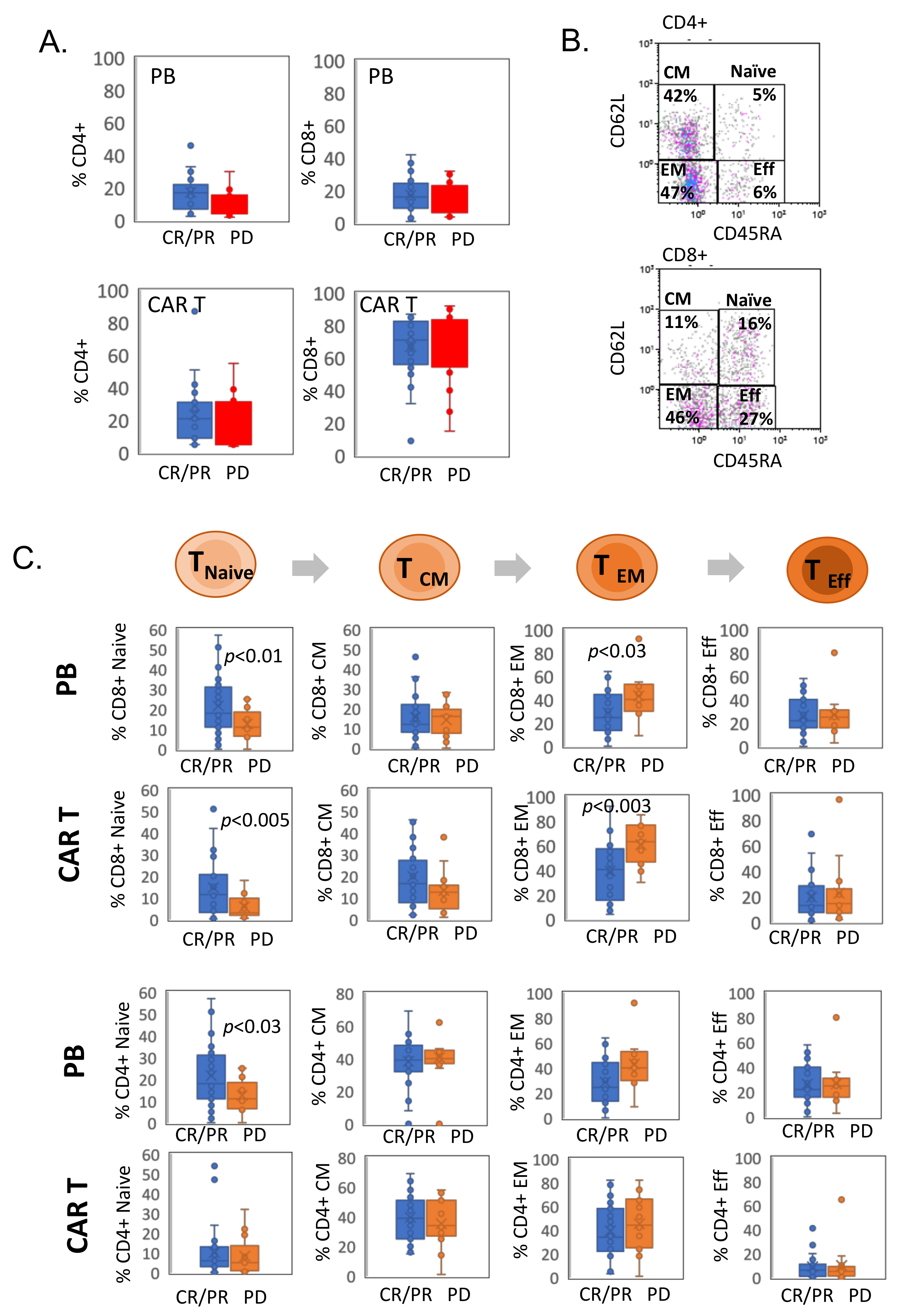
3.2. CAR T Cell Phenotype Characterization
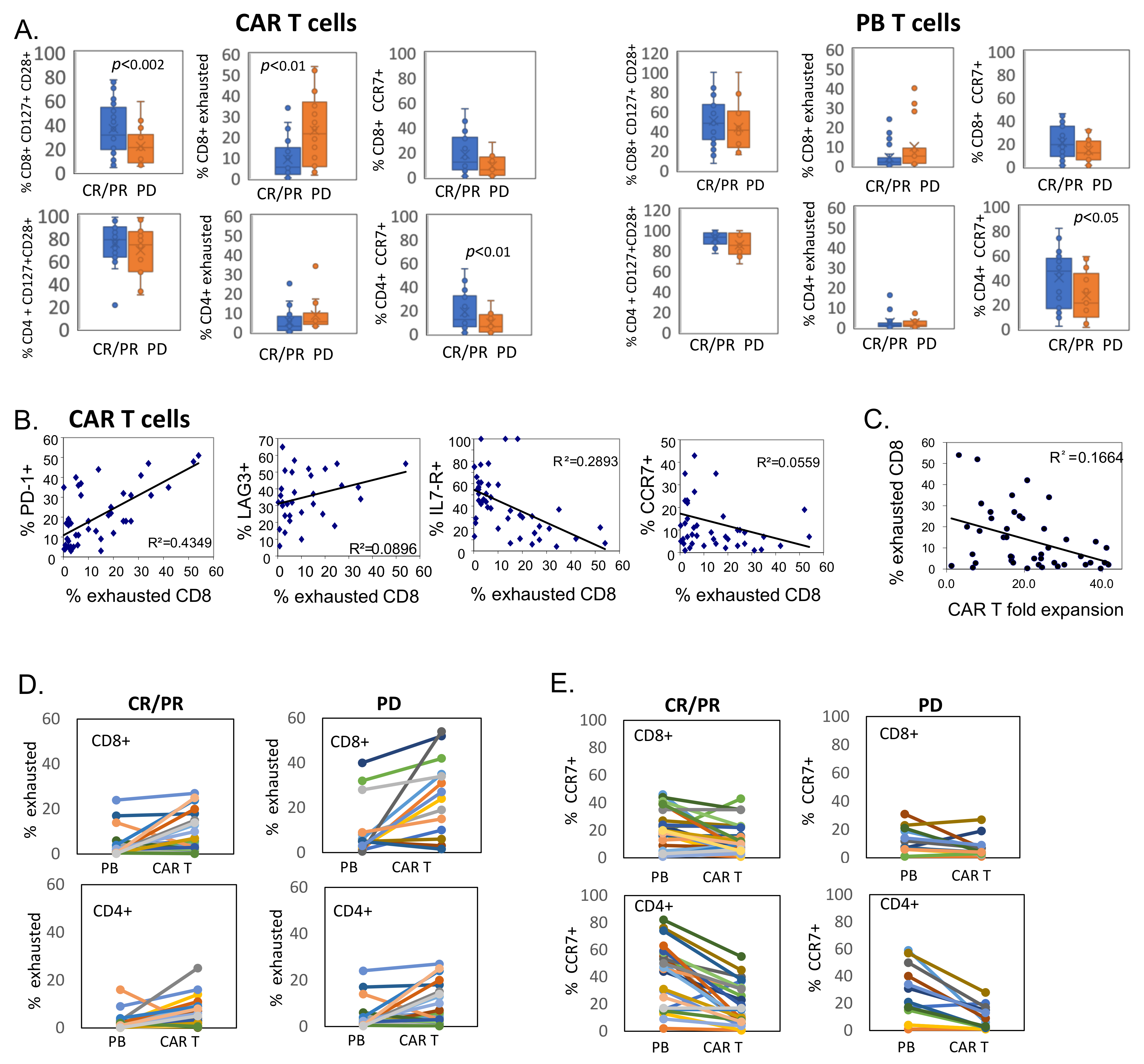
3.3. CD8+ CAR T Cells Exhibit Features of Exhaustion
3.4. Transcriptional Profile of CAR T Products
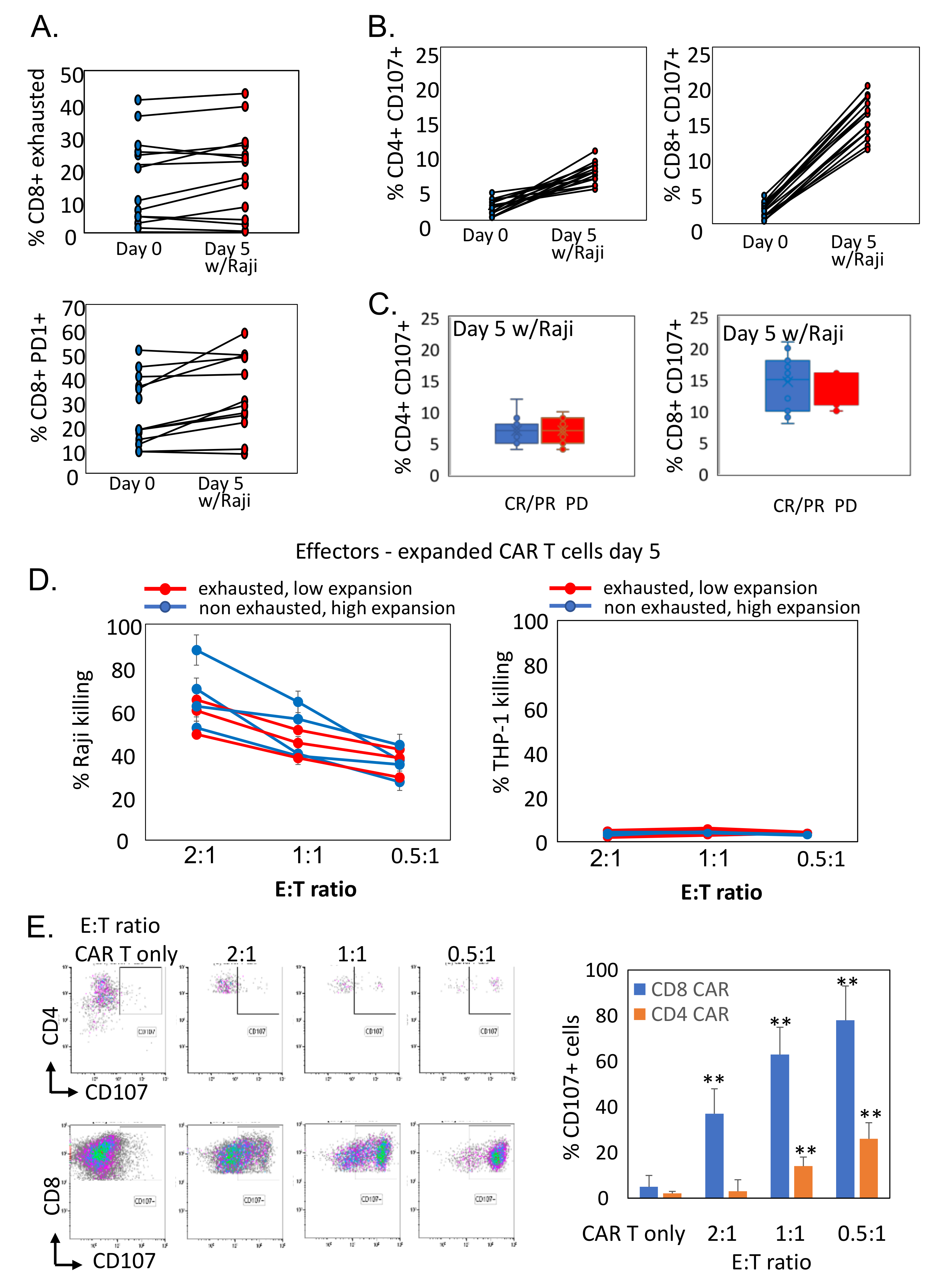
3.5. Reduced Ex Vivo Expansion of CAR T Cells Correlates with Exhausted Phenotype
3.6. Antigen-Exposed CAR T Cells Preserve Cytotoxic Activity against CD19-Expressing Target Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 2018, 15, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Senechal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lym-phoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of re-sponse and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.M.; Hogan, P.G.; Rao, A.; Martinez, G.J. Transcriptional and epigenetic regulation of T cell hyporesponsiveness. J. Leukoc. Biol. 2017, 102, 601–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Amezquita, R.A.; Kaech, S.M. Immunology: The chronicles of T-cell exhaustion. Nature 2017, 543, 190–191. [Google Scholar] [CrossRef]
- Gallimore, A.; Glithero, A.; Godkin, A.; Tissot, A.C.; Plückthun, A.; Elliott, T.; Hengartner, H.; Zinkernagel, R. Induction and Exhaustion of Lymphocytic Choriomeningitis Virus–specific Cytotoxic T Lymphocytes Visualized Using Soluble Tetrameric Major Histocompatibility Complex Class I–Peptide Complexes. J. Exp. Med. 1998, 187, 1383–1393. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor anti-gen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schumacher, T. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, M.; Wang, C.; Cong, L.; Marjanovic, N.D.; Kowalczyk, M.S.; Zhang, H.; Nyman, J.; Sakuishi, K.; Kurtulus, S.; Gennert, D.; et al. A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell 2016, 166, 1500–1511.e9. [Google Scholar] [CrossRef] [Green Version]
- Schietinger, A.; Philip, M.; Krisnawan, V.; Chiu, E.Y.; Delrow, J.J.; Basom, R.S.; Lauer, P.; Brockstedt, D.G.; Knoblaugh, S.E.; Hämmerling, G.J.; et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 2016, 45, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbar, A.N.; Henson, S.M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 2011, 11, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Quigley, M.; Pereyra, F.; Nilsson, B.; Porichis, F.; Fonseca, C.; Eichbaum, Q.; Julg, B.; Jesneck, J.L.; Brosnahan, K.; Imam, S.; et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med. 2010, 16, 1147–1151. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef]
- Björkström, N.K.; Béziat, V.; Cichocki, F.; Liu, L.L.; Levine, J.; Larsson, S.; Koup, R.A.; Anderson, S.; Ljunggren, H.-G.; Malmberg, K.-J. CD8 T cells express randomly selected KIRs with distinct specificities compared with NK cells. Blood 2012, 120, 3455–3465. [Google Scholar] [CrossRef] [Green Version]
- Henson, S.M.; Akbar, A.N. KLRG1—More than a marker for T cell senescence. AGE 2009, 31, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8(+) T cells dif-ferentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Kared, H.; Martelli, S.; Ng, T.P.; Pender, S.L.; Larbi, A. CD57 in human natural killer cells and T-lymphocytes. Cancer Immunol. Immunother. 2016, 65, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Doering, T.A.; Crawford, A.; Angelosanto, J.M.; Paley, M.A.; Ziegler, C.G.; Wherry, E.J. Network Analysis Reveals Centrally Connected Genes and Pathways Involved in CD8+ T Cell Exhaustion versus Memory. Immunity 2012, 37, 1130–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preston, G.C.; Sinclair, L.V.; Kaskar, A.; Hukelmann, J.L.; Navarro, M.N.; Ferrero, I.; Macdonald, H.R.; Cowling, V.H.; Cantrell, D.A. Single cell tuning of Myc expression by antigen receptor signal strength and interleukin-2 in T lymphocytes. EMBO J. 2015, 34, 2008–2024. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [Green Version]
- Faude, S.; Wei, J.; Muralidharan, K.; Xu, X.; Wertheim, G.; Paessler, M.; Bhoj, V.G.; Grupp, S.A.; Maude, S.L.; Rheingold, S.R.; et al. Absolute lymphocyte count proliferation kinetics after CAR T-cell infusion impact response and relapse. Blood Adv. 2021, 5, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, O.; Jacoby, E.; Nissani, A.; Levi, M.; Nagler, A.; Kubi, A.; Brezinger, K.; Brayer, H.; Zeltzer, L.-A.; Rozenbaum, M.; et al. Head-to-head comparison of in-house produced CD19 CAR-T cell in ALL and NHL patients. J. Immunother. Cancer 2020, 8, e000148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Q.; Han, G.; Puebla-Osorio, N.; Ma, M.C.J.; Strati, P.; Chasen, B.; Dai, E.; Dang, M.; Jain, N.; Yang, H.; et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med. 2020, 26, 1878–1887. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Galletti, G.; De Simone, G.; Mazza, E.M.C.; Puccio, S.; Mezzanotte, C.; Bi, T.M.; Davydov, A.N.; Metsger, M.; Scamardella, E.; Alvisi, G.; et al. Two subsets of stem-like CD8+ memory T cell progenitors with distinct fate commitments in humans. Nat. Immunol. 2020, 21, 1552–1562. [Google Scholar] [CrossRef]
- Liu, L.; Bi, E.; Ma, X.; Xiong, W.; Qian, J.; Ye, L.; Su, P.; Wang, Q.; Xiao, L.; Yang, M.; et al. Enhanced CAR-T activity against established tumors by polarizing human T cells to secrete interleukin-9. Nat. Commun. 2020, 11, 5902. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Tan, J.T.; Wherry, E.J.; Konieczny, B.T.; Surh, C.D.; Ahmed, R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 2003, 4, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, A.O.; Pillai, R.N.; Yang, S.; Nasti, T.H.; Akondy, R.S.; Wieland, A.; Sica, G.L.; Yu, K.; Koenig, L.; Patel, N.T.; et al. Prolifera-tion of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 4993–4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legat, A.; Speiser, D.E.; Pircher, H.; Zehn, D.; Fuertes Marraco, S.A. Inhibitory Receptor Expression Depends More Dominantly on Differentiation and Activation than “Exhaustion” of Human CD8 T Cells. Front. Immunol. 2013, 4, 455. [Google Scholar] [CrossRef] [Green Version]
- Schluns, K.S.; Lefrançois, L. Cytokine control of memory T-cell development and survival. Nat. Rev. Immunol. 2003, 3, 269–279. [Google Scholar] [CrossRef]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef]
- Abo, T.; Balch, C.M. A differentiation antigen of human NK and K cells identified by a monoclonal antibody (HNK-1). J. Immunol. 1981, 127, 1024–1029. [Google Scholar]
- Dolfi, D.V.; Mansfield, K.D.; Polley, A.M.; Doyle, S.A.; Freeman, G.J.; Pircher, H.; Schmader, K.E.; Wherry, E.J. Increased T-bet is associated with senescence of influenza virus-specific CD8 T cells in aged humans. J. Leukoc. Biol. 2013, 93, 825–836. [Google Scholar] [CrossRef] [Green Version]
- Brenchley, J.M.; Karandikar, N.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Crotty, L.E.; Casazza, J.P.; Kuruppu, J.; Migueles, S.A.; Connors, M.; et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood 2003, 101, 2711–2720. [Google Scholar] [CrossRef]
- Canale, F.P.; Ramello, M.C.; Nunez, N.; Bossio, S.N.; Piaggio, E.; Gruppi, A.; Rodriguez, E.V.A.; Montes, C.L. CD39 Expression Defines Cell Exhaustion in Tumor-Infiltrating CD8(+) T Cells-Response. Cancer Res. 2018, 78, 5175. [Google Scholar] [CrossRef] [Green Version]
- Shevchenko, I.; Mathes, A.; Groth, C.; Karakhanova, S.; Müller, V.; Utikal, J.; Werner, J.; Bazhin, A.V.; Umansky, V. Enhanced expression of CD39 and CD73 on T cells in the regulation of anti-tumor immune responses. OncoImmunology 2020, 9, 1744946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Usatorre, A.; Carmona, S.J.; Godfroid, C.; Maroun, C.Y.; Labiano, S.; Romero, P. Enhanced Phenotype Definition for Precision Isolation of Precursor Exhausted Tumor-Infiltrating CD8 T Cells. Front. Immunol. 2020, 11, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knaus, H.A.; Berglund, S.; Hackl, H.; Blackford, A.L.; Zeidner, J.; Montiel-Esparza, R.; Mukhopadhyay, R.; Vanura, K.; Blazar, B.R.; Karp, J.E.; et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight 2018, 3, e120974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef] [Green Version]
- Salerno, F.; Guislain, A.; Freen-Van Heeren, J.J.; Nicolet, B.P.; Young, H.A.; Wolkers, M.C. Critical role of post-transcriptional regulation for IFN-gamma in tumor-infiltrating T cells. Oncoimmunology 2019, 8, e1532762. [Google Scholar] [CrossRef] [Green Version]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Ep-igenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Liu, R.; Wang, P.; Yuan, Z.; Yang, J.; Xiong, H.; Zhang, N.; Huang, Q.; Fu, X.; Sun, W.; et al. CD8+CD57+ T cells exhibit distinct features in human non-small cell lung cancer. J. Immunother. Cancer 2020, 8, e000639. [Google Scholar] [CrossRef]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Frey, N.V.; Engels, B.; Barrett, D.M.; Shestova, O.; Ravikumar, P.; Cummins, K.D.; Lee, Y.G.; Pajarillo, R.; Chun, I.; et al. Antigen-independent activation enhances the efficacy of 4-1BB-costimulated CD22 CAR T cells. Nat. Med. 2021, 27, 842–850. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabo-lism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Kohler, M.E.; Chien, C.D.; Sauter, C.T.; Jacoby, E.; Yan, C.; Hu, Y.; Wanhainen, K.; Qin, H.; Fry, T.J. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci. Transl. Med. 2017, 9, eaag1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creelan, B.C.; Wang, C.; Teer, J.K.; Toloza, E.M.; Yao, J.; Kim, S.; Landin, A.M.; Mullinax, J.E.; Saller, J.J.; Saltos, A.N.; et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: A phase 1 trial. Nat. Med. 2021, 27, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; Van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [Green Version]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [Green Version]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef]
- Weber, E.W.; Parker, K.R.; Sotillo, E.; Lynn, R.C.; Anbunathan, H.; Lattin, J.; Good, Z.; Belk, J.A.; Daniel, B.; Klysz, D.; et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 2021, 372, eaba1786. [Google Scholar] [CrossRef]
- Garfall, A.L.; Dancy, E.K.; Cohen, A.D.; Hwang, W.T.; Fraietta, J.A.; Davis, M.M.; Levine, B.L.; Siegel, D.L.; Stadtmauer, E.A.; Vogl, D.T.; et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv. 2019, 3, 2812–2815. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Value |
|---|---|
| Median age, years (range) | 48.01 (27–73) |
| Gender | |
| Male, n (%) | 24 (57%) |
| Female, n (%) | 18 (43%) |
| Diseases | |
| DLBCL n, (%) | 24 (57%) |
| Mantle n, (%) | 3 (7%) |
| Follicular n, (%) | 5 (12%) |
| ALL n, (%) | 5 (12%) |
| CLL n, (%) | 5 (12%) |
| Response | |
| CR n, (%) | 24 (57%) |
| PR n, (%) | 4 (10%) |
| PD n, (%) | 14 (33%) |
| Toxicity (CRS) | |
| No n, (%) | 5 (12%) |
| Grade 1–2 n, (%) | 32 (76) |
| Grade 3–4 n, (%) | 5 (12%) |
| Neurotoxicity | |
| No n, (%) | 22 (53%) |
| Grade 1–2 n, (%) | 12 (27%) |
| Grade 3–4 n, (%) | 8 (20%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beider, K.; Itzhaki, O.; Schachter, J.; Grushchenko-Polaq, A.H.; Voevoda-Dimenshtein, V.; Rosenberg, E.; Ostrovsky, O.; Devillers, O.; Shapira Frommer, R.; Zeltzer, L.-a.; et al. Molecular and Functional Signatures Associated with CAR T Cell Exhaustion and Impaired Clinical Response in Patients with B Cell Malignancies. Cells 2022, 11, 1140. https://doi.org/10.3390/cells11071140
Beider K, Itzhaki O, Schachter J, Grushchenko-Polaq AH, Voevoda-Dimenshtein V, Rosenberg E, Ostrovsky O, Devillers O, Shapira Frommer R, Zeltzer L-a, et al. Molecular and Functional Signatures Associated with CAR T Cell Exhaustion and Impaired Clinical Response in Patients with B Cell Malignancies. Cells. 2022; 11(7):1140. https://doi.org/10.3390/cells11071140
Chicago/Turabian StyleBeider, Katia, Orit Itzhaki, Jacob Schachter, Ania Hava Grushchenko-Polaq, Valeria Voevoda-Dimenshtein, Evgenia Rosenberg, Olga Ostrovsky, Olivia Devillers, Ronnie Shapira Frommer, Li-at Zeltzer, and et al. 2022. "Molecular and Functional Signatures Associated with CAR T Cell Exhaustion and Impaired Clinical Response in Patients with B Cell Malignancies" Cells 11, no. 7: 1140. https://doi.org/10.3390/cells11071140
APA StyleBeider, K., Itzhaki, O., Schachter, J., Grushchenko-Polaq, A. H., Voevoda-Dimenshtein, V., Rosenberg, E., Ostrovsky, O., Devillers, O., Shapira Frommer, R., Zeltzer, L.-a., Toren, A., Jacoby, E., Shimoni, A., Avigdor, A., Nagler, A., & Besser, M. J. (2022). Molecular and Functional Signatures Associated with CAR T Cell Exhaustion and Impaired Clinical Response in Patients with B Cell Malignancies. Cells, 11(7), 1140. https://doi.org/10.3390/cells11071140